

Abstract
Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV8) displays two life modes, latency and lytic reactivation in the infected host cells which are equally important for virus mediated pathogenesis. During latency only a small number of genes are expressed. Under specific conditions, KSHV can undergo lytic replication with the production of viral progeny. One immediate-early gene RTA, encoded by open reading frame 50 of KSHV, has been shown to play a critical role in switching the viral latency to lytic reactivation. Over-expression of RTA from a heterologous promoter is sufficient for driving KSHV lytic replication which results in production of viral progeny. In the present study, we show that RTA can activate the expression of the ORF59 which encodes the processivity factor essential for DNA replication during lytic reactivation. We also show that RTA regulates ORF59 promoter through interaction with RBP-Jκ as well as a cis-acting RTA responsive element within the promoter. In the context of KSHV infected cells, the upregulation of ORF59 is a direct response to RTA expression. Taken together, our findings provide new evidence to explain the mechanism by which RTA can regulate its downstream gene ORF59, further increasing our understanding of the biology of KSHV lytic replication.
Keywords: KSHV, RTA, ORF59
Introduction
Kaposi’s sarcoma (KS)-associated herpesvirus (KSHV), also known as human herpesvirus 8, was discovered in a KS lesion from an AIDS patient in 1994 (Chang et al., 1994). Since then, extensive epidemio-logic studies with molecular and serologic assays have provided convincing data to support the idea that KSHV is the etiological agent of KS, including classic, endemic, iatrogenic and AIDS associated KS (Buonaguro et al., 1996; Chang and Moore, 1996). KSHV has also been associated with several other lymphoproliferative diseases including primary effusion lymphoma (or body cavity-associated lymphoma) (Cesarman et al., 1995) and a subset of Multicentric Castleman’s disease (MCD) (Soulier et al., 1995). KSHV has a DNA genome of 165 kb that encodes up to 90 viral genes (Moore et al., 1996; Russo et al., 1996). Based on sequence homology KSHV has been classified in the gamma 2 herpesvirus subfamily, rhadinovirus genus, closely related to rhesus monkey rhadinovirus (RRV), murine herpesvirus 68 (MHV-68) and herpesvirus saimiri (HVS) (Albrecht et al., 1992; Alexander et al., 2000; Virgin et al., 1997).
As a member of the γ-herpesviruses family, KSHV displays two distinct phases in its life cycle, known as latency and lytic reactivation. During latency, viral gene expression is highly controlled in which a limited number of viral genes including LANA, vFLIP, vCyclin, kaposin and K15 are expressed (Dittmer et al., 1998; Sharp et al., 2002). Upon lytic induction such as hypoxia, most of the viral genes are sequentially expressed and the virus undergoes lytic reactivation (Davis et al., 2001; Haque et al., 2003). The mechanisms that control the switch between viral latency and lytic reactivation have been extensively studied (Deng et al., 2007; Lan et al., 2004; Liang et al., 2002; Lukac et al., 1999). The replication and transcription activator (RTA) encoded by ORF50, the homolog of the BRLF1 gene of Epstein–Barr virus, has been shown to play a critical role in inducing the viral lytic cycle. RTA functions as an essential and sufficient lytic switch transcription factor in latently infected cells (Lukac et al., 1998; West and Wood, 2003; Xu et al., 2005). Upon treatment with tetradecanoyl phorbol acetate (TPA) or sodium butyrate, a lytic cascade of viral gene expression is activated, resulting in viral genomic replication, virion production, and cell lysis (Renne et al., 1996; Yu et al., 1999). RTA can be detected as early as 1 h after TPA induction, and its synthesis is resistant to inhibition by cycloheximide (Lukac et al., 1999). Similarly, ectopic expression of RTA also can selectively activate the expression of downstream delayed-early or late genes, leading to the successful production of infectious virions (Gradoville et al., 2000; Lukac et al., 1998). However, deletion of the ORF50 gene from the genome (Xu et al., 2005) or inhibition by a dominant negative mutant (Lukac et al., 1999) will prevent lytic replication. Thus, RTA is critical for initiation of the latent-lytic switch transition of KSHV.
Identification of the viral genes that RTA activates and characterization of the mechanisms mediating these activations is an important step in understanding the virus life cycle and pathogenesis. Using transient transfection of promoter/reporter constructs along with RTA expression constructs, a panel of downstream genes has been identified and characterized, such as ORF57, K8, vIL-6, Kaposin and poly-adenylated nuclear (PAN) RNA (Chang et al., 2002; Deng et al., 2002; Duan et al., 2001; Lukac et al., 2001; Song et al., 2001). RTA activates its target genes through either directly binding with high affinity to its own response element or in combination with other cellular factors, such as RBP-Jκ (LANA), C/EBP-α (PAN) and Ap1 (K8) (Lan et al., 2005a, b; Liang et al., 2002; Wang et al., 2003a,b). Given the large number of RTA target genes, additional cellular partners and alternative binding sites for RTA may have a role in the reactivation process.
KSHV encodes eight proteins that participate in origin-dependent DNA replication. They include DNA polymerase (Pol-8, ORF9), processivity factor (PF-8, ORF59), helicase (HEL, ORF44), primase (PRI, ORF56), primase-associated factor (ORF40/41), single-strand DNA binding protein (SSB, ORF6), RTA (ORF 50), and replication-associated protein (RAP, K8) (AuCoin et al., 2004; Chen et al., 2005; Wu et al., 2001). Of particular interest is ORF59, which was first identified by monoclonal antibody (mAb) 11D1 generated against body cavity-based B cell lymphoma cell line (BCBL-1) harboring latent KSHV episome (Chan et al., 1998). It is a delayed-early protein which localizes to KSHV-infected cell nuclei and whose expression is induced by TPA treatment (Chan et al.,1998). It has been shown that PF-8 encoded by ORF59 interacts with KSHV DNA polymerase (Pol-8, encoded by ORF9) in vitro and stimulates DNA synthesis activity of Pol-8 on singly primed M13 template in DNA polymerase assays (Chan and Chandran, 2000; Lin et al., 1998). It was also observed that in DNA polymerase assays Pol-8 alone incorporates only three nucleotides, whereas when combined with ORF59 it can incorporate thousands of nucleotides. This suggests ORF59 encoded PF-8 is a processivity factor for KSHV Pol-8 (Lin et al., 1998). Compared to the amino acid sequences of other known herpesvirus processivity factors, PF-8 displays the greatest homology to ORF59 (31.5% identity) of HVS and to BMRF1 (28.5% identity) of EBV (Lin et al., 1998).
In this study, we show that RTA can activate ORF59 promoter in a dose-dependent manner. We also show that RTA regulates ORF59 promoter through interaction with RBP-Jκ and a cis-acting RTA responsive element within the promoter. In the context of KSHV infected cells, the ORF59 promoter is responsive to RTA expression. Taken together, this study provides new clues as to the mechanisms by which RTA activates the processivity factor ORF59 important for lytic replication of KSHV.
Results
RTA activates ORF59 promoter in a dose-dependent manner
Previous studies showed that ORF59 was not detected earlier than RTA protein upon TPA induction and cannot be detected when RTA was disrupted from the genome. To explore if RTA operates as a transcriptional modulator of the ORF59 promoter encoded by KSHV, we cloned the ORF59 upstream promoter sequence (1909 bp) into the pGL3-enhancer reporter vector, which has the coding sequence for firefly luciferase. This reporter construct was then transfected into human embryonic kidney (HEK) 293T cells or DG75 cells (a KSHV-negative lymphoma cell line) along with increasing amounts of pCR3.1-RTA expression construct. Also, pRL-Tk, a reporter plasmid encoding the Rellina luciferase under the control of a constitutively active herpes simplex virus thymidine kinase promoter, was cotransfected to normalize transfection efficiencies. Fold activation was calculated by comparing the normalized firefly luciferase activity of pCR3.1-RTA-transfected cells to that of the vector control-transfected cells. Cotransfection of the RTA expression vector with the pGL3-59pFL reporter construct consistently resulted in activation relative to reporter construct alone (Fig. 1). Moreover, increasing the amount of the RTA expression construct resulted in activation of the ORF59 promoter in a dose-dependent manner (Fig. 1). These data indicate that RTA activates the ORF59 promoter and the activation of ORF59 promoter was directly proportional to the quantity of RTA expressed as demonstrated by Western blot (Fig. 1).
Fig. 1.
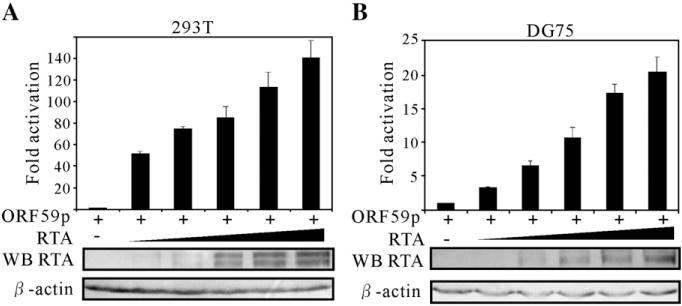
Transcriptional activation of RTA on the ORF59 promoters in a dose-dependent manner. The reporter plasmid pGL3-ORF59pFL contains a 1909-bp sequence upstream of the start codon of ORF59 that drives the expression of firefly luciferase. A fixed amount (1 μg) of the reporter plasmids was cotransfected into 10 million 293T (A) and DG75 (B) cells with 0, 0.5, 1, 2.5, 5 and 10 μg of pCR3.1-RTA and 10, 9.5, 9, 7.5, 5 and 0 μg of pCR3.1 control vector. 50 ng pRL-Tk was also included in each transfection, and served as an internal control for transfection efficiency. 24 h posttransfection, cell lysate of each transfection was assayed with the dual-luciferase system. Firefly luciferase activities were normalized to the corresponding Renilla luciferase activities. Fold activation by RTA was calculated by comparing the normalized firefly luciferase activity stimulated by pCR3.1-RTA to that stimulated by pCR3.1. The means and standard deviations from three independent transfections are shown.
The RBP-Jκ binding sites within the ORF59 promoter are important for RTA-mediated transactivation
The experiments above showed that RTA up-regulates ORF59 promoter in a dose-dependent manner. To elucidate the potential mechanism of the regulation by RTA, we analyzed the ORF59 promoter by scanning the full length promoter. A number of potential transcriptional factor binding sites were predicted. Interestingly, there are 3 RBP-Jκ potential binding sites (designated Jκ1, Jκ2 and Jκ3), located at −1477, −932 and −871 positions respectively. As shown previously, interaction between RTA and RBP-Jκ is critical for viral lytic reactivation (Liang et al., 2002). Additionally, RTA can activate its downstream genes such as ORF57 and LANA through interactions with RBP-Jκ (Lan et al., 2005a,b; Liang et al., 2002). Based on these findings, it is possible that RBP-Jκ may also play a role in regulation of the ORF59 promoter. To test this hypothesis, we constructed three truncated versions of the ORF59 promoters in which RBP-Jκ binding sites were removed sequentially until all sites deleted to determine the contribution of each site in the regulation of ORF59 expression. In a luciferase assay, equivalent amounts pGL3-ORF59pFL or pGL3-ORF59pmt were transfected into 293T or DG75 cells, in combination with a fixed amount of the RTA expression construct. pRL-Tk was also included in each transfection and served as an internal control for transfection efficiency. As we expected, deletion of the first potential RBP-Jκ recognition site (Jκ1) caused an obvious reduction of RTA-mediated transactivation of ORF59 promoter compared to the levels of wild type promoter in both 293T and DG75 cells (Figs. 2A and B, lane 2). The average reduction of fold activation was about 30%. Additionally, the more potential RBP-Jκ recognition sites were deleted, the more luciferase activity was diminished. As shown, deletion of all 3 potential RBP-Jκ binding sites led to the maximum reduction of RTA-mediated transactivation by over 50% in these cell lines (Figs. 2A and B, lane 4). Taken together, these data clearly indicate that RBP-Jκ plays a role in the RTA-mediated transactivation of ORF59 promoter.
Fig. 2.
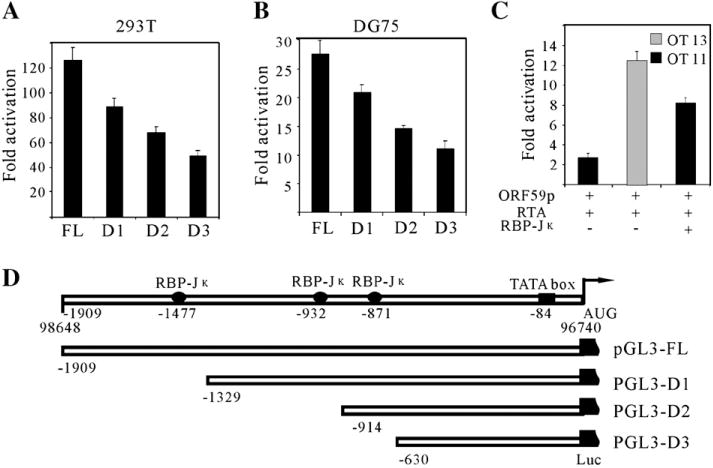
RBP-Jκ binding sites within ORF59 promoter are essential for regulation by RTA. A series of truncated promoters were cloned into a luciferase reporter plasmid pGL3-enhancer (D). Fixed amount (1 μg) of reporter plasmids were cotransfected into 293T (A) and DG75 (B) cells with either an RTA expression plasmid pCR3.1-RTA (5 μg) or pCR3.1 (5 μg). (C) ORF59 full length promoter reporter plasmid (1 μg) was transfected into a RBP-Jκ knockout cell line (OT11) and a wild type cell line (OT13) with 5 μg pCR3.1-RTA expression vector; an extra rescue experiment was carried out by introducing 5 μg of RBP-Jκ expression vector into OT11. To normalize the total amount of DNA, pcDNA 3.1 control vector was used. All the cells were harvested at 24 h posttransfection, dual-luciferase assays and fold activations were performed as described above. The means and standard deviations from three independent transfections are shown. (D) Schematic diagram of serial deletions of potential RBP-Jκ binding sites in the ORF59 promoter. Potential RBP-Jκ binding sites and TATA box are shown with the numbers referring to the nucleotide position upstream of the translation initiation site. The mapped translation initiation site is shown by the arrow.
To corroborate the results and confirm the role of RBP-Jκ in ORF59 regulation, we performed the reporter assays in a RBP-Jκ knockout cell line OT11 along with its isogenic wild type counterpart OT13 cells. OT11 and OT13 were transfected with equivalent amounts of pGL3-59pFL and RTA expression vector, respectively. 24 h post-transfection, cells were harvested for analysis using a Dual-Luciferase™ reporter system. The results showed that the activation level of the ORF59 promoter by RTA in RBP-Jκ−/− OT11 was much lower than that in RBP-Jκ+/+ OT13 (Fig. 2C, lanes 1 and 2). To further support this observation, we included an RBP-Jκ rescue expression construct into our analysis. The results showed that when RBP-Jκ was provided exogenously from a heterologous promoter into the system that approximately 75% of the RTA response was rescued (Fig. 2C, lane 3). This suggests that RTA cannot activate the ORF59 promoter in the absence of RBP-Jκ as efficiently as in the presence of RBP-Jκ and confirms that RBP-Jκ plays an essential role in regulation of ORF59 promoter by RTA.
RBP-Jκ binds to its cognate sequences within ORF59 promoter
The results of the luciferase reporter assays above indicated that the presence of the RBP-Jκ binding sites within the ORF59 promoter is essential for RTA to activate this promoter. To address whether RBP-Jκ could bind to the potential cis-acting DNA element in vitro, EMSAs were performed. We designed 3 double-stranded oligonucleotides (named Jκ 1, Jκ 2 and Jκ 3) containing RBP-Jκ consensus binding sequence as well as the flanking sequences within the ORF59 promoter. Nuclear extracts, from RBP-Jκ expression vector transfected 293T cells, were incubated with 32P-end-labeled probes and the binding mixtures were resolved on a native polyacrylamide gel. As shown, a specific RBP-Jκ shift was observed (Fig. 3A, lanes 3, 9 and 15). The specificity of these bands was demonstrated by its disappearance in the presence of 50-fold specific cold competitor probe (Fig. 3A, lanes 4, 10 and 16), while the nonspecific competitor did not (Fig. 3A, lanes 5, 11 and 17). The specificity of these shifts was further verified by their supershifts in the presence of specific antibody (Fig. 3A, lanes 6, 12, 18). These probes also can form a complex with in vitro translated RBP-Jκ protein (Fig. 3B). A similar but robust specific RBP-Jκ shift was observed when in vitro translated recombinant Myc-RBP-Jκ protein used (Fig. 3B, lanes 2, 5 and 8). The binding complex was supershifted when supplemented with a monoclonal anti-Myc-tag antibody (Fig. 3B, lanes 3, 6 and 9). In addition, endogenous RBP-Jκ from OT13 cells also can form a similar shift (Fig. 3B, lanes 11, 13 and 15); however there is no obvious shift formed using the nuclear extracts from OT11 without RBP-Jκ (Fig. 3B, lanes 10, 12 and 14). These results suggest that RBP-Jκ can bind to its consensus binding sites within the ORF59 promoter, mediating RTA transactivation.
Fig. 3.
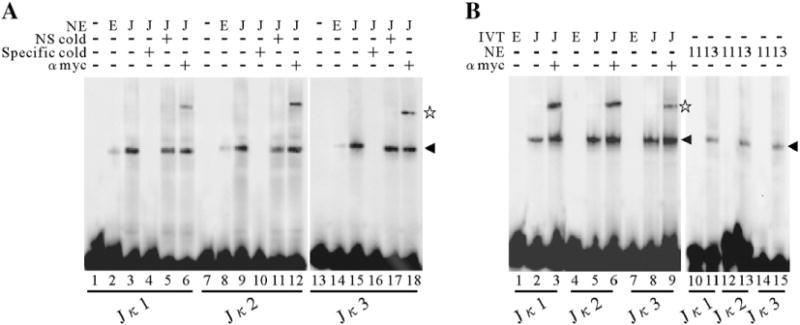
RBP-Jκ binding to its cognate sequences within ORF59 promoter in vitro. (A) EMSA of the 3 putative RBP-Jκ binding sites in the ORF59 promoter with nuclear extracts (NE). Nuclear extracts (100 ng) containing RBP-Jκ protein were incubated with labeled double-stranded oligonucleotides, Jκ1 (lanes 3 to 6), Jκ2 (lanes 9 to 12) and Jκ3 (lanes 15 to 18), respectively, and the binding mixtures were then resolved on a native polyacrylamide gel. A 50-fold excess of unlabeled specific competitors (Jκ1 in lane 4; Jκ2 in lane 10 and Jκ3 in lane 16) and a nonspecific competitor (Jκ1 in lane 5; Jκ2 in lane 11 and Jκ3 in lane 17) was incubated in the presence of RBP-Jκ for the competition assay. (B) EMSA of Jκ1 (lanes 1 to 3), Jκ2 (lanes 4 to 6) and Jκ3 (lanes 7 to 9) with in vitro translated recombinant Myc-RBP-Jκ protein. End-labeled oligonucleotides were incubated with in vitro translated Myc-tagged RBP-Jκ protein (50 ng) and resolved on a native polyacrylamide gel. The DNA–protein complexes were supershifted by a monoclonal anti-Myc-tag antibody (lanes 3, 6 and 9). EMSA of Jκ1 (lanes 10, 11), Jκ2 (lanes 12, 13) and Jκ3 (lanes 14, 15) with nuclear extracts from OT11 or OT13 cells were also performed. The arrowhead denotes the specific binding of RBP-Jκ to DNA and the pentagram denotes the supershift. NE, nuclear extracts; IVT, in vitro translated; NS, nonspecific; E, empty vector; J, RBP-Jκ expression vector; 11, OT11; 13, OT13.
A cis-acting RTA responsive element (RRE) is identified within the ORF59 promoter
The experiments above suggest that interaction between RTA and RBP-Jκ plays an essential role in regulation of ORF59 by RTA. However, when RBP-Jκ binding sites were sequentially removed and more than two thirds of ORF59 promoter was deleted, the activation level of ORF59 promoter by RTA was not completely shut down, indicating that there could be other potential elements critical for RTA regulation. To identify the potential elements within the remaining part that contributes to ORF59 promoter activity, a panel of truncations was generated. These constructs contain 5′ progressive deletions from −630 upstream of the ORF59 promoter. These pGL3-ORF59p reporter plasmids were cotransfected into DG75 cells either with pCR3.1-RTA or pCR3.1, in combination with equal amounts of pRL-Tk. The sequential deletion of 5′ promoter sequences to −212, pGL3-59pD4, D5, and D6 resulted in fold activation levels that were largely the same, fluctuating on average between 12 and 14-fold (Fig. 4B). Interestingly, the response of pGL3-59pD7 to ORF50 protein (Fig. 4B, lane 4) was even higher in terms of luciferase activity when compared with that of pGL3-59pD3, indicating there could be other cellular transcriptional factors acting on RTA regulation. However, when the deletion was up to −110, the responsiveness of pGL3-59pD8 to RTA was sharply decreased and the fold activation was reduced dramatically from 18.6 to 1.7-fold (Figs. 4A and B, lane 5). The results in transformed human embryonic kidney cell line 293T closely resembled that seen in DG75 lymphoma cells. The remarkable drop in fold activation suggests that this activation is highly dependent on the region between position −212 and −110 from the start site. To further narrow down the region, we designed another reporter construct pGL3-59pD9, with a further 5′ deletion between −212 and −159 in pGL3-59pD7 promoter, was able to confer a very higher, near wild type, response to RTA protein (Figs. 4A and B, lane 6). These data indicate that the 50 bp region between −159 and −110 is very critical for RTA-mediated transactivation. Thus, we hypothesized that it represents the most likely location for RTA response elements (RRE), and named it 59pRRE.
Fig. 4.
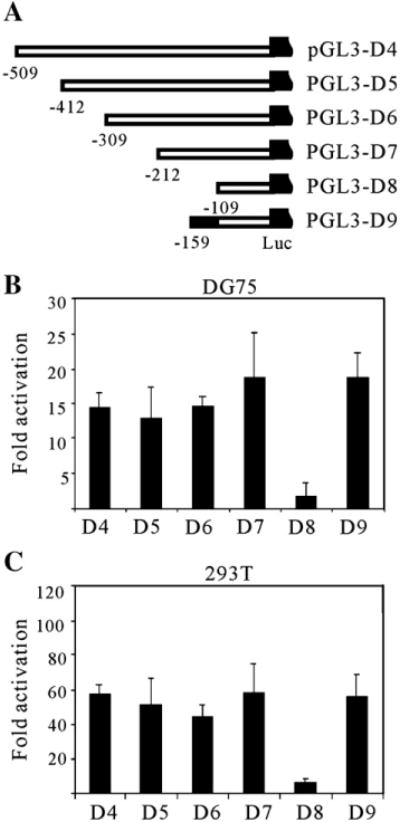
Mapping of the minimal responsive element in ORF59 promoter. (A) Scheme showing a series of truncated promoters (D4 to D9) were cloned into a luciferase reporter plasmid, pGL3-enhancer. Serial deletions were made on the 630-bp fragment from the 5′ end, with the number indicating the nucleotide position upstream of the AUG codon. Fixed amount (1 μg) of reporter plasmids were cotransfected into DG75 (B) and 293T (C) cells with either an Rta expression plasmid pCR3.1-RTA (5 μg) or pCR3.1 (5 μg). All the cells were harvested at 24 h posttransfection, dual-luciferase assays and fold activations were performed as described in Fig. 1. The means and standard deviations from three independent transfections are shown.
The 59pRRE confers RTA responsiveness to a heterologous promoter
Previous studies showed that the DNA element, RRE, found in promoters of KSHV genes, such as PAN RNA, vIL-6 and ORF57, could be transferred to a heterologous promoter, conferring activation in the context of minimal promoter (Chang et al., 2002; Deng et al., 2002; Lukac et al., 2001). To determine whether the 59pRRE contained between −159 and −110 of the ORF59 promoter, we first constructed a reporter plasmid, Hsp70-Luc, by fusing the well-characterized TATA box of the cellular gene hsp70 to pGL3-basic as previously reported (Lukac et al., 2001). Double-stranded oligonucleotides corresponding to the identified 50-bp RRE sequence were then cloned in both orientations into Hsp70-Luc promoter (Fig. 5A). The resulting constructs, 59pRRE-F-Hsp and 59pRRE-R-Hsp, were then cotransfected with increasing amounts of RTA expression plasmids. RTA activated the 59pRRE-F-Hsp and 59pRRE-R-Hsp reporter constructs in a dose-dependent manner in 293T cells, the maximal activities are 51.3- and 42.5-fold respectively. Little or no difference was observed between 59pRRE-F-Hsp and 59pRRE-R-Hsp, suggesting that this activity is orientation-independent. In contrast, the backbone Hsp70-Luc was not be activated by RTA, and resulted in only a weak response (Fig. 5B). These results indicate that the RRE mapped from deletion analysis is sufficient to confer RTA responsiveness to a heterologous promoter, acting in an enhancer-like fashion.
Fig. 5.
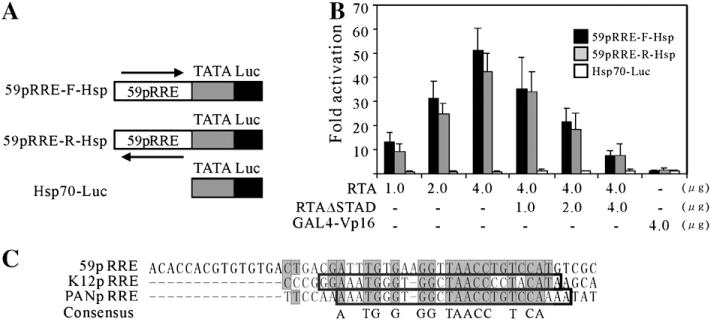
The 59pRRE confers RTA responsiveness to a heterologous promoter and possesses conservation with PAN RNA and K12 RRE. (A) 59pRRE was fused to the hsp70 TATA box (Taylor and Kingston, 1990) in forward and reverse orientations and then cloned into vector pGL3-basic. (B) The resulting plasmids, 59pRR-F-Hsp and 59pRRE-R-Hsp, and backbone alone were tested for RTA activation by reporter assays. Each was cotransfected into 293T cells with increasing amounts of ORF50 expression vector or combined with increasing amounts of the dominant negative mutant ORF50ΔSTAD as indicated. To determine the specificity of RTA activation, GAL4-VP16 was also introduced. All the cells were harvested at 24 h posttransfection, dual-luciferase assays and fold activations were performed as described in Fig. 1. The means and standard deviations from three independent transfections are shown. (C) The alignment depicts the conservation between the RREs of ORF59, PAN RNA and K12 promoter. The nucleotide sequences of three RREs were aligned by the Vector NTI Suite, version 9.0. The grey boxes denote the identical residues shared by the ORF59 promoter with either PAN RNA or K12 promoter, and the consensus is shown at the bottom. The opened boxes refer to the minimal RREs of PAN RNA and K12 promoters.
To examine the specificity of RTA activation, the dominant negative mutant, RTAΔSTAD, was introduced into this experiment. RTAΔSTAD, containing a deletion in the C-terminal activation domain, has been demonstrated to specifically inhibit RTA-mediated transactivation and KSHV replication (Bu et al., 2007; Lukac et al., 1999). Indeed, the increasing amount of RTAΔSTAD abrogated the robust activation of RTA, leading to a progressive decrease of responsiveness (Fig. 5B). In addition, GAL4-VP16 (Chang et al., 2002; Sadowski et al., 1988), another potent transcriptional activator, also failed to activate 59pRRE-F-Hsp and 59pRRE-R-Hsp, supporting the specificity of RTA activation of 59pRRE. Taken together, these data suggest that the mapped RRE is necessary and sufficient for the activation of ORF59 promoter by RTA.
The 59pRRE possesses homology in nucleotide sequence with that of PAN RNA and K12
RTA has been shown to regulate and transactivate a number of downstream viral genes through directly binding to RRE within their promoters. A panel of RREs have been identified, such as ORF57, K8, PAN RNA, K12, vIL-6 and RTA itself (Chang et al., 2002; Deng et al., 2002; Lukac et al., 2001; Sakakibara et al., 2001; Song et al., 2001). A comparison of the 50-bp 59pRRE to these published RREs of KSHV revealed that the 59pRRE possesses a great deal of homology with that of PAN RNA and K12 in primary nucleotide sequence. The aligned sequences shown in Fig. 5C demonstrated that 17 of 34-nt is identical, including 11 consecutive identical nucleotides in the 5′ of PANpRRE (Fig. 5C). The three RREs, when compared with each other simultaneously, share 14 conserved nucleotides, including a highly homologous GGNTAACC block (Fig. 5C). Moreover, all the 14 conserved nucleotides identified were located in the 26-bp minimal RTA responsive element of PAN RNA and K12 (Chang et al., 2002). The remarkable homology with other RREs along with the experimental results above indicates 59pRRE is very likely to be a functional RRE in the ORF59 promoter.
RTA directly binds to the cis-acting 59pRRE DNA element in vitro
RTA has been shown to bind directly to the mapped RREs in the promoters of PAN RNA and vIL-6 etc (Chang et al., 2002; Deng et al., 2002; Song et al., 2001). To determine whether RTA activates ORF59 promoter through direct binding to the potential RTA response element (RRE) mentioned above, an electrophoretic mobility shift assay (EMSA) was performed. 32P-end-labeled 59pA, corresponds to the 50-bp 59pRRE sequence mapped by the reporter assays, was incubated with nuclear extracts of 293T cells transfected with RTA expression vector pEF-RTA, and the binding mixtures were then resolved on a native polyacrylamide gel. A strong RTA binding shift was detected (Fig. 6B, lane 3), as indicated by the arrowhead. To confirm the specificity of RTA binding to its cis-acting elements, a competition assay was used. A 50-fold excess of an unlabeled oligonucleotide 59pA or the nonspecific oligonucleotide was incubated with binding mixtures prior to the adding of labeled oligonucleotide 59pA. Introduction of a specific competitor to the binding mixtures significantly impaired the ability of RTA binding to the labeled oligonucleotide 59pA, whereas the nonspecific competitor did not (Fig. 6B, lanes 4 and 5). Shift assay was repeated with the in vitro translated Myc-tagged RTA protein. Consistent with the results above, incubation of 59pA with in vitro translated RTA protein results in a clear shift (Fig. 6B, lane 8). Finally, the DNA–protein complexes were further shifted by monoclonal anti-Myc-tag antibody (Fig. 6B, lanes 6 and 9), as indicated by the pentagram. These results indicate that RTA directly binds to 59pRRE within the 50-bp oligonucleotide 59pA in a sequence-specific manner.
Fig. 6.
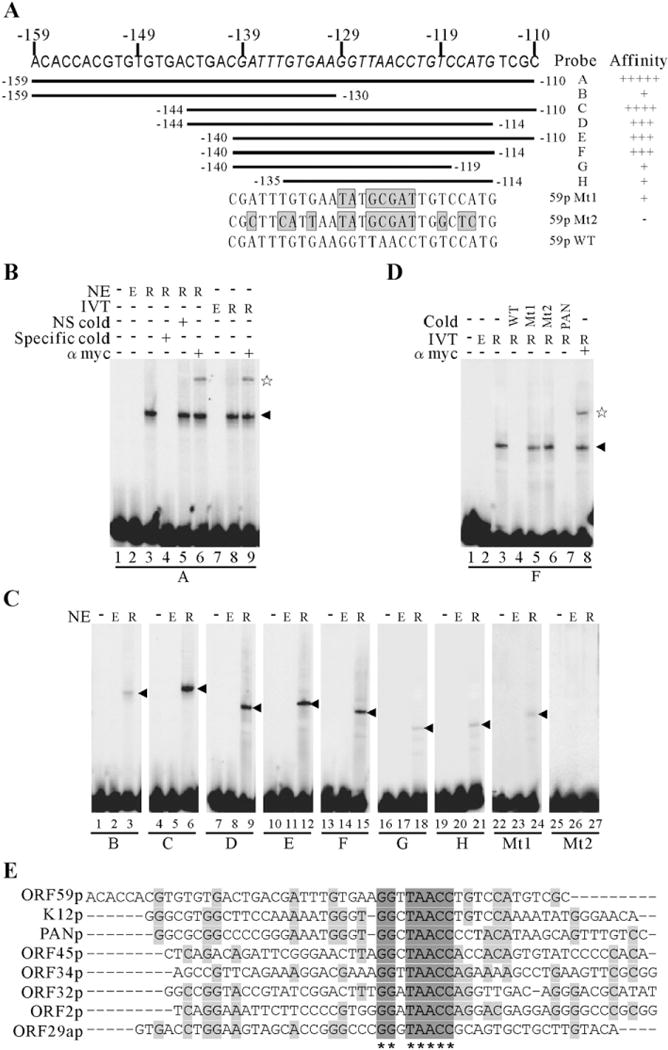
RTA directly binds to the 59pRRE in vitro and mapping of the core ORF59 RRE and the conservation of consensus binding sequences in other promoters. (A) Schematic representation of the oligonucleotides tested for RTA binding by EMSAs. Nucleotide residues of the 27-bp core RRE are italicized and the mutated minimal 59pRREs are shown at the bottom. The grey boxes correspond to the mutation sites. (B) EMSA of probe A with either overexpressed nuclear extracts (lanes 3 to 6) or in vitro translated recombinant Myc-RTA protein (lanes 8, 9). A 50-fold excess of unlabeled specific competitors (lane 4) and a nonspecific competitor (lane 5) was incubated in the presence of recombinant Myc-RTA protein for the competition assay. The RTA-bound complexes were supershifted by a monoclonal anti-Myc-tag antibody (lanes 6, 9). (C) Wild type oligonucleotides B to H and mutated ones were labeled and incubated in the absence or presence of nuclear extracts from empty vector or RTA expression vector transfected 293T cells, and their relative binding affinities are summarized and shown in the right of (A). (D) The specificity of the binding complex formed by 59pF and RTA. 59pF were end-labeled and incubated with in vitro translated Myc-tagged RTA protein. Competition assays were performed by inclusion 50-fold excess of unlabeled specific competitors 59pF (lane 4) or mutated 59pF (lanes 5 and 6) or PANpRRE (lane 7) in the binding reactions before incubation with 1-fold of radiolabeled probe 59pF. The specific binding complex was supershifted by a monoclonal anti-Myc-tag antibody (lane 8). The arrowhead denotes the specific binding of RTA to DNA and the pentagram denotes the supershift. NE, nuclear extracts; IVT, in vitro translated; NS, nonspecific; Mt, mutated 59pRRE; E, empty vector; R, RTA expression vector. (E) The alignment depicts the conservation of consensus binding sequences (GGNTAACC) in other KSHV promoters.
Mapping of a core ORF59 RRE
To determine the minimal or core DNA sequence responsible for binding of RTA to the ORF59 promoter, another 2 oligonucleotides, 59pB and 59pC (Fig. 6A), were designed based on the alignment result above (Fig. 5C). 59pB contains the 5′ 30-bp of 59pA; 59pC contains the 3′ 35-bp of 59pA, and both of them share the region from −144 to −130. They were end-labeled, incubated with nuclear extracts containing RTA protein and then tested by EMSA. In comparison to 59pA, 59pB, lacking the 3′ 20-bp of 59pA, can still bind with RTA, however the binding affinity was significantly decreased (Fig. 6C, lane 3). In contrast, the binding of RTA to 59pC which lacks the 5′ 15-bp of 59pA, is still as efficient as that of 59pA (Fig. 6C, lane 6). These data suggest that the region spanning from position −144 to −110 is sufficient for the specific binding by RTA, while the 5′ 15-bp has only minimal effect. To further narrow down the region responsible for RTA binding, we designed a series of oligonucleotides, from 59pD to 59pH (Fig. 6A). 59pD, 59pE and 59pF was further deleted of 4-bp on the 5′ or 3′ and both end of 59pC, respectively. RTA still bound to 59pD, 59pE and 59pF (Fig. 6C, lanes 9, 12 and 15), although the efficiency is slightly decreased compared to that of 59pC and 59pA. Further deletion of 59pF in either 5′ or 3′ end (i.e. 59pG or 59p H) led to a near complete loss of binding affinity to RTA protein (Fig. 6C, lanes 18 and 21). These results suggest the region from position −140 to −114 contains the core sequence responsible for binding of RTA. Interestingly, the region possesses highest homology with K12 and PAN RRE. To further determine whether the conserved nucleotides were required for the binding of ORF50 protein, point mutations were introduced into 59pF (Fig. 6A). An additional 2 oligonucleotides, 59pMt1 and 59pMt2, were designed and tested by EMSA. As shown, these mutations in homologous block severely abolished the binding affinity of RTA (Fig. 6C, lanes 24 and 27). Taken together, these results suggest that a minimal RTA responsive element in ORF59 promoter is CGATTTGTGAAGGTTAACCTGTCCATG, which shares 14 conserved nucleotides with that of PAN RNA and K12. To further confirm the specificity of RTA to the minimal RRE 59pF, we performed competition and supershift experiments with in vitro translated recombinant Myc-RTA protein. A 50-fold excess unlabeled wild type 59pF competed very efficiently for RTA binding, whereas mutated ones did not (Fig. 6D, lanes 4 to 6). Consistent with the result from the binding assays, 59pMt1 and 59pMt2 had only minimal interaction with RTA protein. In addition, unlabeled oligonucleotides corresponding to the PAN minimal RRE efficiently competed for RTA binding with 59pF (Fig. 6D, lane 7). Moreover, inclusion a monoclonal antibody against anti-Myc-tag in the binding reaction supershifted the RTA-bound complexes formed on 59pF (Fig. 6D, lane 8). Collectively, these data demonstrated that the 27-nt oligonucleotide 59pF, ranging from position −140 to −114 of the ORF59 promoter, or 96879 to 96853 according to the viral genome sequences (GenBank: U75698), contains a minimal RRE.
The above experiments showed that the mapped minimal RTA responsive element in ORF59 promoter possesses significant homology with that of the PAN RNA as well as that of the K12. Mutation in the highly homologous block (GGNTAACC) severely affected the interaction between RTA and this RRE (Fig. 6C). These data suggests that GGNTAACC could be a highly conserved binding sequence which RTA would utilize to regulate the expression of other downstream genes. Therefore, we performed genome wide scan to look for such sequence in other promoters. Very interestingly, there are totally 5 putative RREs are found which contains this sequence in the promoters of ORF45, ORF34, ORF32, ORF2 and ORF29a. The locations of these potential RREs were summarized in Table 1 and the alignment demonstrating the homology between these RREs and those of ORF59, K12 as well as PAN was shown in Fig. 6E.
Table 1.
Potential RTA consensus binding sequences located in other promoters
| Seed sequence | Location | Orientation | Downstream gene | CDS | Putative function |
|---|---|---|---|---|---|
| ggttaacc | 54232–54239 | F | ORF34 | 54675–55658 | EBV BGLF3-like protein |
| 96861–96868 | R | ORF59 | 95549–96739 | processivity factor | |
| ggataacc | 51065–51072 | F | ORF32 | 51404–52768 | EBV BGLF1-like protein |
| 19042–19049 | R | ORF2 | 17921–18553 | DHFR-like protein | |
| ggctaacc | 28604–28611 | F | PAN | / | nut-1 RNA |
| 69231–69238 | R | ORF45 | 67353–68576 | acidic domain transactivation | |
| 118842–118849 | R | K12 | 117919–118101 | kaposin | |
| gggtaacc | 55218–55225 | R | 29a | 53738–54676 | similar to packaging protein |
The ORF59 promoter contains a functional TATA box
Using TF search program, a potential TATA-like box, TATTTA, 23 nt or 27 nt upstream from the transcription initiation sites was predicted. To determine the role of this putative TATA box in directing RTA-mediated expression of ORF59 protein, we mutated it from TATTTA to GCGCGC by site-directed mutagenesis. As shown, changes from A/T to G/C should inhibit the TATA transcriptional activation (Patikoglou et al., 1999). The resulting construct, pGL3-TATAmt (Fig. 7A), was cotransfected with either pCR3.1-RTA or pCR3.1 and tested the responsiveness to RTA in comparison to that of wild type pGL-FL. As shown in Fig. 7B, mutation of TATA box alone led to a dramatic reduction of RTA response in both 293T and DG75 cell lines. The RTA activations were diminished to 35.1% and 29.2% in 293T and DG75, respectively. These results indicate that this TATA box contributed to the RTA-mediated activation of the ORF59 promoter.
Fig. 7.
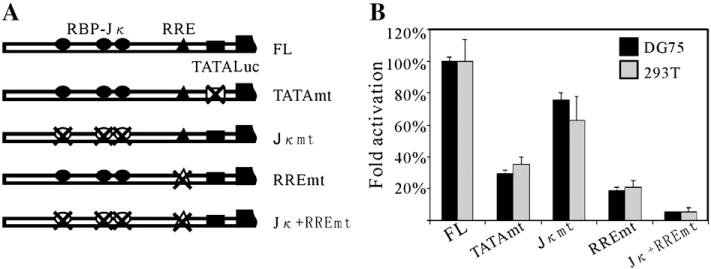
The contribution of TATA box, RBP-Jκ binding sites and RRE to the activation of ORF59 promoter by RTA. (A) Diagram of the mutation constructs using site-directed mutagenesis in the ORF59 promoter. All the mutated promoters are cloned into the pGL3-enhancer reporter vector and the resulting constructs are named according to the mutation sites. (B) The mutated constructs were assayed for their responsiveness to RTA in reporter assays with DG75 and 293T cell lines, as described in Fig. 2. Activity of reporter plasmids containing mutated ORF59 promoter is expressed as % activity of wild type promoter-containing reporter vector (pGL3-FL). Results are the averages of at least three experiments ±standard deviations.
Both RBP-Jκ binding sites and RRE contribute to the activation of ORF59 promoter by RTA
The experiments above definitely show that RBP-Jκ binding sites and RRE have roles in the activation of the ORF59 promoter by RTA separately. To determine their contributions to the activation by RTA in the context of full length ORF59 promoter, we designed three additional mutants: pGL3-Jκmt, pGL3-RREmt and pGL3-Jκ+RREmt. pGL3-Jκmt contains mutations where all the three RBP-Jκ binding sites changed to a EcoRI site (GAATTC); pGL3-RREmt mutations mapped in the 27-nt minimal RRE region, same as 59pMt2 (Fig. 6A); and pGL3-Jκ+RREmt combined both mutations. Their transcriptional activities by RTA protein were tested relative to that of their parental plasmid, pGL3-59pFL. As shown, when all 3 RBP-Jκ binding sites are mutated, approximately 30% reduction of fold activation was observed in DG75 lymphoma cells, slightly lower than that of pGL3-59pD3, which was deleted for all three RBP-Jκ binding sites. The mutations in the minimal RRE resulted in a striking decrease; with more than 70% of RTA responsiveness ablated. When both of RBP-Jκ binding sites and RRE were mutated, RTA-mediated activation was further reduced to 6.9% or 7.1% in DG75 and 293T, respectively (Fig. 7B). These results strongly suggest that (i) RBP-Jκ is indispensable and (ii) the mapped RRE is critical for the activation of the ORF59 promoter by RTA, although other cellular factors are likely to contribute to activation of the ORF59 promoter.
RTA activates ORF59 transcription in vivo
The data above strongly suggest that RTA may have a direct role in upregulation of ORF59 during viral lytic reactivation. To test this hypothesis, we took advantage of a RTA-inducible BCBL-1 cell line generated by Jae Jung group (Nakamura et al., 2003). In this cell line, RTA can be induced using tetracycline and facilitates the detection of RTA responsive genes (Nakamura et al., 2003). As shown in Fig. 8A, expression of ORF59 as well as ORF57 both are responsive to RTA induction. 4 hours post induction of RTA, mRNA levels of ORF59 and ORF57 started to accumulate. Previously, it has been reported that little transcripts of ORF59 were present in the butyrate-induced cells, harbouring an ORF57 deleted recombinant KSHV compared to the wild type counterpart (Majerciak et al., 2007). This indicated that ORF57 may play a predominant role in the regulation of ORF59 during lytic reactivation. However another report showed that in same system in which RTA instead of ORF57 was deleted ORF59 was not expressed in TPA treated cells (Xu et al., 2005), however, the expression of ORF57 in this system was highly upregulated. This suggests that the expression of ORF59 may be independent from ORF57. These controversial observations led us to determine whether the expression of ORF59 is dependent on ORF57 or if RTA plays a predominant role in regulation of ORF59 in KSHV infected cells. We took advantage of a shRNA expression vector pLL3.7 and its lentiviral packaging system, which have been widely used to inhibit gene expression in mammalian cells (Rubinson et al., 2003). Pseudotyped lentivirus particles bearing either ORF57 shRNA expression vector, ORF57shRNA-pLL3.7, or control vector, pLL3.7, were used to infect RTA-inducible BCBL1, TRE-BCBL1-RTA. More than 90% of the cells were infected and express the enhanced green fluorescent protein (EGFP) (data not shown). The resulting stable cell lines, TRE-BCBL1-RTA-57shRNA and TRE-BCBL1-RTA-ctrl, were induced with 5 μg/ml tetracycline and harvested at different time points to prepare RNA. The relative mRNA levels of RTA, ORF57 and ORF59 over the time course of induction was monitored by real-time PCR analysis. The results show that the expression of ORF57 in TRE-BCBL1-RTA-57shRNA cells were silenced and knocked down to a very low level in comparison to their counterpart (Fig. 8B). Importantly, the transcripts of ORF59 were detected at similar levels as control, suggesting that the expression of ORF59 is responsive to ORF50, but not toORF57 expression in this system (Fig. 8B).
Fig. 8.
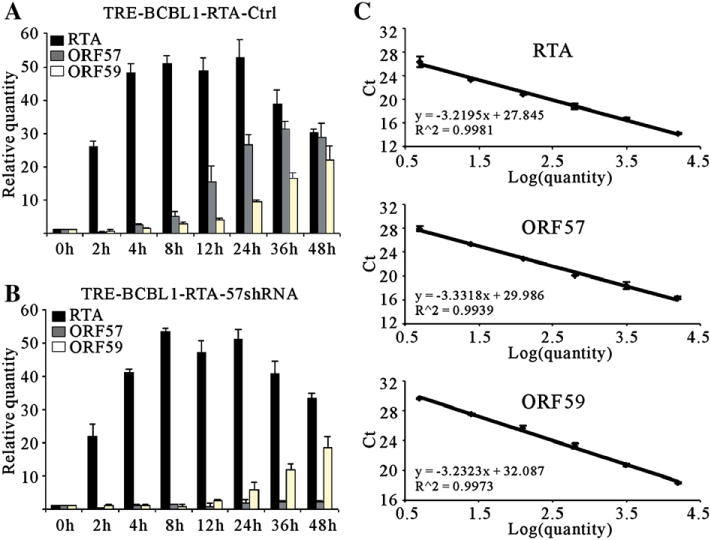
Real-time RT-PCR analysis of RTA, ORF57 and ORF59 transcripts in TRE-BCBL1-TRE cells. The RTA-inducible BCBL-1 cells, infected with lentivirus carrying empty control pLL3.7 or ORF57 shRNA expression vectors, were treated with 5 μg/ml tetracycline and then harvested at various time points. Total RNA was extracted by TRIzol reagent. A total of 5 μg of RNA was used with the First Strand cDNA Synthesis Kit to construct cDNA. The real-time PCR was performed using the SYBR green real-time PCR master mix kit with β-actin as the standard. The relative quantitative comparison of RTA, ORF57 and ORF59 mRNA levels over the time course of induction of the TRE-BCBL1-RTA-Ctrl (A) and TRE-BCBL1-RTA-57shRNA (B) cells infected with lentivirus control or shRNA expression vectors respectively. The standard curves are shown to the right (C). Each time point was tested in triplicate for the calculation of the mean and standard deviation.
Discussion
Similar to other herpesviruses, KSHV is also characterized by its ability to establish two distinct mode of infection: latent infection and lytic reactivation (Staudt and Dittmer, 2007). Latency is established soon after primary lytic infection and is characterized by the persistence of the viral genome in the cell nucleus and expression of a small subset of viral genes. Lytic reactivation of latent virus involves the sequential activation of immediate-early, early, and late viral genes (Sarid et al., 1998). In KSHV infected cells, this gene expression cascade is initiated by the immediate-early viral transactivator RTA (Sun et al., 1998, 1999). The ectopic expression of RTA in latently infected B cells is sufficient to reactivate the latent virus and initiate replication (Lukac et al., 1998, 1999; Nakamura et al., 2003). It has been shown that RTA can directly activate several downstream genes such as ORF57, K8, vIL-6 and PAN (Chang et al., 2002; Deng et al., 2002; Duan et al., 2001; Lukac et al., 2001; Song et al., 2001), through either directly binding with high affinity to its own responsive elements or in combination with other cellular factors, such as RBP-Jκ, C/EBP-α, Oct-1 and Ap1 (Lan et al., 2005a,b; Sakakibara et al., 2001; Wang et al., 2003a,b).
KSHV encoded ORF59, encodes a viral DNA polymerase processivity factor, which has been proposed to play an important role in viral lytic replication and pathogenesis (Chan et al., 1998). Expression of ORF59 encoding processivity factor is essential for DNA replication during lytic reactivation (Chan and Chandran, 2000; Chen et al., 2004, 2005). However, the regulation of ORF59 during lytic reactivation remains unclear. Since RTA is the master molecule switching KSHV from a latent to a lytic replication mode, and its expression precedes that of ORF59, we were interested in examining whether RTA can directly activate the promoter of ORF59. Indeed, there is indirect evidence showing that ORF59 expression could be responsive to RTA induction using a Bacmid system in which RTA is knocked out (Xu et al., 2005). In this study, we first explored whether RTA operated as a transcriptional modulator of the ORF59 promoter using transient reporter assays. The results showed that RTA alone was able to strikingly activate high levels of ORF59 promoter activity in a dose-dependent manner. In addition, this effect is not cell type dependent. This experiment convincingly established a link between RTA and ORF59 expression. We next sought to elucidate the mechanisms by which RTA regulates ORF59 promoter. Since RTA usually regulates its downstream gene promoter through either direct binding or indirect binding mechanisms, we firstly checked the ORF59 promoter sequence to identify any potential binding sites of transcriptional factors with which RTA can interact to access the promoter. Indeed, we found three RBP-Jκ binding sites within the ORF59 promoter. As shown previously, RBP-Jκ is a major downstream target of Notch signaling pathway and RTA can bind to this molecule to gain access to promoters regulating gene expression (Panin and Irvine, 1998; Weinmaster, 2000). This provided the possibility that RTA also utilized a similar mechanism to regulate ORF59 promoter. Using deletion analysis of the putative RBP-Jκ binding sites, we found that RBP-Jκ has an important role in RTA-mediated activation of ORF59 promoter. We also repeated the reporter assay in OT11 cells, in which the RBP-Jκ gene was knocked out as well as its isogenic wild type counterpart OT13 cells. Expectedly, we obtained similar results in that the RBP-Jκ binding sites were at least in part responsible for activation of ORF59 by RTA. The role of RBP-Jκ binding sites in RTA induced ORF59 activation was further supported by the fact that all the three RBP-Jκ putative binding sites within the ORF59 promoter can bind directly to RBP-Jκ from nuclear extracts or in vitro translated protein. These data convincingly demonstrated that RBP-Jκ is important for activation of the ORF59 promoter. However, deletion of all the three RBP-Jκ binding sites has not completely shut down the RTA-mediated activation in both epithelial and B cell lines. This indicated there was some other mechanism regulating this activation. We further identified a cis-acting RRE within the promoter spanning from position −159 to −110. Interestingly, this RRE shares significant homology with the RREs in PAN and K12 promoter. This suggests that the interaction between RTA and its responsive DNA sequences could be a highly conserved mechanism by which RTA regulates the expression of its downstream genes in the context of KSHV genome. Through site mutagenesis, we identified that a GGNTAACC block within the ORF59 promoter is the core element responsible for RTA binding. Very interestingly, we found that the promoters of ORF45, ORF34, ORF32, ORF2 and ORF29a contain this sequence. It is highly possible that RTA could regulate the expression of these genes through binding to the similar RREs. It is definitely important to prove this possibility in the future studies. Furthermore, we showed that 59pRRE confers responsiveness to a heterologous promoter in an orientation-independent and RTA-specific manner. This new RRE was further narrowed down to a 27-bp region and was directly bound by either overexpressed nuclear extracts containing RTA or in vitro translated RTA protein, suggesting that RTA activated the expression of ORF59 gene during lytic cycle through direct binding of the cis-acting RRE. We have further determined the contribution of RBP-Jκ binding sites and 59pRRE to the activation by RTA in the context of full length promoter, data show that mutation of the 59pRRE resulted in a dramatic reduction of luciferase activity, whereas mutation of all the three RBP-Jκ binding sites led to only 30% decrease compare to their wild type counterpart. These results illustrate that RBP-Jκ binding sites are indispensable but 59pRRE is more critical for the activation of ORF59 promoter by RTA. We do not have a definite explanation for this observation at this time but we cannot rule out the possibility that 59pRRE other than RBP-Jκ binding sites is proximal to the transcriptional start site, which may offer more chances to interact with transcriptional complexes. In addition, we also identified a potential TATA box in the ORF59 promoter. Mutation of this TATA box severely impaired RTA-mediated activation suggesting this TATA box is functional for transcription induced by RTA.
Previously, ORF59 was shown to be responsive to ORF57, another immediately early gene of KSHV during lytic reactivation using a Bacmid system in which ORF57 was deleted (Majerciak et al., 2007). However, another group found that ORF59 is not responsive to ORF57 using the same Bacmid system in which RTA is deleted since ORF57 is upregulated upon chemical induction (Xu et al., 2005). Whether or not ORF59 is responsive to ORF57 remained controversial. To address this issue, we silenced the expression of ORF57 using a pseudotype lentivirus based shRNA system and determined that ORF57 has trivial or no effect on the expression of ORF59. In either ORF57 knockdown cells or control cells, expression of ORF59 is responsive to RTA induction well.
In summary, we explored the mechanisms by which RTA regulates a critical lytic gene ORF59 and showed that RTA can activate the expression of ORF59 through interaction with the cellular RBP-Jκ protein as well as a novel RTA responsive element. A TATA box within the ORF59 promoter was also identified. In addition, we also found that expression of ORF59 was mainly responsive to RTA instead of ORF57 in the context of KSHV infected cells. Taken together, our findings provide valuable evidence which supports a mechanism by which RTA regulates its downstream gene ORF59 and so enhances our understanding of the biology of KSHV lytic reactivation.
Materials and methods
Constructs
To construct the reporter plasmid of the ORF59 promoter, a 1909 bp sequence upstream of the translation initiation codon (nt 98648 to 96740, according to the HHV-8 genomic sequence, GenBank: U75698) was amplified from Bacmid Z8 using primers 59p-FL-F (5′-CGGGTACCTCGATGCTGAGAGC-3′) and 59p-FL-R (5′-ATAGATCTGATTGCGGCCGTAGACGCAC-3′) in which KpnI and BglII site were introduced respectively. The amplification product was then cloned into KpnI and BglII sites of the pGL3-enhancer vector (Promega, Madison, USA). To make truncations, various promoter regions were amplified by using the common primer 59p-FL-R and a specific forward primer. The specific primers were as follows: 59p-D1-F (5′-AAGGTACCCAAGGAGGACAGAGTCAGGA-3′) for plasmid pGL3-D1 (−1329), 59p-D2-F(5′-AAGGTACCCAGCGATGCCGACAAGGACT-3′) for plasmid pGL3-D2 (−914), 59p-D3-F(5′-AAGGTACCTGCGTAAGGCGACCACCAAAG-3′) for plasmid pGL3-D3 (−630), 59p-D4-F(5′-AAGGTACCGTGCCTTGCCAACGATTA-3′) for plasmid pGL3-D4 (−509), 59p-D5-F(5′-AAGGTACCAGTCACGCAGAGGTCCAG-3′) for plasmid pGL3-D5 (−412), 59p-D6-F(5′-AAGGTACCTCCGGAGGTTCCTGGAAG-3′) for plasmid pGL3-D6 (−309), 59p-D7-F(5′-AAGGTACCCATACACCGGCAGTTTCA-3′) for plasmid pGL3-D7 (−212), 59p-D8-F(5′-AAGGTACCACACTTCCACCTCCCCTA-3′) for plasmid pGL3-D8 (−109), 59p-D9-F(5′-TTGGTACCACACCACGTGTGTGACT-3′) for plasmid pGL3-D9 (−159). Each PCR product was then digested with KpnI and BglII and cloned into pGL3-enhancer.
The reporter plasmid Hsp70-Luc was constructed as previously described (Lukac et al., 2001). Hsp70-Luc contains the TATA box and surrounding basal promoter sequences of the cellular hsp70 gene (Taylor and Kingston, 1990) cloned into the BglII/HindIII sites of pGL3-basic. 59pRRE-F-Hsp and 59pRRE-R-Hsp were constructed by annealing oligonucleotides: 59pRRE-F (5′-GATCTACACCACGTGTGTGACTGACGATTTGTGAAGGTTAACCTGTCCATGTCGCA-3′) and 59pRRE-R (5′-GATCTGCGACATGGACAGGTTAACCTTCACAAATCGTCAGTCACACACGTGGTGTA-3′) in annealing buffer (50 mM KCl, 10 mM Tris–HCl [pH 8.3], 1.5 mM MgCl2). Annealed oligonucleotides were then successively extracted with phenol–CHCl3 and CHCl3 and precipitated. The resultant annealed oligonucleotides were ligated into Hsp70-Luc which had been digested with BglII and treated with calf intestinal alkaline phosphatase (CIAP) (Takara Bio Inc.). The orientations of the inserts were confirmed by sequencing.
pGL3-Jκ1mt, pGL3-Jκ12mt, pGL3-Jκ123mt, pGL3-TAmt, pGL3-RREmt and pGL3-Jκ+RREmt were generated through PCR-based site mutagenesis. For pGL3-Jκ1mt, bearing a mutation in the first RBP-Jκ consensus binding site, first step PCR was performed using pGL3-FL as a template with 2 set of primers. The first set of primers was 59p-FL-F and ORF59p-Jκ1mt-R (5′-GAAACGTgaattcCATACTGTCTAACCACCCGA-3′). The second set of primers was ORF59p-Jκ1mt-F (5′-GTATGgaattcACGTTTCCACGTTCCCCAAGC-3′) and ORF59p-FL-R. The lowercased letters represent mutated nucleotides. Both products from first step PCR were purified and used as a template in second-step PCR, using 59p-FL-F and 59p-FL-R as primers. The products were then digested with KpnI and BglII, and cloned into vector pGL3-enhancer. The pGL3-Jκ12mt was constructed in a similar way using pGL3-Jκ1mt as a template in the first step. The first set of primers was 59p-FL-F and 59p-Jκ2mt-R (5′-GTGCACgaattcAACATCAAGGTGCAGGGGTAT-3′). The second set of primers was 59p-Jκ2mt-F (5′-GATGTTgaattcGTGCACCAGCTCAGCGATG-3′) and ORF59p-FL-R. For pGL3-Jκ123mt, first step PCR was performed using pGL3-Jκ12mt as a template. The first set of primers was 59p-FL-F and 59p-Jκ2mt-R (5′-GGCCAgaattcAAAACGTAAACAAAAATCTATAAAAG-3′). The second set of primers was 59p-Jκ2mt-F (5′-CGTTTTgaattcTGGCCGAACGCCTAGTTAACTT-3′) and 59p-FL-R. For pGL3-TAmt, first step PCR was performed using pGL3-FL as a template. The first set of primers was 59p-FL-F and 59p-TAmt-R (5′-CGATAGACTGCCTgcgcgcGAACTTTTAGG-3′). The second set of primers was 59p-TAmt-F (5′-CCTAAAAGTTCgcgcgcAGGCAGTCTATCG-3′) and 59p-FL-R. For pGL3-RREmt, first step PCR was performed using pGL3-FL as a template. The first set of primers was 59p-FL-F and 59p-RREmt-R (5′-CAgaGcCAatcgcAtaTTaAtgAAgCGTCAGTCACACACGTGGTG-3′). The second set of primers was 59p-RREmt-F (5′-CGcTTcaTtAAtaTgcgatTGgCtcTGTCGCACACTTCCACCTCC-3′) and 59p-FL-R. pGL3-Jκ+RREmt, in which all the 3 RBP-Jκ consensus binding sites and RRE are mutated, was constructed in a similar way using pGL3-Jκ123mt as a template.
Expression vectors, including pCR3.1-RTA, pEF-RTA and pA3M-RBP-Jκ, were described previously (Lan et al., 2005a). All constructs and mutations were verified by DNA sequencing.
Antibodies and cell lines
KSHV RTA mouse monoclonal antibody was a kind gift from Koichi Yamanishi (Osaka University, Osaka, Japan). Myc-tagged proteins were detected by supernatants from 9E10 hybridoma, as described previously (Lan et al., 2005a).
DG75 (a KSHV-negative B cell line), BCBL1 (a KSHV-positive B cell line) and human embryonic kidney fibroblast 293T cells transformed with T antigens were provided by Dr. Erle S. Robertson (University of Pennsylvania, Philadelphia, PA). RTA-inducible BCBL1 (TRE-BCBL1-RTA) is a gift from Jae Jung (Harvard University, Boston, MA). The 293T cells were grown in high-glucose DMEM supplemented with 5% bovine growth serum (BGS; HyClone, Logan, UT), 2 mM L-glutamine, 25 U/ml penicillin and 25 μg/ml streptomycin. DG75, BCBL1 and RTA-inducible BCBL1 were grown in RPMI medium 1640 supplemented with 10% BGS, 2 mM L-glutamine, 25 U/ml penicillin and 25 μg/ml streptomycin. Mouse RBP-Jκ−/− (OT11) and wild type (OT13) fibroblast cell lines were kindly provided by T. Honjo (Kyoto University, Kyoto) and were grown in high-glucose DMEM supplemented with 10% BGS and 100 U of mouse gamma interferon (PeproTech, Inc., New Jersey) per ml at 32 °C. All cells were cultured at 37 °C (except mentioned) in the presence of 5% CO2.
Transfection and dual reporter assays
DG75 cells were transfected by electroporation with a Bio-Rad Gene Pulser II electroporator. A total of 10 million cells harvested in exponential phase were collected and washed with phosphate-buffered saline (PBS) and then resuspended in 400 μl of serum-free RPMI 1640 with DNA mixture for transfection. Resuspended cells were transferred to a 0.4-cm cuvette and electroporated at 975 μF and 220 V. The electroporated cells were then transferred to 10 ml of complete medium, followed by incubation at 37 °C with 5% carbon dioxide. Lipofectamine 2000 (Invitrogen, Inc.) was used to transfect 293T, OT11 or OT13 cells, according to the manufacturer’s protocol.
The dual-luciferase reporter assay system from Promega (Madison, USA) was used to test promoter activity, based on the manufacture’s protocol. The results shown represent experiments performed in triplicate.
Electrophoretic mobility shift assay (EMSA)
Oligonucleotides spanning partial or all of the ORF59 RBP-Jκ binding sites or RTA response element were synthesized, annealed and end-labeled with T4 Polynucleotide Kinase (Promega, Madison, USA) following purification with Select-D with G-25 columns (Shelton/IBI, Inc.). Radioactive probes were diluted in water to a final concentration of 80,000 cpm/μl. EMSA was performed following the protocol described previously (Lan et al., 2005a,b). The oligonucleotide pairs used to generate double-stranded probes are listed in Table 2.
Table 2.
Oligonucleotides used in EMSA
| Name | Sequence |
|---|---|
| 59pJκ1F | 5′-AGACAGTATGGTGGGGACGTTTCCAC-3′ |
| 59pJκ1R | 5′-GTGGAAACGTCCCCACCATACTGTCT-3′ |
| 59pJκ2F | 5′-CCTTGATGTTTCCCACGTGCACCAGC-3′ |
| 59pJκ2R | 5′-GCTGGTGCACGTGGGAAACATCAAGG-3′ |
| 59pJκ3F | 5′-TTACGTTTTTGGGAATGGCCGAACGC-3′ |
| 59pJκ3R | 5′-GCGTTCGGCCATTCCCAAAAACGTAA-3′ |
| 59pRRE-AF | 5′-ACACCACGTGTGTGACTGACGATTTGTGAAGGTTAACCTGTCCATGTCGC-3′ |
| 59pRRE-AR | 5′-GCGACATGGACAGGTTAACCTTCACAAATCGTCAGTCACACACGTGGTGT-3′ |
| 59pRRE-BF | 5′-ACACCACGTGTGTGACTGACGATTTGTGAA-3′ |
| 59pRRE-BR | 5′-TTCACAAATCGTCAGTCACACACGTGGTGT-3′ |
| 59pRRE-CF | 5′-CTGACGATTTGTGAAGGTTAACCTGTCCATGTCGC-3′ |
| 59pRRE-CR | 5′-GCGACATGGACAGGTTAACCTTCACAAATCGTCAG-3′ |
| 59pRRE-DF | 5′-CTGACGATTTGTGAAGGTTAACCTGTCCATG-3′ |
| 59pRRE-DR | 5′-CATGGACAGGTTAACCTTCACAAATCGTCAG-3′ |
| 59pRRE-EF | 5′-CGATTTGTGAAGGTTAACCTGTCCATGTCGC-3′ |
| 59pRRE-ER | 5′-GCGACATGGACAGGTTAACCTTCACAAATCG-3′ |
| 59pRRE-FF | 5′-CGATTTGTGAAGGTTAACCTGTCCATG-3′ |
| 59pRRE-FR | 5′-CATGGACAGGTTAACCTTCACAAATCG-3′ |
| 59pRRE-GF | 5′-CGATTTGTGAAGGTTAACCTGT-3′ |
| 59pRRE-GR | 5′-ACAGGTTAACCTTCACAAATCG-3′ |
| 59pRRE-HF | 5′-TGTGAAGGTTAACCTGTCCATG-3′ |
| 59pRRE-HR | 5′-CATGGACAGGTTAACCTTCACA-3′ |
| 59pRRE-Mt1F | 5′-CGATTTGTGAATATGCGATTGTCCATG-3′, |
| 59pRRE-Mt1R | 5′-CATGGACAATCGCATATTCACAAATCG-3′ |
| 59pRRE-Mt2F | 5′-CGCTTCATTAATATGCGATTGGCTCTG-3′ |
| 59pRRE-Mt2R | 5′-CAGAGCCAATCGCATATTAATGAAGCG-3′ |
RNA interference lentivirus system
To knockdown the expression of ORF57, we used siRNA expression vector, pLL3.7 (kindly provided by Dr. Luk Van Parijs). This vector contains a cytomegalovirus promoter driving expression of enhanced GFP and the mouse U6 promoter with downstream restriction sites to allow the efficient introduction of oligonucleotides encoding shRNA. The shRNA sequences of ORF57, 5′-GTAGAGGCATGTAACCTTC-3′ for sense and 5′-GAAGGTTACATGCCTCTA-3′ for antisense, were designed and confirmed using siRNA Selection Program (Whitehead Institute for Biomedical Research). Oligonucleotides including siRNA and hairpin structure were chemically synthesized and listed as follows: forward for 5′-TGTAGAGGCATGTAACCTTCTTCAAGAGAGAAGGTTACATGCCTCTACTTTTTTC-3′, reverse for 5′-TCGAGAAAAAAGTAGAGGCATGTAACCTTCTCTCTTGAAGAAGGTTACATGCCTCTACA-3′. The oligonucleotides were double-stranded and phosphorylated and inserted into pLL3.7 vector using HpaI and XhoI sites. We prepared lentivirus with packaging vectors into 293T cells according to the manufacturer’s instruction (Dull et al., 1998). The viruses were collected from the culture supernatants on day 2 and 3 posttransfection, concentrated by ultracentrifugation for 1.5 h at 25,000 rpm, and resuspended in phosphate-buffered saline (PBS). Titers were determined by infecting HEK293T cells with serial dilutions of concentrated lentivirus and counting EGFP-expressing cells after 48 h under fluorescent microscopy. For a typical preparation, the titer was approximately 4–10×108 infectious units per ml. TRE-BCBL1-TRE cells in 6-well plate were infected with lentivirus carrying empty control pLL3.7 or ORF57 shRNA expression vectors at MOI of 100.
Quantitative real-time PCR analysis
Quantitative real-time PCR (qPCR) was used to make a relative quantitative comparison of RTA, ORF57 and ORF59 levels over the time course of induction of lentivirus-infected TRE-BCBL1-RTA cells with 5 μg/ml tetracycline. At various time points postinduction, cells were harvested and total RNA was collected using Trizol reagent (Invitrogen, Inc.) following the manufacturer’s instructions. cDNA (20 μl) for real-time PCR was generated from 5 μg of total RNA with the First Strand cDNA Synthesis Kit (Fermentas UAB), priming with 1 μl of oligo(dT)18, according to the manufacturer’s instructions. The primer sequences for the real-time PCR system were designed and chosen with Primer Express software, version 3.0 (Applied Biosystems, California, USA). They are listed as follows: for RTA, 5′-AGACCCGGCGTTTATTAGTACGT-3′ and 5′-CAGTAATCACGGCCCCTTGA-3′; for ORF57, 5′-TGGCGAGGTCAAGCTTAACTTC-3′ and 5′-CCCCTGGCCTGTAGTATTCCA-3′; and for ORF59, 5′-TTGGCACTCCAACGAAATATTAGAA-3′ and 5′-CGGGAACCTTTTGCGAAGA-3′. The real-time PCRs were performed with SYBR green real-time PCR master mix kit (Toyobo, Osaka, Japan). Reactions in a total volume of 20 μl consisted of 10 μl of Mastermix, 1 mM each primer and 4 μl of the diluted cDNA product. Following 2 min at 50 °C, the DNA polymerase was activated at 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s and at 60 °C for 1 min. The standard curves for the RTA, ORF50, ORF59, and β-actin cDNAs were generated by serial dilution and a melting curve analysis was performed to verify the specificity of the products. The values for the relative quantification were calculated by the ΔΔCt method. Data were normalized to human β-actin levels for the equal amount of cDNA input in each sample and reported as the increase in mRNA accumulation compared to the untreated TRE-BCBL1-RTA mRNA levels. All reactions were carried out six times with a 7900HT sequence detection systems (Applied Biosystems, California, USA). As a negative control, each plate contained a minimum of three wells lacking template.
Acknowledgments
This work was supported by grants from the Knowledge Innovation Program of the Chinese Academy of Sciences (PCL2006-01) and Natural Science Foundation of China (30770098) to KL. KL is a special fellow and ESR is a scholar of the Leukemia and Lymphoma Society of America.
References
- Albrecht JC, Nicholas J, Biller D, Cameron KR, Biesinger B, Newman C, Wittmann S, Craxton MA, Coleman H, Fleckenstein B, et al. Primary structure of the herpesvirus saimiri genome. J Virol. 1992;66(8):5047–5058. doi: 10.1128/jvi.66.8.5047-5058.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander L, Denekamp L, Knapp A, Auerbach MR, Damania B, Desrosiers RC. The primary sequence of rhesus monkey rhadinovirus isolate 26–95: sequence similarities to Kaposi’s sarcoma-associated herpesvirus and rhesus monkey rhadinovirus isolate 17577. J Virol. 2000;74(7):3388–3398. doi: 10.1128/jvi.74.7.3388-3398.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AuCoin DP, Colletti KS, Cei SA, Papouskova I, Tarrant M, Pari GS. Amplification of the Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 lytic origin of DNA replication is dependent upon a cis-acting AT-rich region and an ORF50 response element and the trans-acting factors ORF50 (K-Rta) and K8 (K-bZIP) Virology. 2004;318(2):542–555. doi: 10.1016/j.virol.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Buonaguro FM, Tornesello ML, Beth-Giraldo E, Hatzakis A, Mueller N, Downing R, Biryamwaho B, Sempala SD, Giraldo G. Herpesvirus-like DNA sequences detected in endemic, classic, iatrogenic and epidemic Kaposi’s sarcoma (KS) biopsies. Int J Cancer. 1996;65(1):25–28. doi: 10.1002/(SICI)1097-0215(19960103)65:1<25::AID-IJC5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Bu W, Carroll KD, Palmeri D, Lukac DM. Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 ORF50/Rta lytic switch protein functions as a tetramer. J Virol. 2007;81(11):5788–5806. doi: 10.1128/JVI.00140-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med. 1995;332(18):1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- Chan SR, Chandran B. Characterization of human herpesvirus 8 ORF59 protein (PF-8) and mapping of the processivity and viral DNA polymerase-interacting domains. J Virol. 2000;74(23):10920–10929. doi: 10.1128/jvi.74.23.10920-10929.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SR, Bloomer C, Chandran B. Identification and characterization of human herpesvirus-8 lytic cycle-associated ORF 59 protein and the encoding cDNA by monoclonal antibody. Virology. 1998;240(1):118–126. doi: 10.1006/viro.1997.8911. [DOI] [PubMed] [Google Scholar]
- Chang Y, Moore PS. Kaposi’s sarcoma (KS)-associated herpesvirus and its role in KS. Infect Agents Dis. 1996;5(4):215–222. [PubMed] [Google Scholar]
- Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266(5192):1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- Chang PJ, Shedd D, Gradoville L, Cho MS, Chen LW, Chang J, Miller G. Open reading frame 50 protein of Kaposi’s sarcoma-associated herpesvirus directly activates the viral PAN and K12 genes by binding to related response elements. J Virol. 2002;76(7):3168–3178. doi: 10.1128/JVI.76.7.3168-3178.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Lin K, Ricciardi RP. Human Kaposi’s sarcoma herpesvirus processivity factor-8 functions as a dimer in DNA synthesis. J Biol Chem. 2004;279(27):28375–28386. doi: 10.1074/jbc.M400032200. [DOI] [PubMed] [Google Scholar]
- Chen Y, Ciustea M, Ricciardi RP. Processivity factor of KSHV contains a nuclear localization signal and binding domains for transporting viral DNA polymerase into the nucleus. Virology. 2005;340(2):183–191. doi: 10.1016/j.virol.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Davis DA, Rinderknecht AS, Zoeteweij JP, Aoki Y, Read-Connole EL, Tosato G, Blauvelt A, Yarchoan R. Hypoxia induces lytic replication of Kaposi sarcoma-associated herpesvirus. Blood. 2001;97(10):3244–3250. doi: 10.1182/blood.v97.10.3244. [DOI] [PubMed] [Google Scholar]
- Deng H, Song MJ, Chu JT, Sun R. Transcriptional regulation of the interleukin-6 gene of human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus) J Virol. 2002;76(16):8252–8264. doi: 10.1128/JVI.76.16.8252-8264.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Liang Y, Sun R. Regulation of KSHV lytic gene expression. Curr Top Microbiol Immunol. 2007;312:157–183. doi: 10.1007/978-3-540-34344-8_6. [DOI] [PubMed] [Google Scholar]
- Dittmer D, Lagunoff M, Renne R, Staskus K, Haase A, Ganem D. A cluster of latently expressed genes in Kaposi’s sarcoma-associated herpesvirus. J Virol. 1998;72(10):8309–8315. doi: 10.1128/jvi.72.10.8309-8315.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Wang S, Liu S, Wood C. Characterization of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus-8 ORF57 promoter. Arch Virol. 2001;146(2):403–413. doi: 10.1007/s007050170185. [DOI] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72(11):8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradoville L, Gerlach J, Grogan E, Shedd D, Nikiforow S, Metroka C, Miller G. Kaposi’s sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire viral lytic cycle in the HH-B2 primary effusion lymphoma cell line. J Virol. 2000;74(13):6207–6212. doi: 10.1128/jvi.74.13.6207-6212.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque M, Davis DA, Wang V, Widmer I, Yarchoan R. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) contains hypoxia response elements: relevance to lytic induction by hypoxia. J Virol. 2003;77(12):6761–6768. doi: 10.1128/JVI.77.12.6761-6768.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan K, Kuppers DA, Verma SC, Robertson ES. Kaposi’s sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: a potential mechanism for virus-mediated control of latency. J Virol. 2004;78(12):6585–6594. doi: 10.1128/JVI.78.12.6585-6594.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan K, Kuppers DA, Robertson ES. Kaposi’s sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J Virol. 2005a;79(6):3468–3478. doi: 10.1128/JVI.79.6.3468-3478.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan K, Kuppers DA, Verma SC, Sharma N, Murakami M, Robertson ES. Induction of Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen by the lytic transactivator RTA: a novel mechanism for establishment of latency. J Virol. 2005b;79(12):7453–7465. doi: 10.1128/JVI.79.12.7453-7465.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Chang J, Lynch SJ, Lukac DM, Ganem D. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jkappa (CSL), the target of the Notch signaling pathway. Genes Dev. 2002;16(15):1977–1989. doi: 10.1101/gad.996502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K, Dai CY, Ricciardi RP. Cloning and functional analysis of Kaposi’s sarcoma-associated herpesvirus DNA polymerase and its processivity factor. J Virol. 1998;72(7):6228–6232. doi: 10.1128/jvi.72.7.6228-6232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukac DM, Renne R, Kirshner JR, Ganem D. Reactivation of Kaposi’s sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology. 1998;252(2):304–312. doi: 10.1006/viro.1998.9486. [DOI] [PubMed] [Google Scholar]
- Lukac DM, Kirshner JR, Ganem D. Transcriptional activation by the product of open reading frame 50 of Kaposi’s sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J Virol. 1999;73(11):9348–9361. doi: 10.1128/jvi.73.11.9348-9361.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukac DM, Garibyan L, Kirshner JR, Palmeri D, Ganem D. DNA binding by Kaposi’s sarcoma-associated herpesvirus lytic switch protein is necessary for transcriptional activation of two viral delayed early promoters. J Virol. 2001;75(15):6786–6799. doi: 10.1128/JVI.75.15.6786-6799.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majerciak V, Pripuzova N, McCoy JP, Gao SJ, Zheng ZM. Targeted disruption of Kaposi’s sarcoma-associated herpesvirus ORF57 in the viral genome is detrimental for the expression of ORF59, K8alpha, and K8.1 and the production of infectious virus. J Virol. 2007;81(3):1062–1071. doi: 10.1128/JVI.01558-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore PS, Gao SJ, Dominguez G, Cesarman E, Lungu O, Knowles DM, Garber R, Pellett PE, McGeoch DJ, Chang Y. Primary characterization of a herpesvirus agent associated with Kaposi’s sarcomae. J Virol. 1996;70(1):549–558. doi: 10.1128/jvi.70.1.549-558.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, Jung JU. Global changes in Kaposi’s sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J Virol. 2003;77(7):4205–4220. doi: 10.1128/JVI.77.7.4205-4220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panin VM, Irvine KD. Modulators of Notch signaling. Semin Cell Dev Biol. 1998;9(6):609–617. doi: 10.1006/scdb.1998.0263. [DOI] [PubMed] [Google Scholar]
- Patikoglou GA, Kim JL, Sun L, Yang SH, Kodadek T, Burley SK. TATA element recognition by the TATA box-binding protein has been conserved throughout evolution. Genes Dev. 1999;13(24):3217–3230. doi: 10.1101/gad.13.24.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. Lytic growth of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med. 1996;2(3):342–346. doi: 10.1038/nm0396-342. [DOI] [PubMed] [Google Scholar]
- Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Rooney DL, Zhang M, Ihrig MM, McManus MT, Gertler FB, Scott ML, Van Parijs L. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet. 2003;33(3):401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore PS. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8) Proc Natl Acad Sci U S A. 1996;93(25):14862–14867. doi: 10.1073/pnas.93.25.14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski I, Ma J, Triezenberg S, Ptashne M. GAL4-VP16 is an unusually potent transcriptional activator. Nature. 1988;335(6190):563–564. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- Sakakibara S, Ueda K, Chen J, Okuno T, Yamanishi K. Octamer-binding sequence is a key element for the autoregulation of Kaposi’s sarcoma-associated herpesvirus ORF50/Lyta gene expression. J Virol. 2001;75(15):6894–6900. doi: 10.1128/JVI.75.15.6894-6900.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarid R, Flore O, Bohenzky RA, Chang Y, Moore PS. Transcription mapping of the Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1) J Virol. 1998;72(2):1005–1012. doi: 10.1128/jvi.72.2.1005-1012.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp TV, Wang HW, Koumi A, Hollyman D, Endo Y, Ye H, Du MQ, Boshoff C. K15 protein of Kaposi’s sarcoma-associated herpesvirus is latently expressed and binds to HAX-1, a protein with antiapoptotic function. J Virol. 2002;76(2):802–816. doi: 10.1128/JVI.76.2.802-816.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MJ, Brown HJ, Wu TT, Sun R. Transcription activation of polyadenylated nuclear RNA by rta in human herpesvirus 8/Kaposi’s sarcoma-associated herpes-virus. J Virol. 2001;75(7):3129–3140. doi: 10.1128/JVI.75.7.3129-3140.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d’Agay MF, Clauvel JP, Raphael M, Degos L, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood. 1995;86(4):1276–1280. [PubMed] [Google Scholar]
- Staudt MR, Dittmer DP. The Rta/Orf50 transactivator proteins of the gamma-herpesviridae. Curr Top Microbiol Immunol. 2007;312:71–100. doi: 10.1007/978-3-540-34344-8_3. [DOI] [PubMed] [Google Scholar]
- Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. A viral gene that activates lytic cycle expression of Kaposi’s sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A. 1998;95(18):10866–10871. doi: 10.1073/pnas.95.18.10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R, Lin SF, Staskus K, Gradoville L, Grogan E, Haase A, Miller G. Kinetics of Kaposi’s sarcoma-associated herpesvirus gene expression. J Virol. 1999;73(3):2232–2242. doi: 10.1128/jvi.73.3.2232-2242.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor IC, Kingston RE. Factor substitution in a human HSP70 gene promoter: TATA-dependent and TATA-independent interactions. Mol Cell Biol. 1990;10(1):165–175. doi: 10.1128/mcb.10.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin HWt, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, Speck SH. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J Virol. 1997;71(8):5894–5904. doi: 10.1128/jvi.71.8.5894-5904.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SE, Wu FY, Fujimuro M, Zong J, Hayward SD, Hayward GS. Role of CCAAT/enhancer-binding protein alpha (C/EBPalpha) in activation of the Kaposi’s sarcoma-associated herpesvirus (KSHV) lytic-cycle replication-associated protein (RAP) promoter in cooperation with the KSHV replication and transcription activator (RTA) and RAP. J Virol. 2003a;77(1):600–623. doi: 10.1128/JVI.77.1.600-623.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SE, Wu FY, Yu Y, Hayward GS. CCAAT/enhancer-binding protein-alpha is induced during the early stages of Kaposi’s sarcoma-associated herpesvirus (KSHV) lytic cycle reactivation and together with the KSHV replication and transcription activator (RTA) cooperatively stimulates the viral RTA, MTA, and PAN promoters. J Virol. 2003b;77(17):9590–9612. doi: 10.1128/JVI.77.17.9590-9612.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinmaster G. Notch signal transduction: a real rip and more. Curr Opin Genet Dev. 2000;10(4):363–369. doi: 10.1016/s0959-437x(00)00097-6. [DOI] [PubMed] [Google Scholar]
- West JT, Wood C. The role of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus-8 regulator of transcription activation (RTA) in control of gene expression. Oncogene. 2003;22(33):5150–5163. doi: 10.1038/sj.onc.1206555. [DOI] [PubMed] [Google Scholar]
- Wu FY, Ahn JH, Alcendor DJ, Jang WJ, Xiao J, Hayward SD, Hayward GS. Origin-independent assembly of Kaposi’s sarcoma-associated herpesvirus DNA replication compartments in transient cotransfection assays and association with the ORF-K8 protein and cellular PML. J Virol. 2001;75(3):1487–1506. doi: 10.1128/JVI.75.3.1487-1506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, AuCoin DP, Huete AR, Cei SA, Hanson LJ, Pari GS. A Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J Virol. 2005;79(6):3479–3487. doi: 10.1128/JVI.79.6.3479-3487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Black JB, Goldsmith CS, Browning PJ, Bhalla K, Offermann MK. Induction of human herpesvirus-8 DNA replication and transcription by butyrate and TPA in BCBL-1 cells. J Gen Virol. 1999;80(Pt 1):83–90. doi: 10.1099/0022-1317-80-1-83. [DOI] [PubMed] [Google Scholar]