

ABSTRACT
Eukaryotic cells are sensitive to mechanical forces they experience from the environment. The process of mechanosensation is complex, and involves elements such as the cytoskeleton and active contraction from myosin motors. Ultimately, mechanosensation is connected to changes in gene expression in the cell, known as mechanotransduction. While the involvement of the cytoskeleton in mechanosensation is known, the processes upstream of cytoskeletal changes are unclear. In this paper, by using a microfluidic device that mechanically compresses live cells, we demonstrate that Ca2+ currents and membrane tension-sensitive ion channels directly signal to the Rho GTPase and myosin contraction. In response to membrane tension changes, cells actively regulate cortical myosin contraction to balance external forces. The process is captured by a mechanochemical model where membrane tension, myosin contraction and the osmotic pressure difference between the cytoplasm and extracellular environment are connected by mechanical force balance. Finally, to complete the picture of mechanotransduction, we find that the tension-sensitive transcription factor YAP family of proteins translocate from the nucleus to the cytoplasm in response to mechanical compression.
KEY WORDS: Ca2+ channels, Cell cortex, Mechanosensation, Tension-activated channel
Highlighted Article: Tissues can sense external mechanical forces. By using a microfluidic compression device, we found that Ca2+ is an important messenger during the early phase of mechanical sensation, and regulates RhoA activity.
INTRODUCTION
Mechanotransduction, the conversion of physical force into biochemical information inside the cell, is a complex process that regulates a large variety of physiological processes such as embryogenesis and tissue growth (Ingber, 2003; Orr et al., 2006). Dysregulation of mechanotransduction is implicated in the development of major human diseases, such as cancer and arteriosclerosis (Jaalouk and Lammerding, 2009; Paszek et al., 2005; Etienne-Manneville and Hall, 2002; Garcia-Cardena et al., 2001; Haga et al., 2007). The most upstream process in mechanotransduction is mechanosensation: the earliest step in which the cell senses changes in the external mechanical environmental and/or forces. It is known that the actin cytoskeleton and myosin contractility are required for mechanosensation and mechanotransduction. Although there are many studies that examine how forces regulate cytoskeletal dynamics in vitro (Parekh et al., 2005; Kovar and Pollard, 2004), precisely how external forces transmit a signal to the F-actin network and myosin in live cells during mechanosensation is unclear. In this paper, by mechanically compressing live cells, we identify that transmembrane Ca2+ currents and membrane tension-sensitive cation channels are responsible for activating RhoA GTPase, which regulates non-muscle myosin II assemblies in the cell cortex and cytoplasm. These experimental results, together with a mechanical model of the cell cortex, suggest that the cell maintains a homeostatic value of membrane tension, and activates myosin contraction in response to tension changes. This feedback loop leads to a dynamic adjustment of active stress generated by the cell, and ultimately can explain the main features of mechanosensation.
Cortical tension and myosin contraction in tissue cells are biochemically controlled by the Rho family of small GTPases, especially RhoA (Etienne-Manneville and Hall, 2002). RhoA switches between a GTP-bound, active state and a GDP-bound, inactive state, which signals to the Rho-associated kinases ROCK1 and ROCK2 (hereafter ROCK). ROCK phosphorylates myosin light chain (MLC), which then controls mini-filament assembly and generation of active contractile stress. Externally applied mechanical forces can trigger this RhoA-mediated response. For example, the amount of the active form of RhoA increases when cells are mechanically pulled by magnetic tweezers (Zhao et al., 2007; Scott et al., 2016). High shear stress (65 dyn/cm2) on bovine aortic endothelial cells leads to a decrease in RhoA activity (Liu et al., 2014), whereas low shear stress induces an initial increase in RhoA activity, which is followed by it returning to control levels after 10 min (Wojciak-Stothard and Ridley, 2003).
To investigate mechanosensation in live cells in real time, we established a microfluidic-based mechanical compression system in which the live cells can dynamically switch between a confined and un-confined status. A fluorescence resonance energy transfer (FRET)-based sensor is used to monitor the real-time response of RhoA activity in cells when they are subjected to different environments. We find that the mechanical compression leads to an immediate drop in RhoA activity as indicated by the RhoA FRET sensor. The decreased RhoA activity is maintained while the cell is compressed. Upon decompression, RhoA activity resumes to the original level. Either depriving cells of Ca2+ or blocking transient receptor potential cation channel subfamily V member 4 (TRPV4) significantly decreases the change in RhoA activity in response to the mechanical shock. Moreover, inhibiting myosin activity by use of blebbistatin does not affect RhoA activity change during compression. These results can be recapitulated in a computational mechanical model of cell mechanosensation where membrane and cortical tensions are explicitly connected to an externally applied force. Conceptually, the results and the model suggest that mechanosensation partly arises from a negative feedback control system that maintains a homeostatic membrane tension. To connect mechanosensation with downstream mechanotransduction, we further reveal that the Yes-associated protein (YAP) family of transcription factors largely left the nucleus and distributed more in the cytoplasm upon compression. This suggests that there is a direct link between physical forces, cell cortical tension and YAP transcriptional activity, as revealed by parallel studies in other settings (Dupont et al., 2011; Low et al., 2014).
Our results are relevant for understanding how cells respond to external mechanical forces and interact with physically confined environments. When metastatic cancer cells leave their primary tumor sites and migrate from a metastasis tumor, they move within and between three-dimensional tissues, capillaries and lymph nodes, the properties of which cannot be fully recapitulated by 2D Petri dishes (Fraley et al., 2010; Giri et al., 2013; Cukierman et al., 2001; Lu et al., 2012). Similarly, cells of the immune system, such as dendritic cells, also migrate within tissues to sample different environments (Alvarez et al., 2008). Cells also experience mechanical forces from the surrounding matrix (Wirtz et al., 2011) as well as from other cells in different environments (Humphrey et al., 2014; Swartz et al., 2001; Thiam et al., 2016; Le Berre et al., 2012). Our work identifies the most-upstream signals that allow cells to respond to mechanical forces and physical changes. Identification of Ca2+ as the primary signal is also consistent with observations in cells under mechanical stretch (Kim and Sun et al., 2014), and opens doors to manipulate the mechanosensation properties of live cells.
RESULTS
The air-driven microfluidic device to compress mammalian cells
To investigate the effects of mechanical compression on RhoA activity in mammalian cells, we designed an air-driven microfluidic compression device. A similar device was previously used to compress bacterial cells to investigate cell growth (Si et al., 2015). As illustrated in Fig. 1A, the device comprised an upper and a lower chamber separated by a polydimethylsiloxane (PDMS) layer of ∼200 μm in thickness. The upper chamber can be inflated by air pressure, which deforms the PDMS membrane downwards and applies mechanical compression on the mammalian cells cultured in the lower chamber. To precisely control the compression on the cells, micropillars were introduced to the device. Micropillars were made of PDMS and assembled onto the bottom of the lower chamber. The pillars support the PDMS membrane, and provide a maximum limit to the downward membrane movement, and thereby control the degree of compression of the mammalian cells. The height of the pillar is ∼4 µm, which is smaller than the typical height of the adherent HT1080 cells on 2D substrates (∼8–15 µm). The size of the pillar is 500 µm by 500 µm, with 200 µm distance between them.
Fig. 1.

Response of HT1080 cells to mechanical compression. (A) Experimental setup for the compression device. Fabrication of the device follows the method in Si et al. (2015). Cells in the device are compressed by the application of air pressure above the PDMS layer. The compression depth is limited by the support pillars with height 4–6 µm. (B) Epifluorescence image of cell (YFP channel) before and after compression, and after the release of compression. The cross-section area of the cell increases under compression, and decreases after the release of compression. Scale bars: 20 µm. (C) Scaled FRET-to-CFP ratio (FRET/CFP) and cell spreading area over time. Compression takes place at t=0 min (indicated by the black arrow). Cells are then released at t=38 min (indicated by the red arrow). Results are mean±s.e.m. (n=40). (D) Summary of mean±s.e.m. FRET/CFP when cell is uncompressed, compressed and released from compression. All results are scaled to the mean value of uncompressed cells and represent three biological replicates (n=40 cells). For each biological replicate, there is one technical replicate. ***P<0.001; n.s., not significant (Student's t-test).
To investigate the effects of mechanical compression on RhoA activity in mammalian cells, we used HT1080 cells stably expressing a RhoA FRET sensor. The intracellular sensor comprised two fluorescent proteins, RhoA GTPase bound to fluorescent protein, and another fluorescent protein conjugated to the RhoA-binding domain of the Rho effector PKN (van Unen et al., 2015). When RhoA-GDP is converted into RhoA-GTP, a PKN1 moiety binds RhoA-GTP, giving a high FRET state that is detected as an increase in the ratio of sensitized emission to CFP fluorescence. We tested the sensor by applying a Rho activator and inhibitor. Results showed that the activity of RhoA sensor changed correspondingly as the activator or inhibitor was added to the cells (Fig. S1B).
Before loading cells to the compression device, the cell culture chamber of the device was coated by 0.2 µg/ml collagen I to ensure the proper adherence and spreading of the cells onto the PDMS substrate. Then the cells were flown into the culture chamber through tubing. After cells adhered and spread onto the collagen-coated PDMS surface, a moderate pressure (∼10 psi or 68 kPa) was applied through the tubing to the air chamber. The pressure was kept constant by a pressure regulator. The downward movement of PDMS layer between the air chamber and the cell culture chamber stopped when the layer contacted micropillars, which applied a defined mechanical deformation on the HT1080 cells (Fig. 1A,B). During compression, a temperature of 37°C was maintained and fresh medium was supplied in a constant flow.
Instantaneous and reversible RhoA activity change upon compression
Previous work has shown that RhoA activity increases and then decreases when cells are pulled by magnetic beads mechanically (Zhao et al., 2007). It is similarly interesting to probe the change of RhoA activity when live single cells are compressed vertically. We observed that the morphology of HT1080 cells changed instantaneously upon compression, as shown by a sudden increase in the observable size of the cell (Fig. 1B, middle panel; Fig. 1C). The ratio of the FRET channel to the donor CFP channel, which is an indicator of the RhoA activity, dropped significantly after the cells were compressed by the air-driven PDMS layer (Fig. 1C,D). The decrease in Rho activity remained constant throughout compression. To investigate whether such changes are reversible, we released the compression by turning off the air flow after 30 min of compression. We found that the RhoA activity increased quickly (within minutes) back to the original value prior to compression, while there was also a simultaneous recovery in cell size (Fig. 1B–D). These results showed that changes in cell size and the RhoA activity are both instantaneous and reversible under compressive mechanical deformation. We also monitored and quantified the fluorescence intensity of YFP channel during the experiment, which should remain constant as long as the expression of RhoA sensor remains unchanged. Results showed that compression and release of compression did not affect the YFP intensity (Fig. S1C), which further validates that changes in the ratio between the fluorescence intensities of FRET and the CFP channel during compression and de-compression are not due to optical artifacts.
Response of RhoA activity to mechanical compression is dependent on Ca2+
In the C-terminal region of RhoA, there is a binding site for calmodulin, which is a ubiquitous transducer of Ca2+ second messenger. A fusion protein was previously designed in which the activity of RhoA was controlled by Ca2+ through calmodulin (Mills et al., 2010). Moreover, previous research showed that Ca2+ influx is necessary for RhoA activation during human umbilical vein endothelial cell spreading on type IV collagen (Masiero et al., 1999). To investigate whether Ca2+ is required for the response of RhoA activity to vertical compression, we incubated cells in Ca2+-free medium after they attached and spread onto the substrate in the compression device.
After cells were incubated in Ca2+-free culture medium for 10 min, they were subjected to compression within the device. We observed that cells in Ca2+-free medium for 10 min had slightly reduced RhoA activity (Fig. 2A,B). After the cells were compressed, RhoA activity still was reduced significantly. However, the relative decrease in RhoA activity for cells incubated in Ca2+-free medium was smaller compared to the change for cells incubated in normal culture medium (Fig. 2B,C). We also treated cells with Ca2+-free medium for 30 min before compression. We find that longer incubation of cells in Ca2+-free medium significantly decreased the activity of RhoA in cells without compression (Fig. 2B,C). The relative decrease in RhoA activity after compression was much smaller compared with that in cells in control medium and in Ca2+-free medium for a shorter incubation time (Fig. 2B). To check that these changes are indeed related to reduced intracellular Ca2+, we imaged HT1080 cells treated with a fluorescent Ca2+ dye (see Fig. 4). Results show that cells in Ca2+-free medium indeed have a lower total intracellular Ca2+. Taken together, our results on HT1080-RhoA cells in the compression device suggest that both the baseline RhoA activity and the response of RhoA to mechanical compression are Ca2+ dependent.
Fig. 2.

Response of cells to mechanical compression when they are incubated in Ca2+-free medium. (A) Epifluorescence images (YFP channel) of cells before and after compression. When cells are incubated in Ca2+-free medium, there is a significant increase in the number of membrane blebs even without compression. Scale bars: 20 µm. (B) The scaled FRET-to-CFP ratio (FRET/CFP) before and during the mechanical compression, for the three conditions in A. Compression takes place at t=0 min. The FRET/CFP ratio is scaled to the average FRET/CFP before compression for each respective control experiment. (C) Time averaged plots of FRET/CFP corresponding to panel B. Each set of results is scaled to its respective control (uncompressed) mean FRET/CFP value. For each set, there are three biological replicates, and for each biological replicate, there is one technical replicate. Results are mean±s.e.m.; n=50 cells for the control data, n=47 cells for 10 min Ca2+-free medium, n=27 cells for 30 min Ca2+-free medium. *P<0.01; **P<0.001; ***P<0.0001 (Student's t-test).
Fig. 4.

Intracellular Ca2+ dye signal after cells are treated with TRPV4 inhibitor or incubated in Ca2+-free medium for 30 min. (A) Image of a cell in the DIC channel and the fluorescence channel showing Ca2+ dye in the cytoplasm. Scale bar: 20 µm. (B) Total intracellular Ca2+ dye signal over time for HT1080 cells with no treatment (control), TRPV4 inhibitor treatment and Ca2+-free medium treatment. The Ca2+ dye signal is generally stable for 15 min after imaging. The slight decrease in signal is due to dye leakage out of the cell, and occurs for all conditions. (C) Population and time average of total intracellular Ca2+ dye signal for control, TRPV4 inhibitor treatment and Ca2+-free medium treatment. Results are mean±s.e.m.; the number of cells in each set of experiments is labeled in C; there were two biological repeats and two technical repeats for each biological repeat in all conditions. ***P<10−9 (Student's t-test).
TRPV4 channels mediate the change of RhoA activity during mechanical compression
We then set out to identify the mechanosenstive element that mediates the response of RhoA activity to mechanical compression. Recent studies have shown that mechanosensitive membrane ion channels can regulate the activity of RhoA (Simoes Sde et al., 2010; Pare et al., 2014; Kolesnikov and Beckendorf, 2007). We are especially interested in TRPV4, a member of the TRP nonselective cation channel superfamily, because of its Ca2+ permeability (Sokabe et al., 2010; Seminario-Vidal et al., 2011). TRPV4 channels are expressed in both neuronal and non-neuronal cells, including HT1080 cells. Channel activation allows cation influx into cells, leading to various Ca2+-dependent processes. We have identified the role of Ca2+ in the response of RhoA activity to vertical compression, which suggests the potential involvement of TRPV4 channel in regulating RhoA activity.
To investigate the role of TRPV4 in RhoA activity and the response to mechanical compression, we treated cells with a TRPV4 inhibitor before compressing them. We observed that incubating cells in TRPV4 inhibitor for 10 min did not affect RhoA activity before compression (Fig. 3A-C). Compressing the cells in the presence of TRPV4 inhibitor also led to an instantaneous drop in RhoA activity; however, the magnitude of the change in RhoA activity due to compression was significantly smaller than that in control cells (Fig. 3B,C). RhoA activity in cells was reduced before compression after being treated with TRPV4 inhibitor for a longer period of time, i.e. 30 min (Fig. 3B,C). There was essentially no change in RhoA activity after the cells are compressed in the presence of TRPV4 inhibitor. Imaging these cells treated with Ca2+ dye shows that they have reduced intracellular Ca2+ (Fig. 4). These results are consistent with those from the cells incubated in Ca2+-free medium, suggesting that TRPV4 channels are indeed regulating the change of RhoA activity to mechanical compression through regulating Ca2+ influx.
Fig. 3.

The response of cells to mechanical compression when they are incubated with TRPV4 inhibitor. (A) Epifluorescence images (YFP channel) of cells before and during compression. Scale bars: 20 µm. (B) The scaled FRET-to-CFP ratio (FRET/CFP) before and during mechanical compression, for all three conditions in A. Compression takes place at t=0 min. The FRET/CFP ratio is scaled to the average FRET/CFP before compression for each respective control experiment. (C) Time averaged plot of FRET/CFP corresponding to panel B. Each set of results are scaled by its respective control, uncompressed mean FRET/CFP value For each set, there are three biological replicates, and for each biological replicate, there is one technical replicate. Results are mean± s.e.m.; n=57 cells for the control data, n=34 cells for 10 min TRPV4 inhibitor; and n=30 cells 30 min TRPV4 inhibitor. ***P<0.0001 (Student's t-test).
Mathematical model of cell response to mechanical compression
The observed changes in Rho activation and the role of Ca2+ flux can be recapitulated in a mathematical model of the cell response to mechanical compression. When an external compression force, Fext, is applied to the cell, the applied force alters the overall force balance at the cell surface (Fext is negative if it is a compressive force). Mathematically, this force balance in the surface normal direction is expressed as (Tao and Sun, 2015; Tao et al., 2017):
| (1) |
where σa is the active contractile stress in the cell cortex, h is the cortical thickness, T is the membrane tension, ΔP is the hydrostatic pressure difference across the membrane, H is the cell surface mean curvature, and A is the area over which the external force is applied. Fext is the external mechanical force experienced by the cell, which is negative if it is a compressive force. When the force is negative, the membrane tension is immediately lowered because Fext subtracts from ΔP, especially at the apical surface. Therefore, the myosin tension and membrane tension needed to balance the reduced pressure is lowered. Mechanically, compressive Fext directly influences the membrane tension, T, and results in reduced opening of TRPV4 and activity of RhoA. Changes in the amount of active RhoA leads to changes in σa, which re-adjusts membrane tension back to the homeostatic value. The opposite scenario is when a pulling force is applied to the cell as in Zhao et al. (2007) and Kim and Sun et al. (2014). In this case, the membrane tension must increase, resulting in an increase in opening of TRPV4 and activity of RhoA. This feedback control loop can be expressed using kinetic equations as:
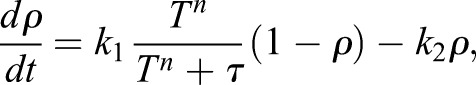 |
(2) |
where ρ is the proportion of active RhoA, and k1,2 are rate constants modeling activation and de-activation. The RhoA activation rate also depends on membrane tension and Ca2+ through a Hill function, and τ is an activation threshold parameter. This Hill function explicitly describes the activity of TRPV4 as a function of membrane tension. As shown in Fig. 5A, inset, the Hill function, Λ(T)=Tn/(Tn+τ), can be approximated by a piecewise linear function. The activation threshold parameter, τ, determines the critical membrane tension, Ts, above which the channel is fully open. The active contraction is modeled as directly proportional to the amount of active RhoA [σa(t)=Γρ(t)], in which Γ is the contractile stress in the situation where RhoA is fully activated. Parameters of the model are given in Table 1.
Fig. 5.

Theoretical prediction of cells under mechanical compression. (A) Mechanistic diagram of our model. Membrane tension, T, and the active contractile stress from myosin contraction, σa, are combined to balance the hydrostatic pressure difference across the cell membrane ΔP. Here, H is local mean curvature and Fext is an externally applied force over the apical region of the cell, A. Membrane tension changes influence Ca2+ influx through the TRPV4 channel, which influences Rho activity. The opening probability of TRPV4 channel is described by a Hill function, which can be approximated by a piecewise linear function. (B) Model prediction of RhoA activity in a cylindrical cell in response to vertical compression when the cell maintains a constant cross-sectional area and constant volume. Depending on the extent of vertical compression, reduction of RhoA activity is observed when the cell reduces volume. If the volume remains constant, RhoA activity generally increases. In the experiment, there is a wide range of vertical compression. The observed average RhoA activity decrease suggests that there is a slight reduction in cell volume.
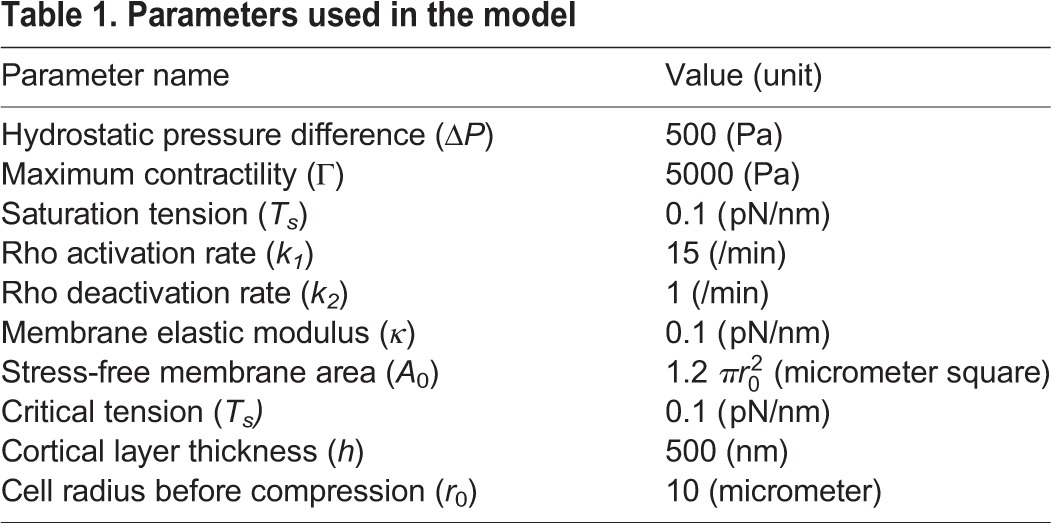
Table 1.
Parameters used in the model
This minimal model can approximately capture the behavior seen in experiments. Moreover, since mammalian cells can actively control the cytoplasmic pressure by adjusting their osmolyte and water content, water flows out of the cell when the hydrostatic pressure is increased, which reduces volume and membrane tension (Tao and Sun, 2015; Tao et al., 2017; Jiang and Sun, 2013). When active control of σa and ΔP are both incorporated, the model is able to capture the rate-dependent response of cells to external mechanical forces. Note that the force balance condition in Eqn 1 assumes that the membrane does not detach from the cortex and there is no membrane blebbing. If there is blebbing, membrane tension changes are more dramatic, and the dynamic remodeling of the cortex and adjustment in contractility are complex.
Based on this model, we can examine two extreme scenarios when the cell is under mechanical compression: cells that maintain a constant cross-sectional area and cells that maintain a constant volume (i.e. constant ΔP, no water flow across the membrane). When cells maintain a constant cross-sectional area, the lateral membrane tension decreases as the volume of the cell decreases. In addition, the apical membrane surface of the cell in contact with the compression surface will experience lower tension. These factors combined suggest that RhoA activity goes down. When cells maintain a constant volume, lateral membrane tension increases as the cell is compressed in the vertical direction. Therefore, RhoA activity would increase on the lateral surfaces and the overall measured RhoA activity would increase.
In Fig. 5, we compare and contrast these two scenarios for a model cylindrical cell under compression. Different degrees of compression and computed RhoA activities are plotted as functions of time. We find that the experimental results are best explained by the case where water can flow out of the cell (Fig. 6). In the next section, we also use the same model to compute the cell response for a more realistic cell geometry and find a similar result. We can conclude that when the cell is under vertical compression, the behavior is somewhere in between the model extremes discussed above: the overall cell volume decreased, despite the fact that the cell cross-sectional area increased, leading to a lower membrane tension and lower RhoA activity. In the next section, we also discuss the case where the cell surface area remains constant during compression. In this case, the model predicts that Rho activity would remain constant and the cell internal hydrostatic pressure would adjust to mechanical compression through osmotic regulation (Fig. 6).
Fig. 6.

Model predictions with realistic cell geometry. (A) Schematic description of the cell geometry before and after compression. (B) Corresponding RhoA activity, overall membrane area and membrane tension in response to vertical compression from the model. With realistic cell geometry, results are similar to the predictions from the simplified model. (C) Model prediction of the corresponding hydrostatic pressure difference in response to vertical compression, when the cell surface area is assumed to be constant. Here, osmotic regulation by the cell ensures hydrostatic pressure difference remains constant at steady state before and after compression. Compression takes place at t=0 min and the cell is released from compression at t=17 min.
Model with realistic cell geometry
In the compression device, most of the cells are not cylindrical in shape. Instead, they form an adhesion with the substrate and the shape of the apical surface can vary. Therefore, we examine the model with a general assumption on cell geometry. Because of the complexity in the cell shape during the dynamic compression-release process, we are only able to compute the steady state results before and after compression.
Before compression takes place, we assume the cell is a hemisphere, as shown in Fig. 6A. Therefore, the force balance equation (Eqn 1) becomes:
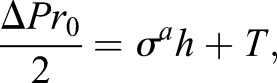 |
(3) |
where r0 is the radius of the cell. To close the system, we can assume a linear constitutive relation for the membrane tension:
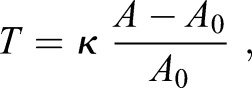 |
(4) |
where κ is the effective stretching modulus of plasma membrane, and A0 is the reference membrane area when the membrane is not under any forces.
When compression takes place, we assume the cell, with height L, is symmetric about both the x (horizontal) and y (vertical) axis (shown in Fig. 6A). The top and bottom surfaces (z=L/2, z=−L/2) are almost flat, and therefore, the membrane tension at top and bottom surfaces is zero. As a result, what really contributes to RhoA signal pathway is the membrane tension at the apical surface. Based on these assumptions, we can parametrize the cell surface using the cross-sectional radius, r, and vertical axis, y (in which y is ranging from –L/2 to L/2). The force balance equation (Eqn 1), then becomes:
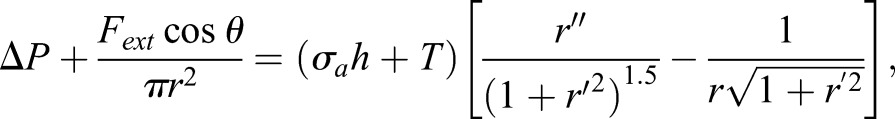 |
(5) |
in which 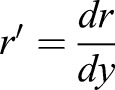 and
and 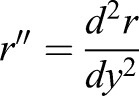 . At contact area (y=L/2),
. At contact area (y=L/2), 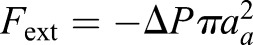 ; in which aa is the radius of contact area between the cell and PDMS layer. aa may depend on the externally applied vertical strain, as discussed in the previous study (Jiang and Sun, 2013). θ is the angle between cell's local normal unit vector and y axis:
; in which aa is the radius of contact area between the cell and PDMS layer. aa may depend on the externally applied vertical strain, as discussed in the previous study (Jiang and Sun, 2013). θ is the angle between cell's local normal unit vector and y axis:  (Fig. 6), and
(Fig. 6), and 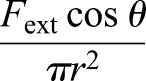 gives the normal component of vertical compression. One boundary condition for Eqn 5 is: r′=0 at y=0, because of symmetry. Based on the experimental findings, the maximum cross-sectional area increased ∼50% after the cell is compressed, or,
gives the normal component of vertical compression. One boundary condition for Eqn 5 is: r′=0 at y=0, because of symmetry. Based on the experimental findings, the maximum cross-sectional area increased ∼50% after the cell is compressed, or, 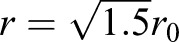 , at y=0 (the reference coordinate is shown in Fig. 6A).
, at y=0 (the reference coordinate is shown in Fig. 6A).
Theoretically, the exact cell geometry, membrane tension and proportion of active RhoA can be solved by coupling Eqn 5, with Eqn 4 and Eqn 2. [Eqn 4 becomes: 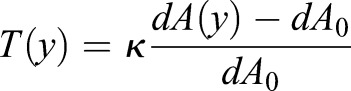 , for spatially varying mean curvature; where dA is local area element as a function of y:
, for spatially varying mean curvature; where dA is local area element as a function of y:
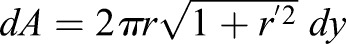 ; and dA0 is local area element of stress-free membrane: dA0=2πr0dy]. However, solving these coupled equations proves to be difficult. This degree of complexity is added when we are dealing with spatially varying membrane tension and σa. We can simplify the model by assuming membrane tension and active contraction is spatially independent. With this assumption, we can examine how cells are going to behave under a 50% vertical compressive strain. We find that the overall cell volume and membrane area decreased, even though the maximum cross-sectional area increased by ∼40%, as we observed in the experiment (Fig. 1). Therefore, membrane tension decreased and RhoA activity goes down as a result.
; and dA0 is local area element of stress-free membrane: dA0=2πr0dy]. However, solving these coupled equations proves to be difficult. This degree of complexity is added when we are dealing with spatially varying membrane tension and σa. We can simplify the model by assuming membrane tension and active contraction is spatially independent. With this assumption, we can examine how cells are going to behave under a 50% vertical compressive strain. We find that the overall cell volume and membrane area decreased, even though the maximum cross-sectional area increased by ∼40%, as we observed in the experiment (Fig. 1). Therefore, membrane tension decreased and RhoA activity goes down as a result.
Cylindrical cells with constant surface area
In the above section, we discussed the behavior of cylindrically shaped cells with constant cross-sectional area and constant volume during compression and release. In both cases, we assumed that the membrane tension is linearly proportional to areal strain. There is, however, another possibility where cells keep a constant surface area. In this scenario, the membrane tension must be a constant during compression and therefore, the active contraction remains unchanged. If we assume cells are cylindrical in shape both before and after compression, keeping a constant surface area implies an increase in cell volume when the cells are compressed. According to our previous studies (Tao and Sun, 2015), the pressure difference across the cell membrane are: 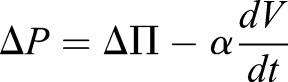 , where ΔΠ, the osmotic pressure difference across the cell membrane, is proportional to solute concentration difference, and α is membrane permeation constant. Since immediately after compression, the solute content does not change, a sudden increase in cell volume means a decrease in hydrostatic pressure difference. The mechanical force balance condition, ΔP=(σah+T)/R, predicts that the radius of curvature, R, will increase to compensate the decrease in hydrostatic pressure difference. Active and passive ion flows across the cell membrane (Tao and Sun, 2015) will occur to adapt to the sudden change in pressure difference. At steady state, the pressure difference recovers to the pre-compression state, as shown in Fig. 6.
, where ΔΠ, the osmotic pressure difference across the cell membrane, is proportional to solute concentration difference, and α is membrane permeation constant. Since immediately after compression, the solute content does not change, a sudden increase in cell volume means a decrease in hydrostatic pressure difference. The mechanical force balance condition, ΔP=(σah+T)/R, predicts that the radius of curvature, R, will increase to compensate the decrease in hydrostatic pressure difference. Active and passive ion flows across the cell membrane (Tao and Sun, 2015) will occur to adapt to the sudden change in pressure difference. At steady state, the pressure difference recovers to the pre-compression state, as shown in Fig. 6.
Change of YAP/TAZ subcellular localization upon mechanical compression
Recent research has revealed the significant role that YAP plays in relaying the mechanical signal in extracellular matrix (ECM) to nucleus (Dupont et al., 2011). Previous work has also shown that the subcellular location of YAP/TAZ is regulated by cell tension and cell geometry (i.e. the cell spreading imposed by the ECM) (Dupont et al., 2011; Wada et al., 2011; Aragona et al., 2013). In our compression device, we observed that the observed area of the compressed cells changes significantly in morphology. Thus, we hypothesized that the subcellular localization of YAP also changes upon mechanical compression.
To test our hypothesis, we fixed the compressed HT1080 cells 30 min or 13 h after subjecting the cells to compression, and then we permeabilized the cells and stained them for YAP. Cells in the uncompressed area showed that the predominant localization of YAP was in cell nucleus (Fig. 7A, top panel). On the other hand, cells under compression showed a much higher level of YAP staining in the cytoplasm (Fig. 7A, bottom panel). With the help of H2B–mCherry, a fluorescent labeled histone protein, we were able to quantify the amount of YAP staining in the nucleus and in the cytoplasm. Results showed that there was a dramatic and quick change in the YAP subcellular localization upon mechanical compression. The ratio between YAP in nucleus and in cytoplasmic decreased by more than 60% after compression, that is, most of the YAP ‘leaked out’ the nucleus. There was no difference in cells compressed for 30 min or 13 h (Fig. 7B). Interestingly, we also found that, while YAP ‘leaks out’ of the nucleus when cells are compressed, cells are expressing more overall YAP, as shown in Fig. S2.
Fig. 7.

YAP expression level before and after compression. (A) Images of fixed and stained cells before (top panel) and after (bottom panel) mechanical compression. Scale bars: 20 µm. (B) Ratio between the nuclear YAP and cytoplasmic YAP before and after compression. ∼70% of nucleus YAP ‘leaked out’ into cytoplasm after compression. The equilibrium is reached at ∼30 min after compression takes place. Results are mean±s.e.m.; n=60 cells before compression, n=70 cells for post-compression after 30 min, n=84 cells for post-compression after 13 h. (C) The change in the localization of YFP during mechanical compression reduces in the Ca2+-deficit condition, such that the mechanosensation of YAP is suppressed. Results are mean±s.e.m.; n=23 cells for control, pre-compression data, n=30 cells for control, post-compression data, n=27 cells for Ca2+-free, pre-compression data and n=36 for Ca2+-free, post-compression data. All data are from three biological replicates; for each biological replicate, there is one technical replicate. *P<0.01; **P<0.001; ***P<0.0001 (Student's t-test).
We further investigated the response of YAP localization in Ca2+-free medium. Results showed that export of YAP from the nucleus to the cytoplasm during compression was suppressed when the cells were deprived of Ca2+, suggesting that the translocation of YAP during mechanical compression is dependent on Ca2+ (Fig. 7C).
DISCUSSION
The physical environment of the cell can influence many aspects of its function, and cells have developed sensory systems to respond to environmental changes. By examining cells under mechanical compression, we discover that the activity of RhoA changes quickly in response to the externally applied force. Under compression, RhoA activity decreases, and it recovers if the compression is removed. We found that RhoA activity is regulated by Ca2+ and the mechanosensitive cation channel TRPV4. When TRPV4 is blocked or Ca2+ currents are diminished, RhoA activity decreased before mechanical compression, and the response to compression is also less pronounced. This behavior suggests that, under normal conditions, the cell membrane is a tension sensor, and possibly through small tension fluctuations, TRPV4 is constantly signaling to RhoA to maintain the appropriate level of contraction. When TRPV4 is partially blocked or Ca2+ currents are absent, the ability to maintain and sense tension changes is reduced. Since RhoA is directly involved in phosphorylation of MLC and generation of active contractile stress, these results implicate Ca2+ as a major regulator of cellular mechanosensation, in agreement with evidence from other lines of investigation (Thiam et al., 2016; Hung et al., 2016). Moreover, these results suggest that the cell membrane and associated channels are major elements in mechanosensation, in agreement with previous suggestions (Haswell et al., 2011; Venkatachalam and Montell, 2007). Our results indicate that on short time scales (in the order of minutes), the RhoA sensor FRET activity decreases first, mediated by a reduction in Ca2+ influx.
The results also suggest that myosin contraction is involved in regulating cell membrane tension. This is consistent with a theoretical model of the cell cortex where cortical contraction and membrane tension are together balancing excess hydrostatic pressure in the cell. The excess hydrostatic pressure, which is of the order of 100–1000 Pa, is generated from excess osmotic pressure inside the cell. Most of this excess pressure is balanced by cortical contraction, and membrane tension is maintained at a low value. During rapid changes in osmolarity or externally applied mechanical force, the membrane tension changes rapidly, which changes the Ca2+ currents and myosin contraction, so that the membrane tension can be restored back to the homeostatic value. Therefore, the cortex acts as an active mechanical surface that dynamically adjusts its tension to changing external osmotic or mechanical perturbations. The active adjustment of cortical contraction is mediated by RhoA activity, signaled by Ca2+ currents. The theoretical model of the cell cortex can fully explain the observed data, and predicts many features of active responses of cells to external forces and varying stiffnesses of the cell substrate.
If we attempt to alter myosin activity through non-Ca2+-related pathways, for example by incubating cells in 25 μM blebbistatin for 1 h, our experimental results show that, on average, blebbistatin only decreases RhoA activity slightly, and the relative drop in RhoA activity due to mechanical compression is similar to that seen in the control experiment (Fig. S3). This means that blebbistatin, although it suppresses myosin contractility, does very little to influence mechanosensitivity of RhoA, which is upstream of myosin. We also find that mechanical compression also directly impacts the YAP nuclear localization on the time scale of hours. YAP has been identified as a mechanosensitive transcription factor directly involved in mechanotransduction. Our results are consistent with the idea that YAP is sensitive to the mechanical state of the cell. In addition, our data reveal that RhoA and myosin contraction should change rapidly (in minutes) in response to changes in mechanical force. Therefore, YAP activity might be directly regulated by Rho or myosin activity.
As an additional observation, we find that the mechanosensitivity of YAP may also depend on the cell shape, as more rounded cells have more YAP ‘leaking out’ of the nucleus compared to more elongated cells (Fig. S2B,C). However, this may be due to the fact that more rounded cells are taller, and, therefore, are more compressed than the elongated cells.
Taken together, our mechanical compression experiments and model reveal that cells sense mechanical changes through changing membrane tension and Ca2+ currents, and dynamically adjust contractile forces to balance external forces. The active regulation of cell contractile forces can also serve as a signal to change transcription factor localization and protein expression changes that completes the process of mechanotransduction.
MATERIALS AND METHODS
Cell culture
Human fibrosarcoma HT1080 (ATCC) cells were cultured in high glucose (4.5 g/l) Dulbecco's modified Eagle's medium (Mediatech), supplemented with 10% fetal bovine serum (Hyclone) and 1% penicillin-streptomycin (Sigma). The cells were maintained in an incubator with 5% CO2 at 37°C. HT1080 cells stably expressing the RhoA FRET sensor was developed by Dr Yi Wu at the University of Connecticut, USA (van Unen et al., 2015), and were recently checked for contamination.
Preparation of microfluidic devices
Molds to print the cell culture chamber and air chamber were fabricated with negative photoresist (SU8-2100, MicroChem). Typical soft lithography procedure was applied to fabricate our microfluidic devices. Briefly, a 200 μm-thick layer of SU8-2100 photoresist (MicroChem Corporation, Newton, MA) was spun-coated onto a silicon wafer and cross-linked by UV light exposure through a photomask. Developer was used to remove non-crosslinked photoresist. Then, a 200 μm thick layer of PDMS (1:10 of agent to base, Sylgard 184, Dow Corning.) was spun onto the mold of culture chamber, and 7 mm-thick PDMS was poured onto the mold of air chamber. Both layers with half-cured PDMS were carefully aligned and then baked until completely cured. The mold for micropillars of 4 μm height was fabricated by patterning negative photoresist following similar procedures to those described above. Then, a 100 μm-thick layer of PDMS was spun onto the mold and baked until fully cured. The PDMS layer with pillars was carefully peeled off the mold and assembled onto a coverglass, with pillars facing the air. The height of micropillars was measured by profilometer (Dektak IIA). The channel device containing both the air chamber and cell culture chamber, a 5-mm circular coverglasses and the micropillar PDMS on glass were treated with air plasma. The circular cover glass was then put in the middle of the air chamber. Finally, the channel device with the cover glass was assembled with the micropillar PDMS, with pillar side facing the air chamber.
Before the experiment, 0.2 µg/ml collagen I solution was added into culture chamber and left to incubate at 37°C for 1 h for coating. Cells were then injected into culture chamber through tubing. After cells adhered and spread onto the collagen-coated PDMS surface, a moderate pressure (∼10 psi or 68 kPa) was applied through the tubing to the air chamber. The pressure was kept constant by a pressure regulator.
Treatment of cells in the compression device
After cells were perfused into the culture chamber of the device, they were allowed to attach and spread to the collagen-coated PDMS bottom for 2 h. Then Ca2+-free medium or normal cell culture medium with drug was perfused into the cell culture chamber through tubing. At least 3 ml of medium was used to ensure the existing medium was all replaced. Cells were returned to incubator. After 10 to 30 min, the compression device containing cells was transferred to the live-cell unit mounted on the microscope, followed by imaging and compression. The concentration of TPRV4 inhibitor (HC-067047, Sigma-Aldrich) was 5 µM. The concentration of blebbistatin was 25 µM.
Fixation and immunostaining cells in the compression device
After cells were compressed for 30 min or overnight, the live-cell unit was turned off. PBS was perfused into the device slowly at room temperature. 3.7% paraformaldehyde (PFA) was then perfused in at 1 ml/h using a syringe pump for 30 min. Mechanical compression on the cells was released by removing air pressure. After permeabilizing cells using 0.1% Triton-X in PBS, primary antibody detecting YAP/TAZ (1:100, cat. no. sc-101199, Santa Cruz Techonology) was perfused into the device. Cells in the device WERE incubated with the primary antibody at 4°C overnight and then incubated with fluorescent secondary antibody for 2 h at room temperature. Unbound antibodies were removed by continuously perfusing PBS through the device for 30 min at 3 ml/h using a syringe pump.
Measurement of Ca2+ content using Ca2+-indicating dye
The Ca2+-indicating dye we use is from the Fluo-4 Direct Ca2+ assay kit (from Thermo Fisher). A 2× stock was prepared according to the manufacturer's recommended protocol. The 2× stock was then diluted to 1× working concentration with normal cell culture medium, normal cell culture medium with TRPV4 inhibitor or Ca2+-free medium according to the ensuing experiments.
Before the media with the Ca2+-indicating dye were used, the cells were pre-incubated in the normal medium for 4 h. During the 4 h of incubation, some cells were treated with TRPV4 inhibitor or incubated with Ca2+-free medium for 30 min, according to the planned experiments. The method of treating cells with TRPV4 inhibitor and Ca2+-free medium is described above. Then, we replaced the media with respective media together with Ca2+-indicating dye. After incubating cells with Ca2+-indicating dye for a further 2 min, we washed out the Ca2+ dye with respective media. Then, the cell culture was mounted on the microscope, ready for imaging. For each experiment, we imaged the cell for 15 min with 1 s exposure time. Pictures are taken for every 2 min. Example of a cell under differential interference contrast (DIC) channel and Ca2+ dye channel is shown in Fig. 4.
The approximate cell boundary was obtained from the DIC channel by measuring the local contrast, which is shown as the blue line in Fig. 4A. The approximated cell boundary is expanded for 10 pixels, in order to capture all the scattered fluorescent light (red line in Fig. 4A). The cell boundary is further expanded for 50 pixels (black line in Fig. 4A). The mean pixel intensity between the red line and the black line is used as approximated background intensity. Then, the total Ca2+ dye signal within one cell is calculated by summing up the pixel intensities within the red contour after subtracting the approximated mean background intensity. The data is normalized to the mean Ca2+ dye signal of the control set at initial time point.
The intracellular Ca2+ dye signal is shown in Fig. 4B. Cells stably express Ca2+ dye during the 15 min period of imaging. The cells being treated with TRPV4 inhibitor or incubated under Ca2+-free medium for 30 min have a significant lower total intracellular Ca2+ dye signal compared to the control set (Fig. 4B,C). Note that the indicator dye does leak out of the cell over time, and the leakage is increased during cell compression. Therefore, we did not use this method to measure Ca2+ level changes during compression.
Image acquisition
All images were acquired with a Nikon TE2000E epifluorescence microscope (Nikon) equipped with a Luca-R CCD camera (Andor Technology), an X-cite illuminator (Excelitas Technologies) and a 40× water immersion objective, NA 1.2, WD 200 μm (Nikon). Each cell was imaged using three scans: donor, FRET and acceptor, with the following band-pass filters: CFP (excitation, 436/20; emission, 480/40); YFP (excitation: 470/40; emission, 525/50); FRET (excitation, 436/20; emission, 535/40). All images were taken with a 1-s exposure time. The pixel to length ratio is 1 pixel equals to 0.2 µm.
Image analysis and data acquisition
Since the pixel intensities within the cell region were much higher than the pixel intensities in the background region, a binary image based on the pixel intensities can be generated for each field of view, where pixels with high intensities in the cell region are marked with 1 and pixels elsewhere are marked with 0. We then used the MATLAB routine, ‘bwboundaries’, to trace the cell boundary from the binary image. Every traced region that had an area that was 1500 pixels square (∼60 µm square) or lower was considered as debris or a cell fragment, and, therefore, was ignored.
For live HT1080 cells stably expressing the RhoA FRET sensor, we used the YFP channel to trace the cell boundary, because of its relatively stable fluorescence regardless of whether the cells were compressed (Fig. S1). For imaging fixed and stained cells, we used the YAP channel to trace the cell boundary and the H2B channel to trace the nuclear boundary. The actual cell boundaries were then expanded by 10 pixels away from the original boundaries, in order to capture all the scattered light from the epifluorescence source. The nuclear boundaries, however, are only expanded by two pixels away from the traced nuclear boundary, in order to avoid excessive overestimation of nuclear YAP (examples are shown in Fig. S1).
The total fluorescent intensity for one cell was computed by adding all the pixel intensities within a traced boundary, after subtracting the mean background intensity. We repeated each set of experiments three times for statistical analysis. For each experiment, we scaled each integrated fluorescent intensity by the mean integrated intensity of the uncompressed control counterpart. The custom software was written in MATLAB, and is available upon request.
Model calculations
Computational results in the modeling section custom were obtained by using custom code written in MATLAB, which is available upon request.
Supplementary Material
Acknowledgements
We acknowledge Kevin Chen for his help in fabricating the compression device.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: L.H., S.S.; Methodology: L.H., J.T., D.M., F.S., S.S.; Software: J.T., S.S.; Validation: L.H., J.T.; Formal analysis: L.H., D.M.; Investigation: L.H., D.M., T.W., V.P.; Resources: Y.W.; Data curation: J.T.; Writing - original draft: L.H., S.S.; Writing - review & editing: L.H., J.T., S.S.; Visualization: J.T., S.S.; Supervision: S.S.; Project administration: D.W., S.S.; Funding acquisition: D.W., S.S.
Funding
The present work has been supported by the National Institutes of Health (grants R01GM114675 and U54CA210172). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.208470.supplemental
References
- Alvarez D., Vollmann E. H. and von Andrian U. H. (2008). Mechanisms and consequences of dendritic cell migration. Immunity 29, 325-342. 10.1016/j.immuni.2008.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragona M., Panciera T., Manfrin A., Giulitti S., Michielin F., Elvassore N., Dupont S. and Piccolo S. (2013). A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 154, 1047-1059. 10.1016/j.cell.2013.07.042 [DOI] [PubMed] [Google Scholar]
- Cukierman E., Pankov R., Stevens D. R. and Yamada K. M. (2001). Taking cell-matrix adhesions to the third dimension. Science 294, 1708-1712. 10.1126/science.1064829 [DOI] [PubMed] [Google Scholar]
- Dupont S., Morsut L., Aragona M., Enzo E., Giulitti S., Cordenonsi M., Zanconato F., Le, Digabel J., Forcato M. et al. (2011). Role of YAP/TAZ in mechanotransduction. Nature 474, 179-183. 10.1038/nature10137 [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S. and Hall A. (2002). Rho GTPases in cell biology. Nature 420, 629-635. 10.1038/nature01148 [DOI] [PubMed] [Google Scholar]
- Fraley S. I., Feng Y., Krishnamurthy R., Kim D. H., Celedon A., Longmore G. D. and Wirtz D. (2010). A distinctive role for focal adhesion proteins in three-dimensional cell motility. Nat. Cell. Biol. 12, 598-604. 10.1038/ncb2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cardena G., Comander J., Anderson K. R., Blackman B. R. and Gimbrone M. A. Jr. (2001). Biomechanical activation of vascular endothelium as a determinant of its functional phenotype. Proc. Natl. Acad. Sci. USA 98, 4478-4485. 10.1073/pnas.071052598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri A., Bajpai S., Trenton N., Jayatilaka H., Longmore G. D. and Wirtz D. (2013). The Arp2/3 complex mediates multigeneration dendritic protrusions for efficient 3-dimensional cancer cell migration. FASEB. J. 27, 4089-4099. 10.1096/fj.12-224352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga J. H., Li Y. S. and Chien S. (2007). Molecular basis of the effects of mechanical stretch on vascular smooth muscle cells. J. Biomech. 40, 947-960. 10.1016/j.jbiomech.2006.04.011 [DOI] [PubMed] [Google Scholar]
- Haswell E. S., Phillps R. and Rees D. C. (2011). Mechanosensitive channels: what can they do and how do they do it. Structure 19, 1356-1369. 10.1016/j.str.2011.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey J. D., Dufresne E. R. and Schwartz M. A. (2014). Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 15, 802-812. 10.1038/nrm3896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung W. C., Yang J. R., Yankaskas C. L., Wong B. S., Wu P. H., Pardo-Pastor C., Serra S. A., Chiang M. J., Gu Z., Wirtz D. et al. (2016). Confinement sensing and signal optimization via Piezo1/PKA and myosin UU pathways. Cell. Rep. 15, 1430-1441. 10.1016/j.celrep.2016.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingber D. E. (2003). Mechanobiology and diseases of mechanotransduction. Ann. Med. 35, 564-577. [DOI] [PubMed] [Google Scholar]
- Jaalouk D. E. and Lammerding J. (2009). Mechanotransduction gone awry. Nat. Rev. Mol. Cell. Biol. 10, 63-73. 10.1038/nrm2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H. Y. and Sun S. X. (2013). Cellular pressure and volume regulation and implications for cell mechanics. Biophys. J. 105, 609-619. 10.1016/j.bpj.2013.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T. J., Sun J., Lu S., Qi Y. X. and Wang Y. (2014). Prologned mechanical stretch initiates intracellular calcium oscillations in human mesenchymal stem cells . PLOS. ONE 9, e109378 10.1371/journal.pone.0109378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesnikov T. and Beckendorf S. K. (2007). 18 wheeler regulates apical constriction of salivary gland cells via the Rho-GTPase-signaling pathway. Dev. Biol. 307, 53-61. 10.1016/j.ydbio.2007.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovar D. R. and Pollard T. D. (2004). Insertional assembly of actin in association with formins produces piconewtons of force. Proc. Natl. Acad. Sci. USA 101, 14725-14730. 10.1073/pnas.0405902101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Berre M., Aubertin J. and Piel M. (2012). Fine control of nuclear confinement identifies a threshold deformation leading to lamina rupture and induction of specific genes. Integr. Biol. 4, 1406-1414. 10.1039/c2ib20056b [DOI] [PubMed] [Google Scholar]
- Liu B., Lu S., Hu Y., Liao X., Ouyang M. and Wang Y. (2014). RhoA and membrane fluidity mediates the spatially polarized Src/FAK activation in response to shear stress. Sci. Rep. 4, 7008 10.1038/srep07008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low B. C., Pan C. Q., Shivashankar G. V., Bershadsky A., Sudol M. and Sheetz M. (2014). YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS. Lett. 588, 2663-2670. 10.1016/j.febslet.2014.04.012 [DOI] [PubMed] [Google Scholar]
- Lu P., Weaver V. M. and Werb Z. (2012). The extracellular matrix: a dynamic niche in cancer progression. J. Cell. Biol. 196, 395-406. 10.1083/jcb.201102147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masiero L., Lapidos K. A., Ambudkar I. and Kohn E. C. (1999). Regulation of the RhoA pathway in human endothelial cell spreading on type IV collagen: role of calcium influx. J. Cell .Sci. 112, 3205-3213. [DOI] [PubMed] [Google Scholar]
- Mills E., Pham E. and Truong K. (2010). Structure based design of a Ca2+-sensitive RhoA protein that controls cell morphology. Cell Calcium. 48, 195-201. 10.1016/j.ceca.2010.08.009 [DOI] [PubMed] [Google Scholar]
- Orr A. W., Helmke B. P., Blackman B. R. and Schwartz M. A. (2006). Mechanisms of Mechanotransduction. Dev. Cell. 10, 11-20. 10.1016/j.devcel.2005.12.006 [DOI] [PubMed] [Google Scholar]
- Parekh S. H., Chaudhuri O., Theriot J. A. and Fletcher D. A. (2005). Loading history determines the velocity of actin-network growth. Nat. Cell. Biol. 7, L1219-L1223. 10.1038/ncb1336 [DOI] [PubMed] [Google Scholar]
- Pare A. C., Vichas A., Fincher C. T., Mirman Z., Farrell D. L., Mainieri A. and Zallen J. A. (2014). A positional Toll receptor code directs convergent extension in Drosophila. Nature 515, 523-527. 10.1038/nature13953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek M. J., Zahir N., Johnson K. R., Lakins J. N., Rozenberg G. I., Gefen A., Reinhart-King C. A., Marqulies S. S., Dembo M., Boettiger D. et al. (2005). Tensional homeostasis and the malignant phenotype. Cancer Cell. 8, 241-254. 10.1016/j.ccr.2005.08.010 [DOI] [PubMed] [Google Scholar]
- Scott D. W., Tolbert C. E. and Burridge K. (2016). Tension on JAM-A activates RhoA via GEF-H1 and p115 RhoGEF. Mol. Biol. Cell 27, 1420-1430. 10.1091/mbc.E15-12-0833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seminario-Vidal L., Okada S. F., Sesma J. I., Kreda S. M., van Heusden C. A., Zu Y., Jones L. C., O'Neal W. K., Penuela S., Laird D. W. et al. (2011). Rho signaling regulates pannexin 1-mediated ATP release from airway epithelia. J. Biol. Chem. 286, 26277-26286. 10.1074/jbc.M111.260562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si F., Li B., Margolin W. and Sun S. X. (2015). Bacterial growth and form under mechanical compression. Sci. Rep. 5, 11367 10.1038/srep11367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoes Sde M., Blankenship J. T., Weitz O., Farrell D. L., Tamada M., Fernandez-Gonzalez R. and Zallen J. A. (2010). Rho-kinase directs Bazooka/Par-3 planar polarity during Drosophila axis elongation. Dev. Cell. 19, 377-388. 10.1016/j.devcel.2010.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokabe T., Fukumi-Tominaga T., Yonemura S., Mizuno A. and Tominaga M. (2010). The TRPV4 channel contributes to intercellular junction formation in keratinocytes. J. Biol. Chem. 285, 18749-18758. 10.1074/jbc.M110.103606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz M. A., Tschumperlin D. J., Kamm R. D. and Drazen J. M. (2001). Mechanical stress is communicated between different cell types to elicit matrix remodeling. Proc. Natl. Acad. Sci. USA 98, 6180-6185. 10.1073/pnas.111133298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J. and Sun S. X. (2015). Active biochemical regulation of cell volume and a simple model of cell tension response. Biophys. J. 109, 1541-1550. 10.1016/j.bpj.2015.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J., Li Y., Vig D. K. and Sun S. X. (2017). Cell mechanics: a dialogue. Rep. Prog. Phys. 80, 036601 10.1088/1361-6633/aa5282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiam H. R., Vargas P., Carpi N., Crespo C. L., Raab M., Terriac E., King M. C., Jacobelli J., Alberts A. S., Stradal T. et al. (2016). Perinuclear Arp2/3-driven actin polymerization enables nuclear deformation to facilitate cell migration through complex environments. Nat. Commun. 7, 10997 10.1038/ncomms10997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Unen J., Reinhard N. R., Yin T., Wu Y. I., Postma M., Gadella T. W. and Goedhart J. (2015). Plasma membrane restricted RhoGEF activity is sufficient for RhoA-mediated actin polymerization. Sci. Rep. 5, 14693 10.1038/srep14693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam K. and Montell C. (2007). TRP channels. Ann. Rev. Biochem. 76, 387-417. 10.1146/annurev.biochem.75.103004.142819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada K., Itoga K., Okano T., Yonemura S. and Sasaki H. (2011). Hippo pathway regulation by cell morphology and stress fibers. Development 138, 3907-3914. 10.1242/dev.070987 [DOI] [PubMed] [Google Scholar]
- Wirtz D., Konstantopoulos K. and Searson P. C. (2011). The physics of cancer: the role of physical interactions and mechanical forces in metastasis. Nat. Rev. Cancer 11, 512-522. 10.1038/nrc3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojciak-Stothard B. and Ridley A. J. (2003). Shear stress-induced endothelial cell polarization is mediated by Rho and Rac but not Cdc42 or PI 3-kinases. J. Cell. Biol. 161, 429-439. 10.1083/jcb.200210135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X. H., Laschinger C., Arora P., Szaszi K., Kapus A. and McCulloch C. A. (2007). Force activates smooth muscle alpha-actin promoter activity through the Rho signaling pathway. J. Cell. Sci. 120, 1801-1809. 10.1242/jcs.001586 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.