

Abstract
Nonequilibrium thermodynamics has shown its applicability in a wide variety of different situations pertaining to fields such as physics, chemistry, biology, and engineering. As successful as it is, however, its current formulation considers only systems close to equilibrium, those satisfying the so-called local equilibrium hypothesis. Here we show that diffusion processes that occur far away from equilibrium can be viewed as at local equilibrium in a space that includes all the relevant variables in addition to the spatial coordinate. In this way, nonequilibrium thermodynamics can be used and the difficulties and ambiguities associated with the lack of a thermodynamic description disappear. We analyze explicitly the inertial effects in diffusion and outline how the main ideas can be applied to other situations.
Concepts of everyday use like energy, heat, and temperature have acquired a precise meaning after the development of thermodynamics. Thermodynamics provides us with the basis for understanding how heat and work are related and with the rules that the macroscopic properties of systems at equilibrium follow (1). Outside equilibrium, most of those rules do not apply and the aforementioned quantities cannot be defined unambiguously. There is, however, a natural extension of thermodynamics to systems away from but close to equilibrium. It is based on the local equilibrium hypothesis, which assumes that a system can be viewed as formed of subsystems where the rules of equilibrium thermodynamics apply. Because of the usual disparity between macroscopic and microscopic scales, most systems fall into this category. This is the case of, for instance, the heat transfer from a flame, the flow through a pipe, or the electrical conduction in a wire. Nonequilibrium thermodynamics then extracts the general features, providing laws such as Fourier's, Fick's, and Ohm's, which do not depend on the detailed microscopic nature of the system (2).
In contrast, there are other situations where the local equilibrium hypothesis does not hold. Many examples are present in the relaxation of glasses and polymers (3–5), in the flow of granular media (6), and in the dynamics of colloids (7). The main characteristic of such systems is the similarity between microscopic and macroscopic scales, which usually involve internal variables with “slow” relaxation times. The so-called inertial effects in diffusion processes are perhaps the simplest and most illustrative example. In this case, the relaxation of the velocity distribution and changes in density occur at the same time scale. Therefore, local equilibrium is never reached. Here we show how nonequilibrium thermodynamics, as already established in the 1960s (2, 8) can be applied to this situation.
Nonequilibrium thermodynamics (2) assumes that the definition of entropy S can be extended to systems close to equilibrium. Therefore, entropy changes are given by the Gibbs equation:
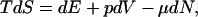 |
1 |
where the thermodynamic extensive variables are the internal energy E, the volume V, and the number of particles N of the system. The intensive variables (temperature T, pressure p, and chemical potential μ) are functions of the extensive variables. Local equilibrium means that the Gibbs equation holds for a small region of the space and for changes in the variables that are actually not infinitely slow. Therefore, the internal state of the system has to relax to equilibrium faster than the variables change. In this way, all variables retain their usual meanings and the functional dependence between intensive and extensive variables is the same as in equilibrium.
Following this approach, nonequilibrium thermodynamics has been applied to study diffusion processes. The simplest case takes place in one dimension at constant temperature, internal energy, and volume. In this case, from Eq. 1 we obtain a Gibbs equation that depends only on the density and the spatial coordinate x:
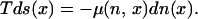 |
2 |
Here s is the entropy per unit volume and n is the density. The chemical potential has the same form as in equilibrium. For instance, for an ideal system, one formed of noninteracting particles, it is proportional to the logarithm of the density plus terms that do not depend on the density (2). Notice that these terms can include thermodynamic variables such as temperature or internal energy, and also the spatial coordinate. In the case of noninteracting Brownian particles, its explicit expression is
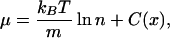 |
3 |
where m is the mass of the particles, kB the Boltzmann constant, and C(x) a function that takes into account possible spatial inhomogeneities. The dynamics of n is restricted by the mass conservation law and therefore follows
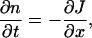 |
4 |
with J being the flux of mass. An additional assumption of nonequilibrium thermodynamics is that this flux is given by
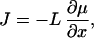 |
5 |
where L is the phenomenological coefficient. From this, we obtain the usual diffusion equation
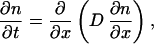 |
6 |
with the diffusion coefficient D ≡ L(∂μ/∂n).
When inertial effects are present, changes in density occur at a time scale comparable with the time the velocities of the particles need to relax to equilibrium. The Gibbs equation as stated in Eq. 2 is no longer valid because local equilibrium is never reached. The entropy production depends also on the particular form of the velocity distribution. Both the spatial coordinate, x, and velocity coordinate, v, are needed to completely specify the state of the system. Therefore, we consider that local quantities are functions of both coordinates. If the system is coupled to other degrees of freedom that relax faster than the velocity and density, a thermodynamic description is still possible. For instance, this is the case of Brownian particles, where the host fluid provides these thermodynamic degrees of freedom. Thus, we consider that diffusion takes place in a two-dimensional space (x, v) instead of in the original one-dimensional space (x). In this case, the chemical potential for an ideal system (e.g., noninteracting Brownian particles) is given by
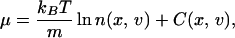 |
7 |
where C(x, v) is a function that does not depend on the density (2). The form of this function can be obtained by realizing that at equilibrium the chemical potential is equal to an arbitrary constant. We can set this constant so that
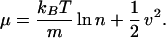 |
8 |
Therefore, the Gibbs equation is now
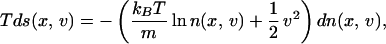 |
9 |
The idea of applying the rules of thermodynamics in an internal space was already proposed by Prigogine and Mazur (9) and has been used in several situations (2, 10). In all of them, however, there was no thermodynamic coupling of these internal degrees of freedom with the spatial coordinate. This is precisely the situation we are considering here.
In the (x, v)-space, the mass conservation law is
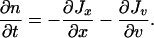 |
10 |
Following the standard thermodynamic approach, the flux of mass is given by
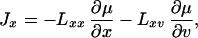 |
11 |
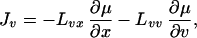 |
12 |
where Lij, with i, j =
{x, v}, are the phenomenological coefficients. There are some
restrictions on the values that Lij can take.
Because the system is at local equilibrium in the
(x, v)-space, Onsager relations imply that
Lxv = −Lvx. In addition, the
flux of mass in real space, J̃x(x) ≡
∫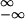 vn(x, v)dv, has to be recovered from
the flux in the (x, v)-space by contracting the velocity
coordinate: J̃x(x) =
∫
vn(x, v)dv, has to be recovered from
the flux in the (x, v)-space by contracting the velocity
coordinate: J̃x(x) =
∫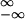 Jx(x, v)dv. Therefore,
Jx(x, v)dv. Therefore,
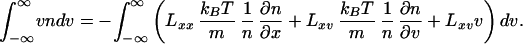 |
13 |
Because n(x, v) can take any arbitrary form, the last equality holds if and only if Lxx = 0 and Lxv = −n. Thus, the only undetermined coefficient is Lvv, which can depend explicitly on n, x, and v.
Previous equations can be rewritten in a more familiar form by identifying the phenomenological coefficients with macroscopic quantities. In this way, with Lvv = n/τ, the fluxes read
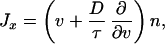 |
14 |
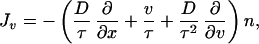 |
15 |
where D ≡ (kBT/m)τ and τ are the diffusion coefficient and the velocity relaxation time, respectively. The equation for the density is given by
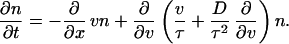 |
16 |
This kinetic equation is equivalent to the Fokker–Planck equation for a Brownian particle with inertia because, in an ideal system, the density is proportional to the probability density, i.e., n(x, v) = mNP(x, v), where P(x, v) is the probability density for a particle to be at x with velocity v, and N is the number of particles of the system. The resulting Fokker–Planck equation could have also been derived by following standard techniques of stochastic processes (11) or kinetic theory (12), which are among the microscopic statistical theories for studying nonequilibrium phenomena.
The approach we have followed, however, explicitly illustrates how thermodynamic concepts can be transferred from equilibrium, through local equilibrium, to far from equilibrium situations. The condition of equilibrium is characterized by the absence of dissipative fluxes (Jx = 0 and Jv = 0). Therefore, from Eq. 14 we obtain that the velocity distribution is Gaussian with variance proportional to the temperature. If deviations from equilibrium are small (Jx ≠ 0 and Jv = 0), the local equilibrium hypothesis holds. This is the domain of validity of Fick's law,
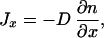 |
17 |
which is obtained directly from the equations for the fluxes. In this case, the distribution of velocities is still Gaussian but now centered at a non-zero average velocity and the variance of the distribution is related to the temperature in the same way as in equilibrium. Beyond local equilibrium (Jx ≠ 0 and Jv ≠ 0), the velocity distribution can take any arbitrary form, from which there is no clear way to assign a temperature. There is, however, a well defined temperature T: that of local equilibrium in the (x, v)-space.
In Fig. 1, we illustrate the concepts discussed previously. We show the velocity profiles obtained from Eq. 16 for two representative situations. For fast relaxation of the velocity coordinate, the velocity distribution is Gaussian and centered slightly away from zero, in accordance with local equilibrium concepts. For slow relaxation, however, the velocity distribution loses its symmetry (and its Gaussian form). In this case, the temperature does not give directly the form of the distribution and one has to resort to local equilibrium in the (x, v)-space to describe the system.
Figure 1.

Velocity profiles obtained from Eq. 16 when a density gradient is applied. The solution has been obtained through a standard numerical algorithm following a first order upwind discretization scheme (16). The system is in a rectangular domain in the (x, v)-space, from x = 0 to x = 1, and from v = −10 to v = 10. The lower and upper dashed curves in the figure represent the boundary conditions applied at x = 0 and x = 1, respectively: n(1, v) = 10n(0, v) = (2π)−0.5 exp(−v2/2). Filled circles correspond to velocity profiles at x = 0.5 for fast relaxation of the velocity coordinate (τ = 0.1), whereas open circles correspond to slow relaxation (τ = 10). In both cases, D/τ ≡ kBT/m = 1. All values are given in arbitrary units.
It is important to emphasize that the temperature T is the one that enters the total entropy changes and therefore the one related to the second principle of thermodynamics. Other definitions of temperature are possible though. To illustrate this point, let us compute the entropy production σ. This quantity is obtained from local changes in entropy, which are given not only by the production but also by the flow:
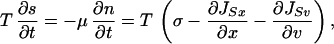 |
18 |
where (JSx, JSv) is the entropy flux. In our case, the expression for the entropy production is
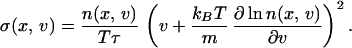 |
19 |
Now, given a Gaussian velocity distribution n(x, v) = n0(x)e−mv2/2kBT̃(x), we can easily understand the meaning of the temperature T̃(x) defined through the variance of the distribution: it is the temperature at which the system would be at equilibrium (σ = 0). The definition of an effective temperature as that giving zero entropy production can be extended to arbitrary velocity distributions. From Eq. 19, we obtain
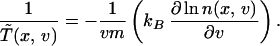 |
20 |
The temperature defined in this way is formally analogous to the equilibrium temperature because the right-hand side term of the preceding equation can be rewritten as the derivative of an entropy with respect to an energy:
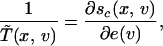 |
21 |
where sc(x, v) = −(kB/m) ln n(x, v) and e(v) = ½v2. The term sc(x, v) and e(v) can be viewed as the configurational entropy and the kinetic energy per unit of mass, respectively. In general, other definitions of effective temperature are possible. For instance, by considering e(v − v̄(x)) instead of e(v) in Eq. 21, the resulting temperature would be that of local equilibrium. In this case, however, this temperature does not give zero entropy production but just that of the macroscopic motion. This temperature is then the one at which, once the macroscopic motion is disregarded, the internal configuration of the system would be at equilibrium.
In general, because T̃(x, v) is a function not only of x but also of v, given a point in space, there is no temperature at which the system would be at equilibrium, i.e., T̃(x, v) ≠ T̃(x). If an effective temperature at a point x were defined, it would depend on the way the additional coordinate is eliminated. Thus, ambiguities in far-from-equilibrium quantities arise when considering a lower-dimensional space than the one in which the process is actually occurring. This is to some extent similar to what happens with effective temperatures defined through fluctuation–dissipation theorems. In such a case, the effective temperature can depend on the scale of observation (13). It is interesting to point out that all of these effective temperatures, despite their possible analogies with the equilibrium temperature, do not have to follow the usual thermodynamic rules because the system is not actually at equilibrium at the temperature T̃.
The idea of increasing the dimensionality of the space were diffusion takes place, so to include as many dimensions as nonequilibrated degrees of freedom the system has, can also be applied to other situations. In a general case, the additional degrees of freedom do not necessarily correspond to the velocity. For instance, let us consider a degree of freedom Θ(x) that at local equilibrium enters the Gibbs equation in the following way:
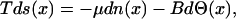 |
22 |
where B ≡ B(n, Θ, T) = T(∂s/∂Θ)n,T. In this case, one usually assumes that given T, n(x), and Θ(x), the function B is completely determined through the equilibrium properties of the system. Far away from equilibrium, we would have to consider explicitly an additional coordinate θ, which is related to the degree of freedom by Θ(x) = ∫θn(x, θ)dθ. The corresponding Gibbs equation
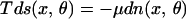 |
23 |
would have to take into account the dependence on the coordinate θ through the chemical potential μ. Once the Gibbs equation has been obtained, the way to proceed would be analogous to the one we followed for the inertial effects. For instance, some systems with both translational and orientational degrees of freedom can be described by the chemical potential
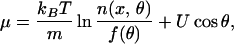 |
24 |
where θ is now an angular coordinate, U cos θ is the orientational energy, and f(θ) is a function accounting for the degeneracy of the orientational states [for rotation in three and two dimensions, f(θ) = sin θ and f(θ) = 1, respectively] (2). This type of systems include, among others, liquid crystals and suspensions of rod-like particles (5), field-responsive suspensions (14), and polarized systems (2). At local equilibrium, some instances of B and Θ are then electric field and polarization, and magnetic field and magnetization. Beyond local equilibrium, by writing the (x, θ) counterpart of Eqs. 10, 11, and 12, one can obtain a kinetic equation that describes the behavior of the system. This equation includes as particular cases the Fokker–Plank equations obtained for those systems by means of microscopic theories (5, 15).
In this paper, we have been assuming ideality and locality. The condition of ideality is that the system consists of noninteracting particles. In this case, the chemical potential is proportional to the logarithm of the density plus terms that do not depend on this quantity. Nonideality can be directly taken into account by considering the right dependence of the thermodynamic quantities on the density and, in general, will give rise to nonlinear partial differential equations. A more difficult aspect to deal with is the presence of nonlocal effects. In such a case the interactions between the different parts of the system will need of integro-differential equations to be incorporated in the description.
The main result of our analysis shows that, in far-from-equilibrium diffusion processes, local equilibrium can be recovered when all of the relevant degrees of freedom are considered at the same level as the spatial coordinate. In the resulting extended space, thermodynamic quantities, such as temperature and the chemical potential, admit a well defined interpretation. The scheme we have developed may then provide the basis for a consistent formulation of thermodynamics far from equilibrium.
Acknowledgments
J.M.R. was supported by DGICYT (Spain) Grant No. PB98-1258. J.M.G.V. is an associate of the Howard Hughes Medical Institute.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Reiss H. Methods of Thermodynamics. New York: Dover; 1997. [Google Scholar]
- 2.de Groot S R, Mazur P. Non-equilibrium Thermodynamics. New York: Dover; 1984. [Google Scholar]
- 3.Götze W Les Houches 1989, editors. In: Liquids, Freezing, and Glass Transition. Hansen J P, Levesque D, Zinn-Justin J, editors. Amsterdam: North–Holland; 1991. [Google Scholar]
- 4.Rubí J M, Pérez-Vicente C, editors. Lecture Notes in Physics. Heidelberg: Springer; 1997. Complex Behaviour of Glassy Systems. [Google Scholar]
- 5.Doi M, Edwards S F. The Theory of Polymer Dynamics. Oxford: Oxford Univ. Press; 1986. [Google Scholar]
- 6.Jaeger H M, Nagel S R, Behringer R P. Rev Mod Phys. 1996;68:1259–1273. [Google Scholar]
- 7.Larson R G. The Structure and Rheology of Complex Fluids. New York: Oxford Univ. Press; 1999. [Google Scholar]
- 8.Prigogine I. Introduction to Thermodynamics of Irreversible Processes. New York: Wiley; 1955. [Google Scholar]
- 9.Prigogine I, Mazur P. Physica. 1953;XIX:241–256. [Google Scholar]
- 10.Pérez-Madrid A, Rubí J M, Mazur P. Physica A. 1994;212:231–238. [Google Scholar]
- 11.van Kampen N G. Stochastic Processes in Physics and Chemistry. Amsterdam: North–Holland; 1992. [Google Scholar]
- 12.McLennan J A. Introduction to Nonequilibrium Statistical Mechanics. Englewood Cliffs, NJ: Prentice–Hall; 1989. [Google Scholar]
- 13.Cugliandolo L F, Kurchan J, Peliti L. Phys Rev E. 1997;55:3898–3914. [Google Scholar]
- 14.Rubí J M, Vilar J M G. J Phys Condens Matter. 2000;12:A75–A84. [Google Scholar]
- 15.Berne B J, Pecora R. Dynamic Light Scattering: With Applications to Chemistry, Biology, and Physics. New York: Wiley; 1977. [Google Scholar]
- 16.Ames W F. Numerical Methods for Partial Differential Equations. New York: Academic; 1997. [Google Scholar]