

Abstract
Skeletal muscle provides the contractile force necessary for movement, swallowing, and breathing and, consequently, is necessary for survival. Skeletal muscle cells are unique in that they are extremely large cells containing thousands of nuclei. These nuclei must all work in concert to maintain skeletal muscle function and thereby maintain life. The nucleus is a major site of signaling integration and gene expression regulation. However, examining nuclear processes in skeletal muscle can be difficult because myonuclei are challenging to isolate. We optimized a protocol to purify myonuclei from whole muscle tissue using ultracentrifugation over a discontinuous sucrose gradient to separate the nuclear fraction. We used these purified nuclei for downstream applications including flow cytometry and mass spectrometry. We used this method to compare the myonuclear proteome of young and old mouse hindlimb muscles ( Cutler et al., 2017 ). This protocol may be applied to isolating myonuclei for a variety of downstream analyses such as flow cytometry, microscopy, Western blot, and proteomics.
Keywords: Fractionation, Sucrose gradient, Magnetic isolation, Ultracentrifugation
Background
Proper skeletal muscle function must be maintained for survival. One component of this maintenance is adjustments in gene expression in response to cellular needs and environmental cues. Nuclear processes modulating gene expression are a critical component in regulating cellular composition and behavior. However, myonuclear proteins involved in these processes are difficult to study because of four technical limitations. First, skeletal muscle is dense, tightly packed with contractile proteins that make up more than 60% of proteins in the tissue ( Deshmukh et al., 2015 ; Cutler et al., 2017 ). These high abundance contractile proteins eclipse the far less abundant nuclear proteins. Second, the dense fibrous structure of skeletal muscle makes it difficult to dissociate without damaging nuclei, making nuclei difficult to isolate. Third, after centrifugation dense debris cosediments with nuclei, compounding the difficulty of isolating nuclei from the tissue. Fourth, skeletal muscle is comprised of multiple cell types, so nuclei isolated and nuclear proteins detected may be from myonuclei or nuclei of other cell types.
Several approaches have been optimized to enrich myonuclei from different organisms for various downstream applications. Ohkawa et al. presented a detailed protocol for isolating myonuclei from mouse tissue that was developed to maximize access of cross-linking reagent for Chromatin Immunoprecipitation (ChIP) analysis ( Ohkawa et al., 2012 ). Wilkie and Shrimer developed a procedure to isolate the myonuclear envelope and sarcoplasmic reticulum for proteomic comparison (Wilkie and Schirmer, 2008). An approach optimized by Dimauro et al. simultaneously collected mitochondrial, nuclear, and cytoplasmic fractions to compare protein localization among different cellular compartments ( Dimauro et al., 2012 ). While each of these approaches to enrich nuclei from skeletal muscle tissue was effective for the intended subsequent analysis, they did not prioritize isolating intact nuclei and not distinguish between myonuclei and nuclei from other cell types. An affinity-based method to selectively isolate nuclei from specific cell types was developed in Arabidopsis thaliana (Deal and Henikoff, 2011) and is now available for mice ( Jankowska et al., 2016 ). However, this affinity-based approach requires genetic labeling of the cell types of interest, which makes it prohibitively cumbersome to examine myonuclei from multiple mouse models. We optimized an ultracentrifugation sucrose gradient-based fractionation approach that requires relatively small sample sizes, no genetic labeling, and is compatible with downstream analysis by flow cytometry and mass spectrometry. The isolated nuclei are intact, biochemically depleted of proteins from non-nuclear organelles, and 85% of nuclei are myonuclei. To isolate myonuclei more quickly we also optimized affinity-based purification using a myonuclear-specific nuclear envelope protein, Transmembrane Protein 38A (TMEM38A) ( Bleunven et al., 2008 ; Cutler et al., 2017 ), and magnetic beads. This approach is more rapid and yields nuclei with comparable population purity to nuclear isolation by ultracentrifugation but with lower biochemical purity. These nuclear isolation techniques can be used to purify myonuclei from any mouse model for diverse downstream analyses.
Materials and Reagents
-
Materials
30 ml ultraclear ultracentrifuge tubes (Beckman Coulter, catalog number: 344058)
10 ml round bottom polypropylene tubes (Corning, Falcon®, catalog number: 352059)
40 µm nylon mesh cell strainer (Biologix, catalog number: 15-1040)
50 ml conical polypropylene tubes (BioExpress, catalog number: C-3394-4)
15 cm glass Pasteur pipets (Fisher Scientific, catalog number: 13-678-20A)
-
20-gauge needle (Fisher Scientific, catalog number: 14-826-5C)
Manufacturer: BD, catalog number: 305176.
10 ml syringe (BD, catalog number: 309604)
1.7 ml low-adhesion hydrophobic tubes (BioExpress, GeneMate, catalog number: C-3302-1)
5 ml conical polystyrene tube with cell strainer cap (Corning, catalog number: 352235)
0.45 µm polypropylene syringe filters (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: F2500-9)
-
Reagents
Ethanol
4,6-Diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, catalog number: D9542)
Protein A-conjugated magnetic beads (Thermo Fisher Scientific, InvitrogenTM, catalog number: 10001D)
Anti-TMEM38A antibody (Merck, catalog number: 06-1005)
Ethylenediaminetetraacetic acid (EDTA) (Fisher Scientific, catalog number: BP120)
Sodium hydroxide (NaOH) (Fisher Scientific, catalog number: S318)
Ethylene glycol-bis(β-aminoethylether)-N,N,N’,N’-tetraacetic acid (EGTA) (Sigma-Aldrich, catalog number: E3889)
Sucrose (Fisher Scientific, catalog number: BP220)
HEPES (Fisher Scientific, catalog number: BP310)
Potassium chloride (KCl) (Fisher Scientific, catalog number: P217)
Magnesium chloride hexahydrate (MgCl2·6H2O) (Fisher Scientific, catalog number: M33)
Spermidine (Sigma-Aldrich, catalog number: 85558)
Dithiothreitol (DTT) (United States Biological, catalog number: D8070)
cOmplete mini protease inhibitors (Roche Diagnostic, catalog number: 11836153001)
Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: A2153)
Spermine tetrahydrochloride (Sigma-Aldrich, catalog number: S2876)
1x phosphate buffered saline (Thermo Fisher Scientific, GibcoTM, catalog number: 21600069)
100 mM EDTA (see Recipes)
100 mM EGTA (see Recipes)
2.1 M sucrose solution (see Recipes)
2.8 M sucrose solution (see Recipes)
Homogenization buffer (see Recipes)
1% BSA homogenization buffer (see Recipes)
Resuspension buffer (see Recipes)
1% BSA resuspension buffer (see Recipes)
Equipment
-
Dissection equipment
Pins (Carolina Biological Supply Company, catalog number: 629122)
4.5 inch pointed dissection scissors (Fisher Scientific, catalog number: 08-940)
Hemostat (GF HEALTH PRODUCTS, catalog number: 2675)
15 ml Dounce homogenizer with PTFE serrated plunger (Cole-Parmer Instrument, catalog numbers: 44468-10 and 44468-16)
UV lamp (optional) (Spectroline, catalog number: ENF-240C)
Centrifuge (Eppendorf, model: 5702)
-
Microcentrifuge (International Equipment Company, model: S139068)
Note: This product has been discontinued.
Ultracentrifuge (Beckman Coulter, model: OptimaTM LE-80K, LLE7)
SW 32 Ti swing bucket rotor (Beckman Coulter, model: SW 32 Ti, catalog number: 369650)
UltraRocker rocking platform (Bio-Rad Laboratories, catalog number: 1660709EDU)
Magnet (optional) (Thermo Fisher Scientific, catalog number: 12320D)
Round ended microspatula (Fisher Scientific, catalog number: 21-401-5)
Procedure
Note: All solutions should be chilled to 4 °C and all steps performed at 4 °C to reduce enzyme activity and preserve sample integrity.
-
Prepare 2.8 M and 2.1 M sucrose solutions (see Recipes) and buffers.
Note: It is easiest to prepare the sucrose gradient if 8 ml of the 2.8 M sucrose solution is added to the bottom of the 30 ml ultracentrifuge tubes before it is chilled to 4 °C.
Prepare dissection equipment by washing thoroughly with ethanol.
-
Dissect gastrocnemius and rectus femoris muscles from mouse (Figure 1 and Videos 1 and 2) and place up to 4 muscles in a 10 ml round bottom tube.
Notes:
The soleus often adheres to the gastrocnemius during dissection. Remove the soleus from the gastrocnemius before proceeding to process the gastrocnemius.
The rectus femoris is one of four muscles that make up the quadriceps.
Video 1. Gastrocnemius dissection.
Download video file (35.7MB, mp4) As shown pictorially in Figure 1 steps 1-5, this video demonstrates gastrocnemius dissection.
As shown pictorially in Figure 1 steps 1-5, this video demonstrates gastrocnemius dissection.Video 2. Rectus femoris dissection.
Download video file (14.8MB, mp4) As shown pictorially in Figure 1 steps 6-8, this video demonstrates rectus femoris dissection.
As shown pictorially in Figure 1 steps 6-8, this video demonstrates rectus femoris dissection. Mince muscles with scissors.
Add 5 ml of chilled homogenization buffer (see Recipes) for 1-2 muscles or 10 ml of chilled homogenization buffer for 3-4 muscles to the minced muscles.
-
Homogenize muscles with Dounce homogenizer on ice about 50 strokes (until all large chunks of muscle have been forced from the bottom of the homogenizer to the top of the plunger) (Figure 2).
Notes:
Warning: a vacuum may form between the plunger and the surface of the homogenization buffer. If the plunger rapidly collides the buffer, this can shatter the homogenizer.
If you are pooling homogenate from more than 4 muscles, homogenize them separately and then pool the homogenate prior to Step 7.
Aged or regenerating muscle samples may take longer to homogenize.
Homogenizer and dissection equipment should be cleaned with ethanol and water.
-
Rinse a 40 µm filter with 1-2 ml of homogenization buffer. Pass the homogenate over this filter into a 50 ml conical.
Note: Fibrous material that cannot pass through the filter may clog the filter. Gently scrape the filter with a round ended microspatula to help the filtrate pass through.
Rinse the filter with 5 ml homogenization buffer.
Centrifuge the filtrate at 1,000 × g for 10 min at 4 °C to obtain a crude nuclear pellet.
During the centrifugation, carefully layer 12 ml of the 2.1 M sucrose solution over the 2.8 M sucrose cushion in the 30 ml ultracentrifuge tube.
-
After the centrifugation is completed, discard the cytoplasmic supernatant and resuspend the crude nuclear pellet in 8 ml homogenization buffer. Carefully layer the resuspended lysate on the top of the sucrose gradient.
Notes:
If DAPI is added at a final concentration of 1 µg/ml to the homogenate at this step, the position of the nuclei in the sucrose gradient can be visualized.
The cytoplasmic fraction can be saved for subsequent biochemical or proteomic analysis. If used immediately, the cytoplasmic fraction should be kept on ice. For longer term storage the cytoplasmic fraction should be aliquoted and stored at -80 °C.
Balance the ultracentrifuge tubes by weight.
Centrifuge the samples at 186,712 max G (32,000 rpm in SW 32 Ti Beckman swing bucket rotor) for 200 min at 4 °C.
-
During the centrifugation, coat a 50 ml conical tube with BSA by incubating it with 20 ml 1% BSA resuspension buffer (see Recipes) for at least 30 min at 4 °C with low speed rocking on a standard laboratory rocker.
Note: Removing the top of a glove box allows the tubes to roll on the rocker resulting in even BSA coating of up to 6 tubes (Figure 3).
-
After the ultracentrifugation is complete, aspirate the upper layers of the sucrose gradient to within a centimeter of the 2.1 M and 2.8 M boundary. Collect the 2.1 M/2.8 M interface into the BSA-coated 50 ml conical tubes using glass Pasteur pipettes (Figure 4 steps 1-4).
Notes:
If the overlying layers have been correctly removed, there is little danger of contaminating the nuclear fraction with material from another interface. Thus collecting 1 cm above and 1 cm below the interface will increase the yield of nuclei without endangering the purity of the fraction.
If DAPI has been added to the homogenate, the position of the nuclei in the gradient can be visualized with UV light. DAPI fluorescence under UV light can be used to confirm that the entire interface containing the nuclei has been collected (Figure 4 step 4).
Nuclei can be collected by puncturing the side of the ultracentrifugation tube with a 20-gauge needle and 10 ml syringe. However, nuclei are sheared passing through the needle and the high viscosity of the sucrose makes collection difficult. Similarly, collection of fractions beginning with the bottom of the gradient is difficult because of the high viscosity of the 2.8 M sucrose.
-
Dilute the collected interface in the 50 ml conical tube 1:10 with resuspension buffer (see Recipes). Mix by inverting 5-10 times (Figure 4 step 5).
Note: The collected interface is much denser than the resuspension buffer. Be careful to completely mix the interface and resuspension buffer.
Centrifuge the diluted interface in the 50 ml conical tube at 1,000 × g for 10 min at 4 °C (Figure 4 step 6).
-
Discard the supernatant and resuspend the nuclear pellet in 1 ml resuspension buffer and transfer to a 1.7 ml tube.
Note: For flow cytometry and microscopy, resuspend the nuclei in chilled resuspension buffer with 1% BSA.
Centrifuge the resuspended nuclei at 800 × g for 10 min at 4 °C (Figure 4 step 8).
-
Discard the supernatant and resuspend the nuclear pellet in appropriate buffer (see Figure 5A for representative images of isolated nuclei).
Notes:
For flow cytometry, resuspend the nuclear pellet in 500 µl resuspension buffer with 1% BSA and pass through a 35 µm cell strainer into a 5 ml polystyrene tube before analyzing.
For microscopy, resuspend the nuclear pellet in 1-3 ml resuspension buffer with 1% BSA.
For Western blot or mass spectrometry, wash the nuclear pellet two more times with resuspension buffer to remove residual BSA, then resuspend in an appropriate denaturing buffer.
Figure 1. Muscle dissection.

. 1. Starting with the mouse lying on its ventral side, use hemostats to retract skin from ankle to hip. 2. Remove the fat pad over the back of the knee. 3. Insert scissors behind Achilles tendon. 4. Open scissors to separate gastrocnemius muscle from underlying tissue. 5. Cut Achilles tendon and the head of the gastrocnemius. If the soleus is still attached to the gastrocnemius, remove it. 6. Turn the mouse over so it is lying on its back. 7. Remove the fat pad over the hip. 8. With scissors parallel to the femur cut the tendon connecting the rectus femoris to the acetabulum (knee) and continue lengthwise to the hip. Finally, with scissors perpendicular to the femur cut the tendon connecting the muscle to the iliac spine and collect the rectus femoris muscle.
Figure 2. Pre-ultracentrifugation preparation.
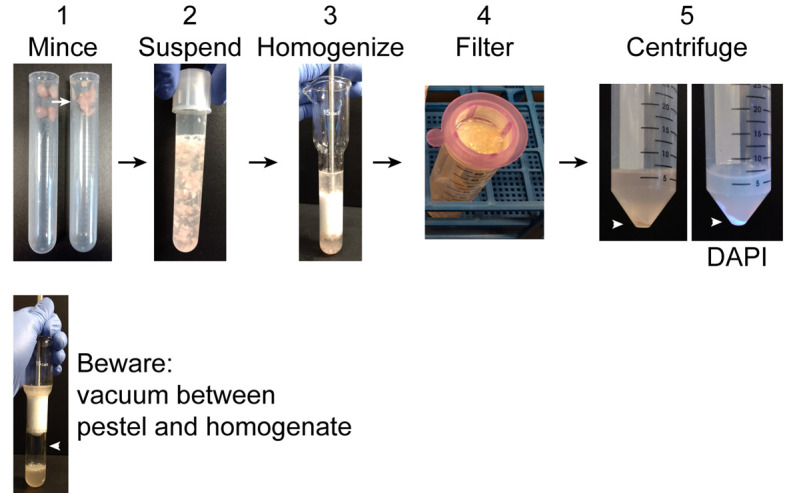
1. Using scissors mince collected muscles in the tube. Intact muscles are shown on the left and minced muscles on the right. 2. Suspend the minced muscles in 10 ml homogenization buffer. 3. Homogenize muscle using a Dounce homogenizer. 4. Pass homogenate through a 40 µm filter into a 50 ml conical tube. Incompletely disrupted tissue and large debris will remain in the filter. 5. Centrifuge the filtrate to obtain a crude nuclear pellet (arrowhead). DAPI clearly labels the nuclear pellet under UV light. Warning: when homogenizing a vacuum may form between the pestle and homogenate. If the pestle is sucked down into the homogenizer, it can strike with sufficient force to shatter the homogenizer.
Figure 3. Loading ultracentrifuge tubes.

Before beginning the protocol, 2.8 M sucrose should be layered into the bottom of the ultracentrifuge tube and chilled. During 1,000 × g centrifugation step of the muscle homogenate, layer chilled 2.1 M sucrose solution over the 2.8 M solution. When the centrifugation of the muscle homogenate is completed, resuspend the crude nuclear pellet in homogenization buffer and carefully layer it over the 2.1 M solution. DAPI labels the nuclei in the lysate. During the ultracentrifugation step coat 50 ml conical tubes with BSA, which is simplified by allowing the tubes to roll in a box on the rocker.
Figure 4. Unloading ultracentrifuge tubes.

1. Identify the interface between the 2.8 M and 2.1 M sucrose solutions which contains the nuclear fraction (white arrow). It is easy to identify by DAPI fluorescence under UV light. 2. Aspirate the overlying layers to a centimeter above the nuclear fraction (white arrow). 3. Collect the nuclear fraction into the BSA-coated 50 ml conical tube. Nuclei in the fraction can be observed by DAPI fluorescence. 4. After collecting the nuclear fraction, the thin gray band of nuclei will no longer be visible and no DAPI fluorescence will be detectable in the ultracentrifuge tube or sucrose collected from the surface of the remaining solution. 5. Resuspend the collected nuclear fraction 1:10 in resuspension buffer by inverting the BSA-coated 50 ml conical tube several times. 6. Pellet the purified nuclei by centrifugation. The nuclear pellet is easy to see both with and without DAPI labeling. 7. Remove the supernatant and resuspend the pellet in 1.5 ml resuspension buffer. 8. Pellet the nuclei by centrifugation. Nuclei can be resuspended for imaging or washed 2 more times for biochemical and proteomic analyses.
Figure 5. Purified myonuclei.
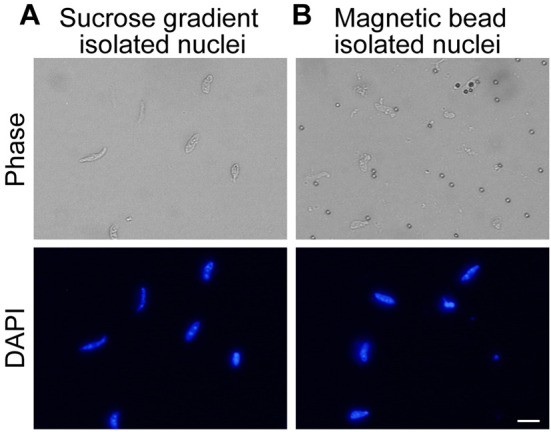
A. Nuclei isolated by ultracentrifugation through 2.1 M sucrose. Nuclei are visible in DAPI channel. B. Affinity purified nuclei isolated by binding of TMEM38A antibody and magnetic beads to nuclei. Nuclei are visible in DAPI channel. Scale bar = 10 µm.
Optional alternative approach
If biochemical purity is not critical, nuclei can be isolated more rapidly by affinity purification. Nuclei isolated by affinity purification are depleted for mitochondrial and cytoplasmic markers but retain substantial amounts of endoplasmic reticulum markers. If specific nuclear markers are known, affinity-based purification can be easily applied to other cell types (Deal and Henikoff, 2011).
Note: All solutions should be chilled to 4 °C and all steps performed at 4 °C.
Wash 1.5 mg Protein A-conjugated magnetic beads by incubating with 500 µl homogenization buffer in a 1.7 ml microcentrifuge tube for 5 min with rocking. Then place the tube in a magnetic separation rack and wait 20 sec for the beads to accumulate on the side of the tube and aspirate the buffer. Repeat 2 times.
-
Add 5 µg anti-TMEM38A antibody and 200 µl homogenization buffer with 1% BSA to the washed beads. Incubate at 4 °C with low speed rocking for 30 min.
Note: Magnetic beads rather than agarose or sepharose beads must be used because debris will cosediment with the agarose or sepharose beads but can be separated from magnetic beads which accumulate on the side of the tube where the magnet is placed.
Process samples as described above for Steps 2-8.
After centrifugation to obtain a crude nuclear pellet (described in Step 8 above), discard the supernatant and resuspend the pellet in 1 ml chilled homogenization buffer with 1% BSA.
Incubate the magnetic beads with the resuspended nuclei from up to 4 muscles at 4 °C for 30 min with gentle rocking.
Place the tubes containing the beads in a magnetic separation rack and wait 20 sec for the beads to accumulate on the side of the tube. Aspirate the buffer and debris at the bottom of the tube.
Wash the magnetic beads once with 500 µl chilled homogenization buffer with 1% BSA (see Recipes).
-
Resuspend the beads in an appropriate buffer for the downstream application (see Figure 5B for representative images of isolated nuclei).
Notes:
For flow cytometry, resuspend the nuclei in 500 µl resuspension buffer with 1% BSA and pass through a 35 µm cell strainer into a 5 ml polystyrene tube before analyzing.
For microscopy, resuspend the nuclei in resuspension buffer with 1% BSA.
For Western blot or mass spectrometry, wash the nuclei twice with PBS to remove residual BSA and resuspend in the appropriate denaturing buffer for downstream application.
Data analysis
Data should be processed according to standard practice for the relevant downstream technique. Appropriate downstream analysis can include microscopy, Western blot, proteomics, and flow cytometry.
The average yield from a single mouse gastrocnemius muscle is 1 x 106 nuclei. Pooling two gastrocnemius and two rectus femoris muscles yields 6 x 106 nuclei and 8 µg of total protein. Isolating nuclei from the tibialis anterior muscle resulted in a very low yield of nuclei that was impractical to analyze. It should be noted that unless care is taken to wash nuclei thoroughly, residual BSA can artificially inflate measurements of total protein. For this reason, it is recommended to first check purity and yield by microscopy and then measure protein concentration if applicable. The yield and purity are consistent between experiments. The greatest variation in yield resulted from failure to keep samples at 4 °C for the duration of the experiment.
For proteomics applications, we had the greatest success when both gastrocnemius and rectus femoris muscles from 3 mice were pooled. Proteomics experiments should be completed with at least 5 biological replicates to detect small changes in protein abundance.
For analysis by flow cytometry nuclei isolated from a single gastrocnemius or rectus femoris muscle provided ample starting material. Flow cytometry experiments should include at least three biological replicates.
Notes
The biochemical purification of myonuclei requires about 5 h to complete. If a large number of samples are being prepared, more time is required. Magnetic bead affinity based preparation requires about 1 h to complete.
Endoplasmic reticulum depletion is variable across preparations.
Recipes
-
100 mM EDTA (50 ml)
Combine:
372 mg EDTA
40 ml ultra-pure water
Adjust pH to 7 with NaOH
Adjust final volume to 50 ml with ultra-pure water
Store at room temperature for up to a year
-
100 mM EGTA (50 ml)
Combine:
1.9 g EGTA
40 ml ultra-pure water
Adjust to pH 8 with NaOH
Adjust final volume to 50 ml with ultra-pure water
Store at room temperature for up to a year
-
2.1 M sucrose solution (50 ml)
Combine the following over high heat with medium stirring:
35.9 g sucrose
2.5 ml HEPES (1 M)
1.25 ml KCl (1 M)
2.5 ml MgCl2 (100 mM)
Ultra-pure water to 50 ml
Note: Because the solution is made over heat, some of the fluid will boil off. Adjust the final volume after the solution has been removed from heat.
Store at 4 °C for up to 1 week
-
2.8 M sucrose solution (25 ml)
Combine the following over high heat with medium stirring:
24 g sucrose
1.25 ml HEPES (1 M)
625 µl KCl (1 M)
1.25 ml MgCl2 (100 mM)
Ultra-pure water to 25 ml
Note: Because the solution is made over heat, some of the fluid will boil off. Adjust the final volume after the solution has been removed from heat.
Prepare fresh for each use
-
Homogenization buffer (50 ml)
Combine the following:
500 µl HEPES (1 M)
3 ml KCl (1 M)
250 µl spermidine (100 mM)
750 µl spermine tetrahydrochloride (10 mM)
10 ml EDTA (10 mM)
250 µl EGTA (100 mM)
2.5 ml MgCl2 (100 mM)
5.13 g sucrose
Ultra-pure water to 50 ml
Filter sterilize and store at 4 °C for up to 1 month
Immediately prior to use add:
100 µl DTT (1 M)
5 Roche cOmplete mini protease inhibitor tablets
-
1% BSA homogenization buffer (50 ml)
Combine the following:
500 mg BSA
50 ml homogenization buffer
Prepare fresh for each use
-
Resuspension buffer (50 ml)
Combine the following:
1 ml HEPES (1 M)
750 µl MgCl2 (100 mM)
500 µl KCl (1 M)
250 µl spermidine (100 mM)
750 µl spermine tetrahydrochloride (10 mM)
1 ml EDTA (10 mM)
45.75 ml ultra-pure water
Store at 4 °C for up to 1 month
-
1% BSA resuspension buffer (50 ml)
Combine the following:
500 mg BSA
50 ml resuspension buffer
Prepare fresh for each use
Acknowledgments
This work was supported by grants to GKP (AR062483), to AAC (AR067645), and a training grant (T32GM008367) from the National Institutes of Health. The described protocol was adapted from the work of Wilkie and Schirmer (2008). No potential conflict of interest was reported by the authors.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
References
- 1.Bleunven C., Treves S., Xia Jinyu X., Leo E., Ronjat M., De Waard M., Kern G., Flucher B. E. and Zorzato F.(2008). SRP-27 is a novel component of the supramolecular signalling complex involved in skeletal muscle excitation–contraction coupling. Biochem J 411(2): 343-349. [DOI] [PubMed] [Google Scholar]
- 2.Cutler A. A., Dammer E. B., Doung D. M., Seyfried N. T., Corbett A. H. and Pavlath G. K.(2017). Biochemical isolation of myonuclei employed to define changes to the myonuclear proteome that occur with aging. Aging Cell 16(4): 738-749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deal R. B. and Henikoff S.(2011). The INTACT method for cell type-specific gene expression and chromatin profiling in Arabidopsis thaliana . Nat Protoc 6(1): 56-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deshmukh A. S., Murgia M., Nagaraj N., Treebak J. T., Cox J. and Mann M.(2015). Deep proteomics of mouse skeletal muscle enables quantitation of protein isoforms, metabolic pathways, and transcription factors. Mol Cell Proteomics 14(4): 841-853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dimauro I., Pearson T., Caporossi D. and Jackson M. J.(2012). A simple protocol for the subcellular fractionation of skeletal muscle cells and tissue. BMC Res Notes 5: 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jankowska U., Latosinska A., Skupien-Rabian B., Swiderska B., Dziedzicka-Wasylewska M. and Kedracka-Krok S.(2016). Optimized procedure of extraction, purification and proteomic analysis of nuclear proteins from mouse brain. J Neurosci Methods 261: 1-9. [DOI] [PubMed] [Google Scholar]
- 7.Ohkawa Y., Mallappa C., Vallaster C. S. and Imbalzano A. N.(2012). Isolation of nuclei from skeletal muscle satellite cells and myofibers for use in chromatin immunoprecipitation assays. Methods Mol Biol 798: 517-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilkie G. S. and Schirmer E. C.(2008). Purification of nuclei and preparation of nuclear envelopes from skeletal muscle. Methods Mol Biol 463: 23-41. [DOI] [PubMed] [Google Scholar]