

ABSTRACT
Human cleft lip with or without cleft palate (CL/P) is a common craniofacial abnormality caused by impaired fusion of the facial prominences. We have previously reported that, in the mouse embryo, epithelial apoptosis mediates fusion at the seam where the prominences coalesce. Here, we show that apoptosis alone is not sufficient to remove the epithelial layers. We observed morphological changes in the seam epithelia, intermingling of cells of epithelial descent into the mesenchyme and molecular signatures of epithelial-mesenchymal transition (EMT). Utilizing mouse lines with cephalic epithelium-specific Pbx loss exhibiting CL/P, we demonstrate that these cellular behaviors are Pbx dependent, as is the transcriptional regulation of the EMT driver Snail1. Furthermore, in the embryo, the majority of epithelial cells expressing high levels of Snail1 do not undergo apoptosis. Pbx1 loss- and gain-of-function in a tractable epithelial culture system revealed that Pbx1 is both necessary and sufficient for EMT induction. This study establishes that Pbx-dependent EMT programs mediate murine upper lip/primary palate morphogenesis and fusion via regulation of Snail1. Of note, the EMT signatures observed in the embryo are mirrored in the epithelial culture system.
KEY WORDS: EMT, Epithelial plasticity, Frontonasal prominences, TALE homeodomain proteins, Pbx1, Cleft lip/palate, Snail1, Snai1
Highlighted Article: Loss of Pbx transcription factors in mouse embryos causes perturbation of epithelial cell plasticity in the facial prominences, resulting in clefting of the lip and primary palate.
INTRODUCTION
The human face is the gateway to social acceptance and a predictor of reproductive success (Pflüger et al., 2012; Wilmer et al., 2014). The mouse offers a suitable model to study human craniofacial development and its abnormalities (Gritli-Linde, 2008). In the mouse, face morphogenesis is heralded by the appearance of the frontonasal prominence (fnp) and paired maxillary prominence (mxp), which arise from branchial arch 1 at gestational day (E) 9.5 (Dixon et al., 2011; Wang et al., 2014). By E10.5, the fnp divides into the medial nasal prominence (mnp) and lateral nasal prominence (lnp), which comprise round, heterogeneous cranial neural crest (CNC)-derived mesenchymal cells within the prominence cores (Bhatt et al., 2013) and tall, tightly associated epithelial cells forming the external sheet. By E11.5, the mnp and lnp merge and fuse with the mxp to form the upper lip and primary palate (Dixon et al., 2011). The three-way seam where the prominences coalesce is named the lambdoidal junction (λ) (Tamarin and Boyde, 1977; Depew and Compagnucci, 2008). In humans, failed fusion of the fnp with the mxp (Mossey et al., 2009; Dixon et al., 2011) causes a unilateral or bilateral gap between the central and lateral upper lip, which can extend into the nostril and/or primary palate, resulting in cleft lip (CL) (Jiang et al., 2006; Wang et al., 2014). Clefting of the secondary palate can accompany CL and presents as CL/P, which is the most common craniofacial birth defect (1/500-1/1000 live births) (Marazita, 2012).
In mice and humans, regulatory networks directed by three amino acid loop extension (TALE) Pbx homeodomain transcription factors (TFs) (Bürglin, 2011; Bürglin and Affolter, 2016; Kamps et al., 1990; Nourse et al., 1990; Longobardi et al., 2014) govern multiple developmental processes (Moens and Selleri, 2006; Slavotinek et al., 2017). We previously described that morphogenesis of the murine midface (comprising upper lip, primary palate, alae of the nose, and nostrils) occurs in part through Pbx-dependent epithelial apoptosis at the λ (Ferretti et al., 2011). We also showed that the fnp epithelium displays high levels of Pbx transcripts, whereas lower levels are detectable in the mesenchyme. Notably, Pbx1 mRNA is more abundant than Pbx2 and Pbx3 mRNAs (Ferretti et al., 2011). Additionally, during embryonic development, the Pbx1 short isoform, Pbx1b, is more prevalent than the long isoform, Pbx1a (Schnabel et al., 2001). Our published research using conditional inactivation of murine Pbx1 in the cephalic epithelium or mesenchyme, on a Pbx2 (or Pbx3)-deficient background, underscored that Pbx TFs are essential in the epithelium and dispensable in the mesenchyme for upper lip/primary palate fusion (Ferretti et al., 2011). Indeed, loss of function of Pbx TFs in the murine cephalic epithelium, but not in the mesenchyme, results in CL/P. Genomic analyses revealed evidence of gene–gene interactions between PBX1/PBX2 and single nucleotide polymorphisms (SNPs) in the ARHGAP29 locus, a candidate for CL/P (Letra et al., 2014). Moreover, haploinsufficiency of the PBX co-factor MEIS2 is associated with orofacial clefting (Crowley et al., 2010; Johansson et al., 2014) and a de novo mutation in MEIS2 was reported in a patient with CP only (CPO) (Louw et al., 2015).
During normal midface development, all epithelial cells must disappear from the seam of fnp and mxp, allowing the mesenchymal cores to form a bridge connecting the prominences at the λ. However, we reported that not all epithelial cells undergo apoptosis at the fusion site (Ferretti et al., 2011). Moreover, mice mutant for CPP32 (CASP3) (Kuida et al., 1996) fail to execute apoptosis but do not exhibit CL or CPO, and mice mutant for Apaf, a proapoptotic factor, display orofacial clefting on a C3H/HeJ strain (Long et al., 2013) but not on a C57BL/6 background (Jin and Ding, 2006). These findings suggest that, in addition to apoptosis, other cellular behaviors mediate prominence fusion at the λ. Multiple studies have addressed the controversial contributions of apoptosis, cell migration, cell extrusion and epithelial-mesenchymal transition (EMT; Acloque et al., 2009; Nieto, 2011; Nieto et al., 2016) to fusion of the secondary palate (Fitchett and Hay, 1989; Carette and Ferguson, 1992; Shuler et al., 1992; Kaartinen et al., 1997; Martinez-Álvarez et al., 2000; Cuervo and Covarrubias, 2004; Jin and Ding, 2006; Ahmed et al., 2007; Kim et al., 2015; reviewed by Bush and Jiang, 2012 and by Lan et al., 2015). However, little is known of the cellular processes that mediate fusion of the upper lip and primary palate.
Here, using mouse embryos and an in vitro epithelial culture system as working models, we demonstrated the presence of EMT during fusion of the facial prominences at the λ. Multiple factors lead to activation of EMT programs in development and disease (Ye and Weinberg, 2015; Santamaria et al., 2017; Voon et al., 2017), including signaling molecules, such as TGFβ (Lamouille et al., 2014), and TFs, such as Snail1/2 (Snai1/2), Smads, Twist and Zeb1/2 (Fuxe et al., 2010). Utilizing mouse lines deficient for Pbx TFs (Selleri et al., 2001, 2004; Kim et al., 2002; Brendolan et al., 2005; Capellini et al., 2006, 2010; Vitobello et al., 2011; Koss et al., 2012; Hurtado et al., 2015; McCulley et al., 2018) with fully penetrant CL/P (Ferretti et al., 2011), we established a novel Pbx–Snail1 regulatory axis that promotes EMT during facial morphogenesis.
RESULTS
Midface morphogenesis is mediated by Pbx-dependent epithelial plasticity at the lambdoidal junction
We previously reported that a Pbx-directed regulatory network mediates frontonasal prominence fusion, at least in part through the control of apoptosis at the λ epithelial seam. We further established that, in all Pbx compound mutant embryos analyzed, orofacial clefting was rescued via Wnt1 ectopic expression in the cephalic epithelium (Ferretti et al., 2011). In the current study, we revealed that apoptosis was restored in Pbx compound mutants with rescued orofacial clefting (n=2) (Fig. S1A), pointing to the direct association of programmed cell death with fusion of the frontonasal prominences. To further investigate this process (Fig. 1), we conducted a spatial and temporal quantitative analysis of apoptosis on wild-type embryonic midfaces from the initiation (E10.5) to the completion (E11.5) of fusion (Fig. S1B,C). Immunofluorescence for active caspase 3 revealed dynamic landscapes of epithelial programmed cell death along the anterior–posterior (A-P) axis of the λ at E10.5 (Fig. S1B). At this time point, immunofluorescence demonstrated that epithelial apoptosis occurred at approximately the same rate in anterior domains (A) throughout the middle-posterior domains (M-P) of the λ (Fig. S1B). Quantitation revealed a broader distribution of the frequency of apoptotic cells in middle-posterior domains and modest differences between the median values of cells undergoing programmed cell death in anterior (15%) versus middle-posterior (12%) domains (Fig. S1D). By contrast, by E11.5, when fusion is complete, marked differences were found in the percentage of apoptotic cells between anterior (<1%), middle (4%) and posterior (16%) domains, with highest levels shifting to posterior-most domains from anterior domains (Fig. S1C,D). In wild-type embryos on a mixed genetic background at both time points and at all A-P levels analyzed, no more than 40% of seam epithelial cells undergo apoptosis. These analyses establish that apoptosis is directly associated with prominence fusion. However, spatial and temporal regulation of apoptosis in less than half of the seam epithelial cells is not sufficient to enable fusion of the facial prominences because all epithelial cells must be removed.
Fig. 1.

Epithelial morphological changes at the lambdoidal junction (λ) are Pbx dependent. Top: sketches of embryonic midfaces of indicated genotypes. Nasal pits are shown in gray. Plane of sections of control and mutant λ are shown below the E11.5 sketches. Dotted lines indicate epithelium; blue rectangles indicate the sectioned region through lnp at the λ (G,H,K,L); red rectangles the sectioned region through lnp/mnp fusion (I,J). (A-F) SEM of control (A) and mutant (B) heads. λ domains (black and green rectangles) are shown at higher magnification in C,E (control) and D,F (mutant). Filled black arrowheads indicate cellular protrusions at the lnp/mnp fusion site (C,E). (G-L) TEM of control (G,I,K) and mutant (H,J,L) λ sections through planes in blue and red rectangles, respectively. (G-J) Empty space enlargements between epithelial cells are shown by unfilled arrowheads in control (G,I). Close contacts between epithelial cells are indicated by filled arrowheads in mutant (H,J). (K,L) In control (K), note break down (unfilled arrowheads) and blebs (dotted line) of BM (yellow line) whereas in the mutant (L) BM is intact (arrowheads). Scale bars: 500 µm (A,B); 20 µm (C-F); 2 µm (G-L). Ep, epithelium; Me, mesenchyme; mx, maxillary process.
Additional cellular behaviors can facilitate tissue remodeling, including cellular plasticity (Nieto, 2013); EMT (Acloque et al., 2009; Nieto et al., 2016); epithelial cell dispersal (Packard et al., 2013); or epithelial cell extrusion (Kim et al., 2015). To investigate the potential contributions of epithelial plasticity and/or EMT to prominence fusion, we analyzed littermate control and Pbx compound mutant embryos that exhibit CL/P (Fig. 1). To obtain conditional inactivation of Pbx1 in the cephalic epithelium on a Pbx2-deficient background, we used Crect deleter mice (Reid et al., 2011). Whole-mount X-gal staining of R26RlacZ/+;CrectCre/+ embryos (Soriano, 1999) showed that Cre activity in the head is localized to the epithelium of facial domains and branchial arches at E10.5-E12.5 (Fig. S2A-D). Pbx1f/+;Pbx2+/−;CrectCre/+ mice are hereafter named as controls, and Pbx1f/f;Pbx2+/−;CrectCre/+ littermates as Pbx1/2 mutants. We confirmed loss of Pbx1 at the Pbx1/2 mutant λ (Fig. S2E-H). We next examined λ epithelial morphology in three controls and three Pbx1/2 mutants at E10.5 and E11.5, respectively, by scanning electron microscopy (SEM) (Fig. 1). In E10.5 controls, λ epithelia revealed changes consistent with epithelial plasticity (Fig. 1A,C,E). Specifically, surface cellular protrusions were observed prior to lnp/mnp fusion, together with cells sloughing off (Fig. 1C,E). Conversely, in E10.5 Pbx1/2 mutants, surface epithelial cells at the presumptive fusion site exhibited a flat and tight appearance lacking pronounced protrusions (Fig. 1B,D,F). Transmission electron microscopy (TEM) of the junction epithelia in E11.5 controls revealed disruption of epithelial organization and seam integrity; space enlargements between epithelial cells at the lnp and lnp/mnp fusion site; and break down of the basement membrane (BM) forming blebs (Fig. 1G,I,K). By contrast, E11.5 Pbx1/2 mutants exhibited tight contacts between epithelial cells and an intact BM (Fig. 1H,J,L). Electron microscopy analyses point to the presence of Pbx-dependent features of epithelial plasticity at the λ.
Presence of ‘transitional’ cells in the λ epithelia and of cells of epithelial descent within the λ mesenchyme is Pbx dependent
To define the fates of the seam epithelial cells and their progeny, we performed genetic lineage-tracing experiments using R26RlacZ/+;CrectCre/+ mice. In embryos resulting from crosses of these mice with Pbx1/2 mutants, lacZ was expressed in cephalic epithelium in both controls and mutants with CL/P. Cells of epithelial derivation were either tracked by immunofluorescence with an antibody (Ab) against β-galactosidase, or by X-gal staining. Immunostaining for β-galactosidase, mesenchymal (vimentin) and epithelial (E-cadherin; also known as cadherin 1) markers on E10.5-E11.5 midface sections (Figs 2 and 3) revealed the presence of cells of epithelial derivation expressing vimentin at the seam (Fig. 2A), hereafter named ‘transitional’ cells. Transitional cells were absent or negligible in Pbx1/2 mutants (Fig. 2B). Immunofluorescence double labeling of cells for vimentin (Vim+) and E-cadherin (Ecadh+) (Fig. 3), together with quantitation of Ecadh+,Vim+ cells, revealed that, in controls, transitional cells are detectable by E11.0 and peak at approximately E11.5. The highest percentage of transitional cells within the central quartiles is approximately 6% of the total number of cells within the control epithelial seam (Fig. 2E). Thus, the emergence of transitional cells is Pbx dependent and precedes, or is concomitant with, frontonasal prominence fusion. Furthermore, immunofluorescence with an active caspase 3 Ab revealed that, in controls, only approximately 2% of the transitional cells undergo apoptosis (Fig. 2E). Consistent with our previous results (Ferretti et al., 2011), mutant embryos did not show significant epithelial apoptosis at the seam (Fig. 2D). Likewise, in Pbx1/2 mutants, cells expressing low levels of vimentin were either absent or negligible within the λ epithelia (Fig. 2D). Additionally, quantitation of cells of epithelial derivation by X-gal staining in E11.5 controls (n=5) revealed the presence of a modest percentage of cells of epithelial descent (blue) within the mesenchymal cores at the λ (Fig. 2F,G; 2H shows quantitation). By contrast, in E11.5 Pbx1/2 mutants (n=3), blue cells were restricted to the epithelium (Fig. 2I-J′). These findings establish the presence of transitional cells at the seam by E11.0-E11.5 and the intermingling of epithelial cells with the mesenchyme of the prominences in a Pbx-dependent manner.
Fig. 2.

Detection and quantitation of transitional cells at the λ of controls and Pbx1/2 mutant embryos together with fate mapping and quantitation of epithelial cells. Top: λ sketches of E11.5 embryos of the indicated genotypes for control reporter (left) and Pbx1/2 mutant (right). Fusion site at the λ is indicated by blue rectangles. (A-D) Immunofluorescence of coronal sections of control and mutant λ with Abs for β-galactosidase (red; A,B and insets), vimentin (green; A-D) and active caspase 3 (red; C,D); DAPI labels nuclei (blue). (A,B) β-galactosidase-positive cells of epithelial derivation. In control (A), seam epithelium β-galactosidase-positive cells express vimentin (filled yellow arrowheads; inset). Negligible numbers of vimentin-positive cells are observed in the mutant (unfilled green arrowheads; B,D). (C) In control λ epithelium, a few cells are either vimentin positive, or active caspase 3 positive, or double positive (filled green, red and yellow arrowheads, respectively). (E) Box-and-whisker plots represent transitional cells expressing E-cadherin (Ecadh+) and vimentin (vim+) over total number of epithelial cells (left) and transitional cells that are also active caspase 3 positive (right). (F-H) Lineage tracing and quantitation (illustrated in box-and-whisker plot; H) of epithelial and epithelial-derived cells (blue) in control (F,G) and Pbx1/2 mutant (I-J′) λ. Scale bars: 50 µm (A-D); 100 µm (F-J′).
Fig. 3.

EMT molecular signatures of epithelial cells at the λ during frontonasal prominence fusion. Top: sketches of λ domains of indicated genotypes. Epithelium is indicated by dashed lines. Blue rectangles correspond to the sectioned region of apposing prominences prior to fusion (A,C,K,L) or rostral to the fusion site (E,G). Red rectangles indicate the sectioned region through lnp/mnp fusion (B,D,F,H-J,M-T). (A-L) Immunofluorescence of coronal sections for control and mutant λ. DAPI (blue) labels nuclei. (A-D) In controls (A,B), claudin 3 (green) in prominence epithelium prior to fusion (A, filled green arrowheads) disappears when fusion occurs (B, unfilled green arrowhead). In Pbx1/2 mutants, epithelial claudin 3 persists both prior to, and during, failed fusion (C,D, filled green arrowheads). (E-H) Double labeling for E-cadherin (red) and vimentin (green). (E,F) In controls, E-cadherin-positive (filled red arrowhead) and E-cadherin-positive, vimentin-positive cells (filled yellow arrowheads) are detected in apposing epithelia (E). At the fusion site, E-cadherin is downregulated (F, unfilled red arrowhead) but some E-cadherin-positive, vimentin-positive cells are still present (filled yellow arrowheads) (F). In mutants, E-cadherin positive-only epithelial cells (filled red arrowheads) are detected; vimentin is not detectable or negligible (G,H). (I,J) Discontinuous laminin (green) is observed in controls (I, inset; unfilled green arrowheads), whereas in mutants, there is a continuous line of laminin (J, inset; filled green arrowhead). (K,L) Smad4 (green) was enriched in cell nuclei at the tip of control lnp (K, inset; filled green arrowheads) whereas in mutants levels of cytoplasmic Smad4 were low (L, inset; unfilled green arrowhead). (M,N) Section in situ hybridization. Snail1 epithelial seam expression (higher in lnp epithelium) was detected only in control (M; filled purple arrowheads) but not dysmorphic mutant epithelium (N; unfilled purple arrowhead). Snail1 mRNA was still present in mutant prominence mesenchyme. (O,P) Snail1/2 (green) was detected in control epithelial cell nuclei (O; filled green arrowheads); in the mutant, Snail1/2 expression was lost in seam epithelia (P; unfilled green arrowhead). BM exhibits nonspecific staining for Smad4 and Snail1/2. (Q-T) Triple staining for Snail1 (green), TUNEL (red) and DAPI (blue). Orange arrowheads indicate cells positive for nuclear Snail1 and TUNEL staining. Green arrowheads indicate cells with nuclear Snail1. See Fig. S3I for quantitation. Scale bars: 50 µm (A-P); 20 µm (Q-T).
Immunofluorescence analysis with molecular markers for epithelial identity and BM integrity showed that the tight-junction component claudin 3 was localized to the apical surface of lnp and mnp apposing epithelia prior to fusion in E10.5 controls (Fig. 3A), but disappeared within the seam during fusion (Fig. 3B; Fig. S3C). Conversely, in Pbx1/2 mutants, claudin 3 was present in the epithelium of the apposing prominences prior to fusion, and persisted once the defective fusion process commenced (Fig. 3C,D; Fig. S3D). Double labeling for E-cadherin and vimentin in E11.5 controls showed that cells within the apposing epithelia were either positive for E-cadherin only, or positive for both E-cadherin and vimentin (Fig. 3E,F). Furthermore, in controls, E-cadherin was markedly downregulated in epithelia at the fusion site (Fig. 3F), consistent with lineage-tracing results and marker analysis (shown in Fig. 2). In mutants, however, all epithelial cells retained high levels of E-cadherin and filamentous vimentin was not detectable either prior to, or after, the formation of the epithelial bridge connecting mutant lnp and mnp (Fig. 3G,H). Lastly, immunofluorescence for laminin revealed that, whereas the BM was disrupted in E11.5 controls, it remained intact in mutants (Fig. 3I,J). All observed changes in cellular markers at the lnp/mnp seam indicate the emergence of a Pbx-dependent plastic cellular landscape, in which epithelial features are lost and mesenchymal properties are gained, associated with the presence of transitional cells and of cells of epithelial descent that migrate into the mesenchymal cores.
Pbx genes control expression of Smad4 and Snail1, crucial effectors of EMT, at the λ during midface morphogenesis
We analyzed levels and localization of Smad4, a key mediator of TGFβ signaling (Lamouille et al., 2014), which interacts with Smad3 to form a complex with nuclear Snail1, a transcriptional repressor of E-cadherin and promoter of EMT (Vincent et al., 2009; Nieto, 2013), in controls and mutant embryos. In E10.5 controls, high levels of nuclear Smad4 within the apposing frontonasal prominences (lnp in Fig. 3K) indicated activation of TGFβ signaling (Heldin and Moustakas, 2012; Macias et al., 2015). By contrast, in Pbx1/2 mutants, negligible Smad4 levels localized only to the cytoplasm (Fig. 3L). Section in situ hybridization in E11.5 controls showed that high levels of Snail1 transcript were present in the mesenchyme of the prominences and, to a lesser extent, at the epithelial seam, where higher expression could be detected in the lnp epithelium (Fig. 3M). Conversely, Snail1 transcript was lost in seam epithelia of Pbx1/2 mutants, although still detectable in the mesenchyme of the dysmorphic mutant λ (Fig. 3N). Immunofluorescence analysis at the same stage confirmed the presence of Snail1/2 in the epithelial seam of controls, with nuclear accumulation in select cells of the apposing lnp and mnp, both anterior to (Fig. S3G) and at the fusion site (Fig. 3O). In Pbx1/2 mutants, Snail1/2 were dramatically decreased in the epithelia of the apposing prominences (Fig. S3H) and were lost from the epithelia at the fusion site (Fig. 3P). These experiments establish Pbx-dependent control of Smad4 and Snail1/2, crucial effectors of EMT, at the λ during midface morphogenesis.
Epithelial cells with nuclear Snail1 do not undergo apoptosis at the λ
Given the presence of apoptosis as well as EMT at the λ epithelia, we conducted double-labeling immunofluorescence experiments in E10.75-E11.5 wild-type embryos to determine whether these two processes are mutually exclusive within these cells. In these experiments, we used a different Snail1-specific antibody that generates less background and appears to provide higher sensitivity, with strong signal in mesenchymal cells and weaker signal in epithelial cells (Fig. 3R,T), consistent with the in situ hybridization results. At both time points, the number of seam epithelial cells that exhibited nuclear Snail1 and were concomitantly TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) positive appeared negligible (Fig. 3Q-T). In particular, at E10.75, when apoptosis is highest, only 1.5% of all epithelial cells displayed nuclear Snail1 and apoptotic markers (quantitation in Fig. S3I). These results suggest the involvement of Snail1 in protecting cells from programmed death in tissues in vivo, in agreement with previous findings in the secondary palate (Martínez-Álvarez et al., 2004; Vega et al., 2004).
Pbx1 directly regulates Snail1 transcription by binding to putative regulatory elements within the Snail1 locus in embryonic midfacial prominences
Sequence analysis of the Snail1 locus across multiple species identified four non-coding regions containing conserved Pbx1-binding sites located within the Snail1 promoter (named hereafter Pro); second intron (SI_A and SI_B); and 5 kb downstream of the Snail1 transcription start site (Down_5 kb) (Fig. 4A). All four elements showed an active chromatin state based on enrichment by p300 and H3K27Ac in mouse embryonic midfaces, as reported elsewhere (Attanasio et al., 2013; Landin Malt et al., 2014; Yue et al., 2014) (Fig. 4A). Whereas the SI_A and Down_5 kb non-coding regions contained a partial Pbx-binding site (TGAT), both Pro and SI_B regions contained hexameric consensus motifs for Pbx–Prep/Meis-binding sites (TGACAG) (Ferretti et al., 2011; Penkov et al., 2013) (Fig. 4B). To test for in vivo interactions between the putative Pbx-binding sites within Snail1 and Pbx proteins, we performed chromatin immunoprecipitation (ChIP) assays with a Pbx1-specific Ab (Ferretti et al., 2011; Amin and Bobola, 2014; Amin et al., 2015; Losa et al., 2017) on midfacial prominences from E11.5 wild-type embryos. The described Pbx1-bound Wnt3 regulatory element (Ferretti et al., 2011) was used as a positive control. ChIP-quantitative (q)PCR showed in vivo binding of Pbx1 to all four elements in midface tissues compared with the negative control (OUT genomic region outside the Snail1 locus; Fig. 4C). In agreement with reports in mouse embryonic limb and face (Attanasio et al., 2013; Landin Malt et al., 2014), we found that all these regions were enriched for H3K27ac and negative for H3K27me3 chromatin marks, indicating an active and open chromatin state at these sites in midfacial tissues (Fig. 4D) (Creyghton et al., 2010; Cheng et al., 2014; Yue et al., 2014; Long et al., 2016). These findings demonstrate that active Snail1 putative regulatory elements, containing Pbx–Prep/Meis-binding sites, are targets of Pbx1 binding in the embryonic midface.
Fig. 4.

Pbx1 regulates Snail1 by binding to conserved noncoding sequences in embryonic midfacial prominences. (A) Mouse Snail1 on chromosome 2 (chr2; mm9). Peaks denote conservation across vertebrates (green); open chromatin (DNAseI hypersensitivity; blue) in mouse mesoderm (Yue et al., 2014); p300-bound sites in mouse embryonic face (pink) (Attanasio et al., 2013); and H3K27ac enrichment in maxillary arch (purple) (Landin Malt et al., 2014). Black rectangles comprise conserved noncoding sequences within Snail1 promoter (Pro), second intron (SI_A and SI_B), and downstream region (Down_5 kb) containing putative Pbx-binding sites. (B) Both Pro and SI_B encompass hexameric consensus Pbx–Prep/Meis-binding motif (TGACAG, blue); SI_A and Down_5 kb contain a partial Pbx-binding site (TGAT, purple). (C) Pbx1 binding to Snail1 in E11.5 midfaces (MF) by ChIP-qPCR. The positive control is a Wnt3 enhancer (Ferretti et al., 2011). OUT, negative control. (D) H3K27Ac and H3K27me3 ChIP-qPCR enrichment at the Snail1 locus. (E) Left top panel: mouse wild-type Snail1 SI_A (SI) and minimal promoter (Pro) cloned into pGL4 Luc reporter vector (SI-Pro). Bottom: mutant pGL4 construct (SI-mutPro) containing a deletion encompassing two Pbx–Prep-binding sites (orange) within the Snail1 promoter (position −557 to −531). Transcription start site (TSS); position 0. Right panel: Pbx-dependent transcription reporter assay in HEK293T cells. Data represented as mean±s.e.m. and calculated as firefly (Renilla) signal ratios normalized to SI-Pro activity upon transfection with empty pcDNA vector (set to 1). Wild-type Snail1 SI-Pro transactivated upon Pbx+Prep overexpression (**P<0.01). SI-mutPro reduces transactivation activity to basal levels (**P<0.01). HIF2α oxygen-insensitive form (HIF2α) transactivates Snail1 SI-Pro (***P<0.001). SI-mutPro does not affect HIF2α transactivation activity. n.s., not statistically significant.
To assess the impact of Pbx binding on Snail1 transcriptional activation, we generated wild-type and mutant reporter constructs encompassing Snail1 Pro and SI_A element (Fig. 4E) and assayed them by luciferase (Luc) reporter assays, as described elsewhere (Brendolan et al., 2005; Capellini et al., 2006, 2010; Ferretti et al., 2011; Koss et al., 2012; Hurtado et al., 2015). A pGL4 Luc reporter vector (SI-Pro) containing the wild-type Snail1 endogenous promoter and the SI_A element upstream of Luc was transfected into HEK293T cells with Pbx1 and its co-factor Prep1 (Pknox1) (Moens and Selleri, 2006). In parallel experiments, a mutant pGL4 construct (SI-mutPro) containing a deletion encompassing the Pbx–Prep-binding sites within the Snail1 promoter was transfected with Pbx1 and Prep1 into HEK293T cells. Multiple replicate experiments showed a consistent 2-fold increase in transcriptional activation of the SI-Pro elements on firefly Luc expression induced by Pbx1–Prep1 relative to empty vector. This moderate but significant (**P<0.01) transcriptional activation by Pbx1–Prep1 is consistent with our previous reports (Brendolan et al., 2005; Koss et al., 2012; Slavotinek et al., 2017) (Fig. 4E, right panel). Deletion of both Pbx–Prep-binding motifs within the Snail1 promoter reduced its transactivation activity to basal levels (**P<0.01), indicating that Snail1 transcription is directly dependent on Pbx–Prep function. In parallel assays in HEK293T cells, we used the HIF2α oxygen-insensitive form, a known transcriptional activator of Snail1 (Luo et al., 2011), and found that, under our experimental conditions, it significantly transactivated firefly Luc driven by Snail1 SI-Pro, which contains multiple hypoxia-responsive elements (***P<0.001). Of note, deletion of the Pbx–Prep-binding sites within Snail1 SI-Pro did not affect Snail1 transactivation by HIF2α (Fig. 4E, right panel). These results point to direct in vivo transcriptional regulation of Snail1 by Pbx–Prep, establishing a novel Pbx-directed network that underpins EMT in midface morphogenesis.
TGFβ-induced EMT in NMuMG cells correlates with upregulation of Pbx1–Prep1/2 transcriptional complexes
We further investigated the roles of Pbx factors in controlling epithelial cell plasticity in mouse epithelial NMuMG cells, a tractable system of TGFβ-induced EMT (Vincent et al., 2009). TGFβ treatment leads to changes in NMuMG cell morphology and molecular markers, with the acquisition of cell motility (Vincent et al., 2009; Netherton and Bonni, 2010). Untreated cells displayed a typical cobblestone epithelial phenotype (Fig. 5A, left panel; bright field) and detectable levels of Smad4, Pbx1 and Pbx co-factors Prep1/2 (Moens and Selleri, 2006) (Fig. 5A, top row). Administration of TGFβ (5 ng/ml) resulted in Smad4 nuclear localization, as reported elsewhere (Vincent et al., 2009), and Prep1/2 upregulation already 1 h after treatment (Fig. 5A, middle row). After 48 h, we observed enlargement of the cells and their nuclei and increased levels of Pbx1 (Fig. 5A, bottom row). These changes were accompanied by the appearance of elongated, fibroblast-like cells and loosening of the cobblestone epithelial cell layer (Fig. 5A, bright field panel; bottom row). Western blot of control and TGFβ-treated cultures (5 ng/ml for 48 h) confirmed downregulation of E-cadherin with upregulation of protein levels of Pbx1b and the EMT markers vimentin, Smad4 and Snail1/2 (Fig. 5B; for quantitation see Fig. 5C). Given the role of TGFβ in inducing apoptosis (Heldin et al., 2009), the reported involvement of Snail1/2 in resistance to apoptosis (Martínez-Álvarez et al., 2004; Vega et al., 2004), and our findings in the embryo, we examined nuclear Snail1 levels and apoptotic markers by double-labeling immunofluorescence in NMuMG cells (Fig. 5D). We quantified nuclear Snail1 levels in TGFβ-treated cultures and defined ‘High-Snail1’ and ‘Low-Snail1’ cells, respectively. The cultured cells that exhibited low levels of Snail1, likely reflecting a suboptimal response to TGFβ treatment, did not undergo apoptosis. Notably, the majority of High-Snail1 cells, fully responsive to TGFβ treatment, did not co-express apoptotic markers. Overall, only 10% of all cultured cells displayed High-Snail1 and were concomitantly either TUNEL or active caspase 3 positive (Fig. 5E). These results demonstrate that: (1) in a cell culture model of EMT, upregulation of Pbx1–Prep1/2 transcriptional complexes correlates with the induction of EMT signatures, including upregulation of Snail1/2; and (2) Snail1 partially protects cells from apoptosis in the context of this in vitro system, whereas full protection is observed in the embryo.
Fig. 5.

TGFβ signaling induces Pbx1 and Prep1/2 in an epithelial culture system. (A) Untreated (Ctrl) mouse epithelial cultures (NMuMG; top row) and cells imaged 1 h and 48 h after TGFβ treatment (5 ng/ml) in bright field (left column) and after immunofluorescence for Smad4, Pbx1 and Prep1/2 (green signal). DAPI is not shown in order to highlight the green signal. Upon treatment, spindle-like cells appear within disrupted epithelial sheets. TGFβ treatment induces Smad4 and Prep1/2 nuclear accumulation after 1 h and Pbx1 upregulation after 48 h (filled green arrowheads). Enlargement of NMuMG cells and their nuclei was observed after 48 h of TGFβ treatment. (B) Western blot of TGFβ-treated NMuMG cells (5 ng/ml for 48 h) reveals decreased E-cadherin with increased levels of Pbx1b, vimentin, Smad4 and Snail1/2. Loading control: actin. (C) Quantitation of western blots. (D) Immunofluorescence of TGFβ-treated NMuMG cells (5 ng/ml for 72 h) showing triple staining for Snail1 (green); active caspase 3 or TUNEL (red); and DAPI (blue). Filled green arrowheads, hue highlights different levels of nuclear Snail1 (light green, Low-Snail1; dark green, High-Snail1). Cells positive for nuclear Snail1 and active caspase 3 or nuclear Snail1 and TUNEL are indicated by light- or dark-orange arrowheads, respectively. (E) Quantitation of the immunofluorescence shown in D (see Materials and Methods). Scale bars: 25 µm (A); 50 µm (DI,II,IV,V); 20 µm (DIII,VI).
Pbx1 is required for TGFβ-induced EMT in epithelial cells in culture
To address the potential requirement for Pbx1 in the control of EMT, small interfering (si)RNA experiments (Elbashir et al., 2001) were performed to knock down (KD) Pbx1 in NMuMG cells. Cell cultures treated with a scrambled siRNA as a negative control (CtrlKD) showed epithelial phenotypes and typical localization of E-cadherin (Fig. 6A,E). TGFβ treatment (CtrlKD+TGFβ) resulted in the appearance of spindle-like cells (Fig. 6B) versus the cobblestone morphology of CtrlKD cultures (Fig. 6A). TGFβ-treated cells also exhibited E-cadherin downregulation and increased filamentous vimentin (Fig. 6F) versus CtrlKD cells (Fig. 6E). Moreover, treated cells appeared enlarged and showed nuclear Smad4 enrichment (Fig. 6J), whereas CtrlKD cells displayed diffused cytoplasmic Smad4 (Fig. 6I). To examine the effects of Pbx1 loss of function, cells were transiently transfected with a Pbx1 siRNA (Pbx1KD) for 48 h, then stimulated with TGFβ (5 ng/ml) for 48 h. Pbx1KD did not result in detectable changes in epithelial cobblestone morphology (Fig. 6C), E-cadherin distribution (Fig. 6G) or Smad4 cellular localization (Fig. 6K), versus controls (Fig. 6A,E,I, respectively). In contrast to CtrlKD+TGFβ cultures (Fig. 6B,F,J), TGFβ treatment of Pbx1KD cells (Pbx1KD+TGFβ) did not yield striking changes in cell morphology, although a few elongated cells were detected (Fig. 6D). Pbx1KD+TGFβ cultures also maintained characteristic epithelial localization of E-cadherin and did not display vimentin or nuclear Smad4 (Fig. 6H,L). These findings establish that Pbx1 is required for TGFβ-induced EMT in this epithelial culture system.
Fig. 6.

Pbx1 is required for TGFβ-induced EMT in cultured NMuMG epithelial cells. (A-D) Bright-field images of NMuMG cells treated with either scrambled siRNA (CtrlKD) or with Pbx1 siRNA (Pbx1KD) knockdown, without (A,C) or with (B,D) TGFβ treatment (5 ng/ml) (+TGFβ) for 48 h. Note the appearance of elongated cells after treatment (filled black arrowhead; B). (E-L) Immunofluorescence of CtrlKD and Pbx1KD cells with or without TGFβ using antibodies for E-cadherin (red), vimentin (green) (E-H) and Smad4 (green) (I-L). Filled green arrowhead (F) indicates vimentin upregulation; unfilled red arrowhead (F), E-cadherin loss; filled red arrowhead (H), E-cadherin persistence; unfilled green arrowhead (H), lack of vimentin induction; unfilled green arrowheads (L), lack of nuclear Smad4. DAPI (blue) stains nuclei (E-L). (M) qRT-PCR of EMT marker expression in cells cultured with or without TGFβ. Values normalized to TBP. All experiments performed three times, each one in triplicate. Data are shown as mean±s.d. *P<0.05; **P<0.01. (N) Western blot of Pbx1a and Pbx1b, E-cadherin, and claudin 3 levels in CtrlKD or Pbx1KD cells, without (−) or with (TGFβ) TGFβ treatment. Protein sizes are shown on the right (kDa). Loading control: actin. For quantification of western blot analyses, see Fig. S4A. Scale bars: 25 µm.
Next, we conducted qRT-PCR analyses of EMT-related gene expression in CtrlKD and Pbx1KD NMuMG cells cultured with or without TGFβ (Fig. 6M). qRT-PCR and western blot analyses confirmed downregulation of Pbx1 mRNA and Pbx1b (Monica et al., 1991) protein (Fig. 6M,N; Fig. S4A) in Pbx1KD cells, and also demonstrated failed upregulation of Snail1 mRNA in TGFβ-treated Pbx1KD cells (Fig. 6M). Furthermore, upon TGFβ treatment, Pbx1KD cells showed significant upregulation of E-cadherin and claudin 3, with downregulation of vimentin versus CtrlKD cells (Fig. 6M,N; Fig. S4A).
We further tested whether we could trigger EMT in this cellular system by decreasing TGFβ doses (1 ng/ml for 48 h) to avoid induction of apoptosis as a consequence of TGFβ treatment. Whereas epithelial morphological changes were not as striking, marker analysis confirmed the onset of EMT in TGFβ-treated CtrlKD cultures under these conditions (Fig. S4B). Pbx-dependent changes in E-cadherin, vimentin and Snail1 levels were corroborated in Pbx1KD cultures treated with low doses of TGFβ (Fig. S4B,C) without a significant increase in the percentage of apoptotic cells (Fig. S4D). These results establish that the molecular signatures of cell fate in TGFβ-induced EMT are Pbx1 dependent even in the absence of an underlying increase in apoptosis.
Forced Pbx1 expression is sufficient to induce EMT in NMuMG cells
We tested whether Pbx1 could sensitize NMuMG cells to undergo EMT after treatment with even lower doses of TGFβ (0.1 ng/ml for 24 h). To this end, we performed gain-of-function experiments by overexpressing a GFP-tagged mouse Pbx1b isoform (mPbx1b-GFP; Monica et al., 1991; Schnabel et al., 2001) via lentiviral vector transduction for 72 h. TGFβ treatment was performed 48 h after viral transduction. Live cell imaging of cultures transduced with control GFP vector showed classic cobblestone cell clusters expressing GFP (Fig. 7A,B). Immunofluorescence analyses after cell fixation revealed the maintenance of adherens junctions in epithelial cells with uniform membrane distribution of E-cadherin and low levels of vimentin at the boundaries of epithelial clusters (Fig. 7C). These results indicated that, under these conditions, NMuMG cells did not undergo EMT. Conversely, cells transduced with mPbx1b-GFP and treated with 0.1 ng/ml TGFβ showed the appearance of elongated cells with loosened intercellular contacts both at the borders of, and within, the epithelial clusters (Fig. 7D). Both control and mPbx1b overexpressing live cells exhibited high levels of GFP, indicating similar transduction efficiency (Fig. 7B,E). Immunofluorescence double labeling for E-cadherin and vimentin in fixed mPbx1b-overexpressing cells revealed E-cadherin thinning and dispersion at cell contacts, and vimentin accumulation versus control cultures (Fig. 7F). These results establish that, in a tractable epithelial cell system, Pbx1b overexpression is sufficient to stimulate cells to undergo EMT even after low-dose TGFβ treatment.
Fig. 7.

Forced Pbx1 expression is sufficient to induce EMT in NMuMG cells. (A-F) Morphological alterations and changes in EMT signatures in TGFβ-treated cells (0.1 ng/ml for 24 h) overexpressing Pbx1b, versus control. (A,D) Bright field shows shape changes and scattering in NMuMG cells transduced with a lentivirus carrying mPbx1b (unfilled black arrowhead) versus control (Ctrl; filled black arrowhead). (B,E) GFP visualizes similar transfection efficiency in live cells with or without mPbx1b overexpression. (C,F) Immunofluorescence shows membrane-associated E-cadherin (filled red arrowhead) and negligible filamentous vimentin (unfilled green arrowhead) in fixed cells transduced with control vector (C). Increase of vimentin (filled green arrowhead) and E-cadherin reduction at cell junctions (unfilled red arrowhead) indicate EMT induction in mPbx1b-lentivirus transduced cells (F). (G) Western blot of Pbx1a, Pbx1b and Snail1 levels in NMuMG cells transduced with Ctrl or mPbx1b. Pbx1a is present at low levels. Loading control: actin. Quantification of western blot is shown on the right. (H,I) Immunofluorescence of fixed cells transduced with lentivirus carrying Ctrl (H) or mPbx1b (I) construct for Pbx1 (red) and vimentin (green). DAPI (blue) stains nuclei. In controls, a small number of cells with low endogenous vimentin were observed (unfilled green arrowhead, H). In cells overexpressing mPbx1b, induction of filamentous cytoplasmic vimentin was observed (green arrowhead, I). Scale bars: 50 µm (A,B,D,E); 25 µm (C,F,H,I).
This finding prompted us to explore whether forced expression of Pbx1b was sufficient to induce EMT without the initial TGFβ trigger. Upon mPbx1b overexpression, quantitation of western blots revealed a 2.2-fold increase in Snail1 levels over control, which coincided with a 2.4-fold increase in Pbx1b levels versus endogenous control (Fig. 7G). Immunofluorescence analyses of NMuMG cells after transduction with control and mPbx1b-expressing lentiviral vector for 72 h, revealed that forced mPbx1b expression resulted in significant cytoplasmic accumulation of filamentous vimentin versus control (Fig. 7H,I). These findings highlight that Pbx1b is sufficient to induce EMT even without exposure to TGFβ, suggesting that, in our system, EMT occurs via a Pbx1-dependent pathway that is not initially triggered by TGFβ. They further support Pbx-dependent control of Snail1 in NMuMG cells (see Fig. 6M) consistent with the observations in the embryonic midface (see Fig. 4C).
DISCUSSION
Although one-third of all human congenital abnormalities affect craniofacial development, including orofacial clefting (Dixon et al., 2011; Zeytinoglu and Davey, 2012; Carlson et al., 2017), our knowledge of the mechanisms that underlie facial morphogenesis and its perturbations is poor. In this study, we revealed that apoptosis is restored in Pbx1/2 mutants with rescued orofacial clefting defects (see Fig. S1A), pointing to a direct association of programmed cell death with fusion of the frontonasal prominences. However, we also demonstrated that epithelial apoptosis at the λ is spatially and temporally regulated with no more than 40% of the seam epithelial cells undergoing programmed cell death. Therefore, apoptosis alone cannot enable prominence coalescence because all seam epithelial cells must be lost to allow fusion. Multiple studies have addressed the contributions of different cellular behaviors, including apoptosis and EMT, to fusion of the secondary palate (reviewed in Bush and Jiang, 2012 and in Lan et al., 2015). Conversely, little is known of the cellular processes that mediate prominence fusion at the λ. Here, we tackled the longstanding controversy of the potential role of EMT in upper lip and primary palate morphogenesis and fusion.
EMT is the most striking manifestation of epithelial plasticity (Nieto, 2013). During this process, cells lose their epithelial properties, acquire mesenchymal characteristics, including motility, and undergo changes in gene expression. EMT occurs in developmental and pathological contexts, such as fibrosis and cancer (Acloque et al., 2009; Lamouille et al., 2014; Ye and Weinberg, 2015; Nieto, 2017). During development, EMT can mediate tissue remodeling at sites of fusion, when two tissue components integrate to form a continuous structure. Disruption of tissue remodeling, which is crucial for the morphogenesis of multiple organs, including the secondary palate, neural tube, heart, and body wall (Ray and Niswander, 2012), leads to devastating birth defects. In 2000, based on morphological observations, Elizabeth Hay and colleagues reported that fusion of the facial prominences occurs via EMT in the chick (Sun et al., 2000). Subsequently, it was suggested that expression of the epithelial tight junction component claudin 6 is increased at the epithelial seam in mouse embryos with spontaneous CL (Nakazawa et al., 2008). However, the underlying mechanisms were not reported.
In this study, we established a temporal sequence of apoptosis and EMT during upper lip/primary palate fusion. Under physiological conditions, apoptosis is detectable by E10.5, prior to the onset of EMT-associated changes, and before the prominences contact each other (see Fig. 8). Subsequently, we documented morphological features of epithelial plasticity at the seam; the presence of a subpopulation of transitional cells expressing epithelial and mesenchymal markers; and migration of cells of epithelial derivation into the mesenchyme of the λ. By E11.5, only 2% of the transitional cells at the seam undergo apoptosis. Accordingly, we concluded that a complete EMT program, including control of the crucial EMT effectors Smad4 and Snail1, mediates murine midfacial prominence fusion in a Pbx-dependent manner. Starting at E10.5, apoptosis might favor the onset of EMT by disrupting the epithelial sheets and facilitating their disintegration, which could lead, in turn, to breakdown of the BM by E11.0. This process is followed by the appearance of transitional cells and a few cells of epithelial descent that migrate into the λ mesenchyme by E11.5 (see Fig. 2E,H), in agreement with a previous study on secondary palate fusion (Martínez-Álvarez et al., 2004).
Fig. 8.
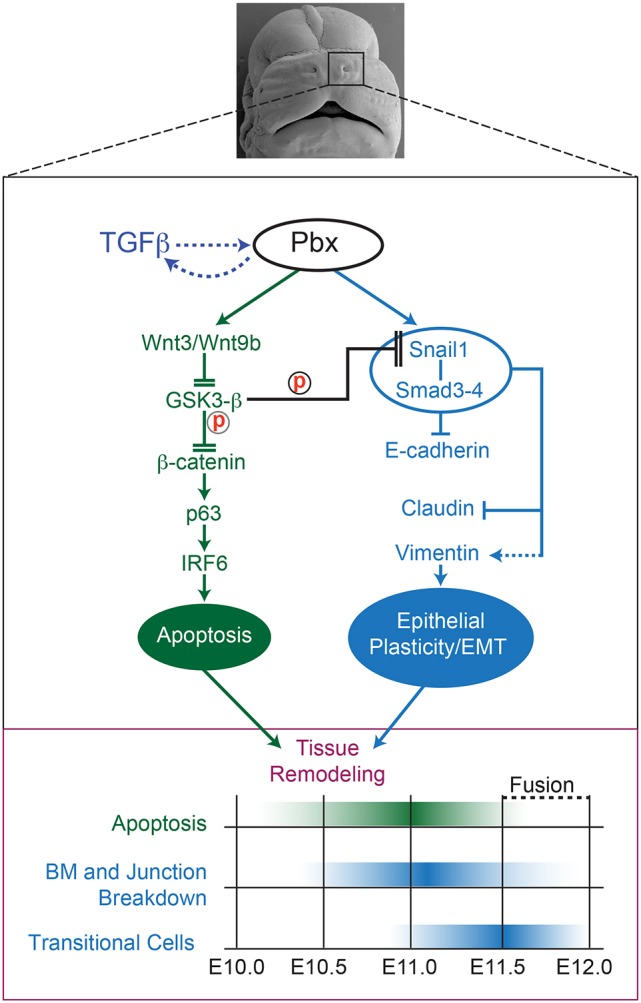
Pbx-dependent crosstalking regulatory axes direct dynamic cellular behaviors controlling epithelial tissue remodeling at the λ. Top: Frontal view of E13.5 murine midface after lnp/mnp/mx fusion. Black rectangle (enlarged in middle panel) highlights Pbx-dependent regulatory networks (active from E10.0 to E12.0). Cross-regulatory loop between TGFβ and Pbx is indicated by dashed arrows. In NMuMG cells, TGFβ promotes Pbx nuclear accumulation (see Fig. 5) and, concomitantly, in the embryo Pbx1/2 loss of function results in loss of Smad4 at the λ epithelia (see Fig. 3). The green axis controls epithelial apoptosis (Ferretti et al., 2011), whereas the blue axis mediates epithelial plasticity/EMT at the λ. These regulatory axes can crosstalk via Snail1 post-transcriptional control by GSK3-β (Zhou et al., 2004; Yook et al., 2006) and converge to execute tissue remodeling. Solid arrows indicate direct transcriptional control; dashed arrows indirect activation; flat heads transcriptional repression; double flat heads reduced protein activity through post-transcriptional mechanisms. Bottom: Developmental timing of epithelial tissue remodeling. Green hue highlights the peak of apoptosis; blue hue the breakdown of the BM and adherens and tight junctions, as well as the appearance of transitional cells. ‘Fusion’ indicates the replacement of the frontonasal epithelial seam by the mesenchymal bridge from E11.5 to E12.0.
Multiple lines of evidence indicate that apoptosis and EMT can be alternative, complementary or sequential processes that secure normal tissue morphogenesis, including: the demonstration that Snail1/2 protect cells from apoptosis (Martínez-Álvarez et al., 2004; Vega et al., 2004); the reports that inhibition of programmed cell death in hepatocytes and epithelial cells induces EMT phenotypes (Franco et al., 2010; An et al., 2015); and our results revealing that the majority of epithelial cells that exhibit nuclear Snail1 do not undergo apoptosis in the embryo. In the embryo, only 1.5% of the λ epithelial cells concomitantly exhibit nuclear Snail1 and apoptotic markers; by contrast, approximately 10% of all TGFβ-treated cells in our in vitro system display High-Snail1 and undergo apoptosis. The mechanisms of TGFβ-induced apoptosis differ between various cell types and systems (Heldin et al., 2009). Also, it cannot be determined to what extent embryonic apoptosis is TGFβ dependent in vivo. These possible scenarios likely account for the discrepancy between our in vivo and in vitro models. Collectively, our results support a model whereby apoptosis and EMT are sequential Pbx-dependent cellular behaviors that are deployed in the embryo at the λ seam to remove epithelial cells. Lastly, although not explored in our study, cellular extrusion (Kim et al., 2015), as well as changes in cell adhesion and migration (Campbell and Casanova, 2016), might also occur at the λ epithelia, contributing to complete fusion of the facial prominences.
TGFβ-induced EMT in a tractable epithelial culture system resulted in increased nuclear levels of Pbx1 and Prep1/2, with the induction of mesenchymal signatures and downregulation of epithelial markers. Loss-of-function experiments in these cells established that Pbx1 is required for TGFβ-induced EMT. Notably, the observed changes were present in this epithelial cell system after low-dose TGFβ-treatment even in the absence of increased apoptosis (see Fig. S4D). In agreement with our findings, it was reported that PBX1 cooperates with its partner PREP1 to trigger TGFβ-induced EMT in human lung adenocarcinoma cells and that PREP1 regulates SMAD3 transcription in this system (Risolino et al., 2014). Moreover, our study demonstrates that forced Pbx1 expression induces EMT in NMuMG cells via a Pbx1-dependent pathway that is not initially triggered by TGFβ. In summary, our work establishes that, in epithelial cells, Pbx1 is required and sufficient to induce EMT, recapitulating most of the cellular and molecular changes observed in the embryo.
Evidence from both our in vivo and in vitro studies proves the direct regulation of Snail1 by Pbx homeoproteins. Snail1, in collaboration with Snail2, has crucial functions in murine craniofacial development (Carver et al., 2001; Murray et al., 2007). Accordingly, the presence of orofacial clefting in compound conditional Pbx1/Snail1 mutants would corroborate their genetic interaction in vivo. However, uncovering Snail1–Pbx1 genetic interactions would likely require Snail2 concomitant loss in the cephalic epithelium, because Snail2 is expressed in ectodermal and EMT territories (Sefton et al., 1998; Acloque et al., 2017). Here, we propose that Pbx1 has either instructive roles in Snail1 transactivation or permissive roles as a ‘poising’ factor by recruiting proteins and/or co-factors that will modify the chromatin environment for activation of target genes, such as Snail1, by other TFs in the midface, in keeping with previous studies (Berkes et al., 2004; Sagerström, 2004; Choe et al., 2009; Grebbin et al., 2016).
We established two Pbx-dependent regulatory networks that control tissue remodeling at the λ (see Fig. 8). Although we did not investigate a potential crosstalk between the two axes here, reports of MCF7 cells undergoing EMT suggest that GSK3-β can control Snail1 by post-transcriptional regulation (Zhou et al., 2004; Yook et al., 2006). Thus, we postulate that the Pbx–Wnt–p63–IRF6 regulatory network that we reported previously (Ferretti et al., 2011) crosstalks with the Pbx–Snail1/Smad–ECad pathway described here. Indeed, absence of Pbx1 causes downregulation of Snail1 via perturbed transcriptional activation, as well as impairment of Wnt signaling (Ferretti et al., 2011), with consequent activation of GSK3-β. Therefore, we envisage that hyperactive GSK3-β might phosphorylate Snail1 at the λ, decreasing its stability and nuclear accumulation, as in MCF7 cells. Consequently, Pbx loss of function at the λ can prevent the execution of Snail1-directed morphogenetic programs by both transcriptional and post-transcriptional mechanisms. In the present study, we showed that restoration of apoptosis at the λ initially mediates rescue of orofacial clefting achieved by overexpressing Wnt1 in Pbx1/2 mutant embryos (Ferretti et al., 2011). Given the potential crosstalk between the two Pbx-dependent pathways, we predict that subsequent rescue of epithelial behaviors and EMT by Snail1 overexpression might also be required to obtain complete repair of orofacial clefting. Of note, EMT-inducing TFs can execute different programs depending on their expression levels, as reported for Twist1 (Beck et al., 2015). Accordingly, modulation of Snail1 levels could coordinate complementary developmental processes, such as apoptosis and EMT, during frontonasal prominence fusion.
In conclusion, we have delineated two cascades that form a robust PBX-dependent regulatory circuit underlying seam tissue remodeling. Genome-wide experiments (van Dijk et al., 2014) will uncover a network of Pbx-directed effectors of apoptosis, epithelial plasticity, EMT, cell adhesion and migration that mediate tissue remodeling at the murine λ. Notably, Pbx1 was recently identified among a group of genes that drive EMT in hepatocellular carcinoma in association with a signature predicting poor survival in affected patients (Kodama et al., 2016). Accordingly, our study brings new knowledge of the interconnected cellular behaviors that execute midface morphogenesis and other biological processes that rely on similar cell behaviors, such as tumor invasion (Ye and Weinberg, 2015; Zheng et al., 2015; Santamaria et al., 2017).
MATERIALS AND METHODS
Mice
Mutant alleles used in this study were previously described and the conditions for genotyping were reported: R26RlacZ reporter (Soriano, 1999); Pbx1 (Selleri et al., 2001; Capellini et al., 2006) and Pbx2 (Selleri et al., 2004; Capellini et al., 2006) knockout; a conditional Pbx1f allele (Koss et al., 2012); the transgenic deleter strain Crect expressing Cre recombinase from the mouse Tfap2a ectoderm-specific enhancer (Ferretti et al., 2011; Reid et al., 2011); and the Rosa–Wnt1 mice used for rescue experiments (Carroll et al., 2005; Ferretti et al., 2011). Experiments on mice were performed following IACUC guidelines and experimental procedures concerning housing, husbandry and welfare. Stages of all embryos analyzed are stated in each figure and figure legend for each assay.
Scanning and transmission electron microscopy
For SEM, after harvesting, embryos were fixed overnight (o/n) in PBS containing 4% (w/v) paraformaldehyde (PFA) and 2% glutaraldehyde at 4°C. Specimens were washed in PBS, dehydrated in ethanol series, dried in carbon dioxide and sputter-coated with gold. Subsequently, they were examined using an FEI Quanta FEG SEM operating at 10 kV. For TEM, embryos were prepared following standard procedures (Venable and Coggeshall, 1965) and imaged with a JEOL JEM 100CX transmission electron microscope. Three embryos for each genotype were analyzed for SEM and TEM.
In situ hybridization
Section in situ hybridization was performed on E11.5 embryos as previously described (Capellini et al., 2006, 2010). The Snail1 cDNA construct was a kind gift from A. Nieto (Sefton et al., 1998). At least three embryos for each genotype were analyzed by section in situ hybridization.
Epithelial lineage tracing and quantitation of β-galactosidase-positive cells
Crect;R26R embryos were collected at specified stages, and stained for β-galactosidase activity in whole mounts and sections using previously described protocols (Ferretti et al., 2011; Van Otterloo et al., 2016). Quantitation of β-galactosidase-positive cells was performed as follows: a total of 25 β-galactosidase-stained sections from five independent E11.5 wild-type embryos were analyzed for an average of five sections per embryo. Sections were photographed at 40× magnification, imported into Adobe Photoshop, and then overlaid with a 6.45 square-cm grid. Each grid contained three cells on average. For each section, 100 grids closest to the seam of the λ junction were counted. The number of grids containing at least one β-galactosidase-positive cell (referred to as ‘blue sectors’) was noted. At the resolution allowed by X-gal staining of embryonic sections, only an estimate of the number of β-galactosidase-positive cells contained in each blue sector could be calculated. Therefore, Fig. 2H shows a box-and-whisker plot illustrating the percentage of ‘blue sectors’. The red line denotes the median value (4%; see Fig. 2H). The box outlines the middle 50% of value range. Whiskers denote maximal and minimal values.
Immunofluorescence and imaging
Embryos were harvested at specified stages from E10.5 to E11.5, fixed o/n at 4°C in PBS containing 4% PFA, rinsed in PBS, and cryoprotected in 30% sucrose o/n at 4°C. Subsequently, they were embedded in OCT Compound and cryosectioned at 10-12 μm per section. Slides were blocked for 1 h with 10% fetal bovine serum (FBS)/PBS and incubated o/n in 0.1% bovine serum albumin (BSA) with primary Abs. Primary Abs and dilutions are listed in the supplementary Materials and Methods. Primary Ab binding was detected by AlexaFluor-conjugated secondary Abs (Molecular Probes). Nuclei were stained with DAPI (Sigma). TUNEL staining was performed according to the manufacturer's instructions (Roche, 12156792910). Fluorescence imaging was performed on a Zeiss Axioplan2 upright microscope, captured using a Hamamatsu Orca camera, and acquired using the Open Lab software (PerkinElmer). Confocal imaging was performed on an inverted Leica DMI 600 microscope, captured using a Leica HyD detector, and acquired using the Leica LAS Software. Sections from at least three embryos for each genotype were analyzed in all immunofluorescence experiments.
Quantitation of apoptosis
Cells undergoing apoptosis at the λ junction of control embryos were detected by immunofluorescence analysis of 10-12 μm cryosections with active caspase 3 (Promega, G7481) Ab (1:200). Algorithms for the quantification of cell death were generated with MATLAB software (MathWorks). To define the λ junction, a line of fixed length was set perpendicular to the mnp and lnp tips on imaged sections, and a second line of fixed length was placed perpendicular to the first. All cells within the enclosed area were then counted for all slides analyzed along the A-P axis. Epithelial and mesenchymal cells were counted separately. The percentage of apoptotic cells was calculated at each developmental stage examined by dividing the number of active caspase 3-positive cells by the total number of counted cells (400-700) at the λ junction on each section. Sections from at least three embryos (approximately 30 sections) were analyzed at each embryonic stage. Box-and-whisker plots were used to illustrate the quantitation of apoptotic cells. Red lines denote median values (see Fig. S1). Boxes outline the middle 50% of the value range. Whiskers denote maximal and minimal values. Assessment of apoptosis in NMuMG cells in culture treated with TGFβ was achieved by using the Annexin V Alexa Fluor 488 kit (Thermo Fisher Scientific, A13201), followed by flow cytometric analysis, according to the manufacturer's instructions.
Quantitation of transitional cells
Box-and-whisker plots were used to illustrate the quantitation of transitional cells at the λ, as described above. The percentage of transitional cells was calculated over the total number of epithelial cells at the λ in E11.5 control embryos (see Fig. 2E). Red lines denote median values for transitional cells and transitional apoptotic cells. Plots were calculated based on the same set of 15 coronal sections obtained from six different embryos in six independent experiments.
Quantitation of cells co-expressing nuclear snail1 and apoptotic markers
Quantitation of nuclear Snail1 levels and colocalization with apoptotic markers was conducted using MATLAB software (MathWorks) on embryonic sections of λ junctions from E10.75 embryos slides of NMuMG cells treated with TGFβ (5 ng/ml for 72 h). For experiments on cultured cells, we calculated the mean value of nuclear Snail1 signal and subsequently used this value as the threshold to identify cells with signal values above or below the threshold (defined as ‘High-Snail1’ and ‘Low-Snail1’ cells, respectively). High-Snail1 cells were approximately 40% of the total number of the cells in culture, whereas Low-Snail1 cells accounted for the remaining 60% of the cells. Among the cells defined as High-Snail1, only approximately 30% were concomitantly positive for TUNEL or caspase 3, which equals approximately 10% of the total number of cultured cells (see Fig. 5E). For experiments on embryonic sections of λ junctions, we calculated the mean value of nuclear Snail1 signal in epithelial cells. This value was used to define signal background levels. Cells with nuclear Snail1 signal values above this threshold were considered as positive. Nuclear Snail1-positive cells were approximately 25% of the total number of epithelial cells at the λ, whereas only approximately 6% of those were concomitantly TUNEL positive. Overall, the total number of cells double positive for nuclear Snail1 and TUNEL accounted for approximately 1.5% of the epithelial cells at the λ junction.
Chromatin immunoprecipitation
Embryonic tissues (midfaces) were isolated from E11.5 mouse embryos, obtained from time matings of Swiss–Webster mice as previously described (Ferretti et al., 2011) and were immediately crosslinked for 10 min in 1% formaldehyde (Electron Microscopy Sciences, #15710). ChIP assays were performed as reported elsewhere (Amin and Bobola, 2014; Amin et al., 2015; Losa et al., 2017). The crosslinked material was sonicated to 200-500 bp fragments with a Diagenode Bioruptor. Immunoprecipitation was performed from 12-14 embryonic midfaces, by overnight incubation with specific Abs (5 µg) at 4°C followed by 30 min incubation with Dynabeads protein A (Invitrogen). After washing, IP and input DNA were de-crosslinked and purified using the QIAquick PCR purification kit (Qiagen, 28106). qPCR was performed to compare enrichment of specific genomic locations after immunoprecipitation on a QuantStudio 6 Flex Real-Time PCR System (Life Technologies). Enrichment of each element following immunoprecipitation with Pbx1 Ab or chromatin mark Abs and negative control Ab (IgG) was calculated as the percentage of the input. The fragment defined as ‘OUT’ (outside the Snail1 locus; see Fig. 4) was located in nonconserved, noncoding regions and was not bound by Pbx proteins, thus serving as an appropriate negative control. Specificity of immunoprecipitation was assessed by using chromatin precipitated with rabbit IgG. Each chromatin immunoprecipitation assay was conducted in at least three independent experiments, each one including technical triplicates. Abs and oligoprimer sequences are listed in the supplementary Materials and Methods.
Cell cultures, siRNA assays and transfections, lentiviral constructions and production
NMuMG mouse mammary gland and HEK293T cells were obtained from ATCC and cultured in D-MEM supplemented with 10% FBS, 10 μg/ml insulin (only for NMuMG cells), and 2 mM L-glutamine (Invitrogen), in humidified 5% CO2. Cell lines were routinely checked for mycoplasm contamination. Purified recombinant TGFβ1 protein was purchased from R&D Systems (240-B-002). NMuMG cells were transiently transfected with Pbx1 siRNA (Dharmacon) using DharmaFECT (Thermo Scientific, T-2001-02). Cells were replated and treated with TGFβ 48 h after transfection, for the duration and dosage indicated in the relative experiments.
For overexpression studies, Pbx1 lentiviral constructs were used. The full-length cDNA of mouse Pbx1b (mPbx1b) was cloned into the lentiviral vector VIRHD-EP42 (Grumolato et al., 2013). For the production of the Pbx1b lentivirus, transient transfections were performed using polyethylenimine (Polysciences) according to the manufacturer's instructions. Confluent HEK293T cells (50-70%) were co-transfected with a combination of plasmids, including lentiviral packaging plasmid pCMV-DR8.9, envelope plasmid pMD2G-VSVG and lentiviral vector VIRHD-EP-mPbx1b. Supernatants containing high-titer viral stocks were collected from HEK293T cells and used to infect NMuMG cells. Puromycin was used for the selection of cell clones expressing mPbx1b. Specifically, approximately 100 mPbx1b-transduced colonies were selected and pooled, while, in parallel, another 100 colonies were infected with the VIRHD-EP vector alone as a negative control. For bright-field microscopy, live imaging and immunofluorescence assays, cells were plated on chamber slides at a density of 2×104 cells/slide following protocols described above for embryonic sections and using Abs listed in the supplementary Materials and Methods.
Western blot analysis
Total protein lysates from NMuMG cells were resolved by 10% SDS-PAGE and blotted to polyvinylidene difluoride (PVDF) membranes (Millipore) as previously described (Risolino et al., 2014). Membranes were first incubated o/n with the primary Ab and then with a peroxidase-conjugated secondary Ab. Peroxidase activity was measured by using an enhanced chemiluminescence (ECL) kit (Pierce/Thermo Fisher Scientific), following the manufacturer's instructions. See supplementary Materials and Methods for details on the primary Abs used. Each western blot assay was performed at least in two independent experiments. Quantitation of western blot analysis was conducted by calculating the relative protein levels from the intensity of the band signals (ImageJ version 1.50i).
Quantitative real-time PCR assays
Total RNA was purified from NMuMG cells using standard procedures. Gene expression was determined by qRT-PCR using QuantiTect SYBR Green PCR master mix (Qiagen) and the 7500 Real-Time PCR System (Applied Biosystems). Oligoprimer sequences are listed in supplementary Materials and Methods. Technical triplicates and biological duplicates were performed for each qRT-PCR assay.
Transcriptional reporter assays
Transient transfections in HEK293T cells were performed using our reported protocols (Koss et al., 2012). The Luc reporter plasmid was constructed by amplifying a conserved 951 bp fragment corresponding to the Snail1 promoter (Pro) (Peiro et al., 2006) and a 368 bp conserved element within the second intron (SI). A mutant construct (SI-mutPro) with a 26 bp deletion, encompassing two conserved Pbx-binding sites, was obtained using the wild-type construct as a template. Strategy and oligoprimers used to clone the Luc constructs are described in supplementary Materials and Methods. All constructs were verified by Sanger sequencing. Cells were transfected with different combinations of empty vector pcDNA and SI-Pro-Luc reporter construct, or pSG5 vectors carrying human Pbx1b and Prep1 and SI-Pro-Luc reporter construct. An oxygen-insensitive form of HIF2α (Addgene), which is known to transactivate the Snail1 promoter in a Pbx1-independent manner, was used as a control of specificity. Firefly values were normalized for Renilla activity. Data are presented as mean±s.e.m. of three technical replicates from three independent biological experiments. Statistical analyses used unpaired Student's t-tests, with P<0.05 considered significant.
Supplementary Material
Acknowledgements
We are grateful to Professor Francesco Blasi (IFOM, Milan, Italy) and Professor Pasquale Verde (CNR, Naples) for discussing unpublished results; we thank Drs Mary Marazita and Elizabeth J. Leslie (University of Pittsburgh) and Dr Juhee Jeong (NYU) for critical discussions. We also thank Dr Hong Li for assistance in lineage-tracing experiments and cell counts.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: M.L., M.R., B.L., C.T.V., A.M.C.B., E.F., T.W., L.S.; Methodology: M.L., M.R., B.L., J.H., L.Q., I.G., H.Y., I.F.C., P.L., S.K., R.A., J.F., E.F., T.W.; Software: R.A.; Formal analysis: M.L., M.R., R.A.; Writing - original draft: M.L., M.R., L.S.; Writing - review & editing: M.L., M.R., L.S.
Funding
This work was funded by the National Institutes of Health [RO1 grant DE024745 to L.S. and RO1 grant DE019843 to T.W.] and by the University of California, San Francisco (UCSF) Program in Craniofacial Biology Dr. Caroline H. Damsky Award [FY 16-17 Exploratory Grants Initiative to M.R.]. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.157628.supplemental
References
- Acloque H., Adams M. S., Fishwick K., Bronner-Fraser M. and Nieto M. A. (2009). Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J. Clin. Invest. 119, 1438-1449. 10.1172/JCI38019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acloque H., Ocaña O. H., Abad D., Stern C. D. and Nieto M. A. (2017). Snail2 and Zeb2 repress P-cadherin to define embryonic territories in the chick embryo. Development 144, 649-656. 10.1242/dev.142562 [DOI] [PubMed] [Google Scholar]
- Ahmed S., Liu C.-C. and Nawshad A. (2007). Mechanisms of palatal epithelial seam disintegration by transforming growth factor (TGF) beta3. Dev. Biol. 309, 193-207. 10.1016/j.ydbio.2007.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin S. and Bobola N. (2014). Chromatin immunoprecipitation and chromatin immunoprecipitation with massively parallel sequencing on mouse embryonic tissue. Methods Mol. Biol. 1196, 231-239. 10.1007/978-1-4939-1242-1_14 [DOI] [PubMed] [Google Scholar]
- Amin S., Donaldson I. J., Zannino D. A., Hensman J., Rattray M., Losa M., Spitz F., Ladam F., Sagerström C. and Bobola N. (2015). Hoxa2 selectively enhances Meis binding to change a branchial arch ground state. Dev. Cell 32, 265-277. 10.1016/j.devcel.2014.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J., Lv J., Li A., Qiao J., Fang L., Li Z., Li B., Zhao W., Chen H. and Wang L. (2015). Constitutive expression of Bcl-2 induces epithelial-mesenchymal transition in mammary epithelial cells. BMC Cancer 15, 476 10.1186/s12885-015-1485-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attanasio C., Nord A. S., Zhu Y., Blow M. J., Li Z., Liberton D. K., Morrison H., Plajzer-Frick I., Holt A., Hosseini R. et al. (2013). Fine tuning of craniofacial morphology by distant-acting enhancers. Science 342, 1241006 10.1126/science.1241006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck B., Lapouge G., Rorive S., Drogat B., Desaedelaere K., Delafaille S., Dubois C., Salmon I., Willekens K., Marine J.-C. et al. (2015). Different levels of Twist1 regulate skin tumor initiation, stemness, and progression. Cell Stem Cell 16, 67-79. 10.1016/j.stem.2014.12.002 [DOI] [PubMed] [Google Scholar]
- Berkes C. A., Bergstrom D. A., Penn B. H., Seaver K. J., Knoepfler P. S. and Tapscott S. J. (2004). Pbx marks genes for activation by MyoD indicating a role for a homeodomain protein in establishing myogenic potential. Mol. Cell 14, 465-477. 10.1016/S1097-2765(04)00260-6 [DOI] [PubMed] [Google Scholar]
- Bhatt S., Diaz R. and Trainor P. A. (2013). Signals and switches in mammalian neural crest cell differentiation. Cold Spring Harbor Perspect. Biol. 5, a008326 10.1101/cshperspect.a008326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendolan A., Ferretti E., Salsi V., Moses K., Quaggin S., Blasi F., Cleary M. L. and Selleri L. (2005). A Pbx1-dependent genetic and transcriptional network regulates spleen ontogeny. Development 132, 3113-3126. 10.1242/dev.01884 [DOI] [PubMed] [Google Scholar]
- Bürglin T. R. (2011). Homeodomain subtypes and functional diversity. Subcell. Biochem. 52, 95-122. 10.1007/978-90-481-9069-0_5 [DOI] [PubMed] [Google Scholar]
- Bürglin T. R. and Affolter M. (2016). Homeodomain proteins: an update. Chromosoma 125, 497-521. 10.1007/s00412-015-0543-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush J. O. and Jiang R. (2012). Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development 139, 231-243. 10.1242/dev.067082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell K. and Casanova J. (2016). A common framework for EMT and collective cell migration. Development 143, 4291-4300. 10.1242/dev.139071 [DOI] [PubMed] [Google Scholar]
- Capellini T. D., Di Giacomo G., Salsi V., Brendolan A., Ferretti E., Srivastava D., Zappavigna V. and Selleri L. (2006). Pbx1/Pbx2 requirement for distal limb patterning is mediated by the hierarchical control of Hox gene spatial distribution and Shh expression. Development 133, 2263-2273. 10.1242/dev.02395 [DOI] [PubMed] [Google Scholar]
- Capellini T. D., Vaccari G., Ferretti E., Fantini S., He M., Pellegrini M., Quintana L., Di Giacomo G., Sharpe J., Selleri L. et al. (2010). Scapula development is governed by genetic interactions of Pbx1 with its family members and with Emx2 via their cooperative control of Alx1. Development 137, 2559-2569. 10.1242/dev.048819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette M. J. and Ferguson M. W. (1992). The fate of medial edge epithelial cells during palatal fusion in vitro: an analysis by DiI labelling and confocal microscopy. Development 114, 379-388. [DOI] [PubMed] [Google Scholar]
- Carlson J. C., Taub M. A., Feingold E., Beaty T. H., Murray J. C., Marazita M. L. and Leslie E. J. (2017). Identifying genetic sources of phenotypic heterogeneity in orofacial clefts by targeted sequencing. Birth Defects Res. 109, 1030-1038. 10.1002/bdr2.23605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll T. J., Park J.-S., Hayashi S., Majumdar A. and McMahon A. P. (2005). Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev. Cell 2, 283-292. 10.1016/j.devcel.2005.05.016 [DOI] [PubMed] [Google Scholar]
- Carver E. A., Jiang R., Lan Y., Oram K. F. and Gridley T. (2001). The mouse snail gene encodes a key regulator of the epithelial-mesenchymal transition. Mol. Cell. Biol. 21, 8184-8188. 10.1128/MCB.21.23.8184-8188.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y., Ma Z., Kim B.-H., Wu W., Cayting P., Boyle A. P., Sundaram V., Xing X., Dogan N., Li J. et al. (2014). Principles of regulatory information conservation between mouse and human. Nature 515, 371-375. 10.1038/nature13985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe S.-K., Lu P., Nakamura M., Lee J. and Sagerström C. G. (2009). Meis cofactors control HDAC and CBP accessibility at Hox-regulated promoters during zebrafish embryogenesis. Dev. Cell 17, 561-567. 10.1016/j.devcel.2009.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton M. P., Cheng A. W., Welstead G. G., Kooistra T., Carey B. W., Steine E. J., Hanna J., Lodato M. A., Frampton G. M., Sharp P. A. et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 107, 21931-21936. 10.1073/pnas.1016071107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley M. A., Conlin L. K., Zackai E. H., Deardorff M. A., Thiel B. D. and Spinner N. B. (2010). Further evidence for the possible role of MEIS2 in the development of cleft palate and cardiac septum. Am. J. Med. Genet. A 152A, 1326-1327. 10.1002/ajmg.a.33375 [DOI] [PubMed] [Google Scholar]
- Cuervo R. and Covarrubias L. (2004). Death is the major fate of medial edge epithelial cells and the cause of basal lamina degradation during palatogenesis. Development 131, 15-24. 10.1242/dev.00907 [DOI] [PubMed] [Google Scholar]
- Depew M. J. and Compagnucci C. (2008). Tweaking the hinge and caps: testing a model of the organization of jaws. J. Exp. Zool. B Mol. Dev. Evol. 310B, 315-335. 10.1002/jez.b.21205 [DOI] [PubMed] [Google Scholar]
- Dixon M. J., Marazita M. L., Beaty T. H. and Murray J. C. (2011). Cleft lip and palate: understanding genetic and environmental influences. Nat. Rev. Genet. 12, 167-178. 10.1038/nrg2933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir S. M., Harborth J., Lendeckel W., Yalcin A., Weber K. and Tuschl T. (2001). Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494-498. 10.1038/35078107 [DOI] [PubMed] [Google Scholar]
- Ferretti E., Li B., Zewdu R., Wells V., Hebert J. M., Karner C., Anderson M. J., Williams T., Dixon J., Dixon M. J. et al. (2011). A conserved Pbx-Wnt-p63-Irf6 regulatory module controls face morphogenesis by promoting epithelial apoptosis. Dev. Cell 21, 627-641. 10.1016/j.devcel.2011.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitchett J. E. and Hay E. D. (1989). Medial edge epithelium transforms to mesenchyme after embryonic palatal shelves fuse. Dev. Biol. 131, 455-474. 10.1016/S0012-1606(89)80017-X [DOI] [PubMed] [Google Scholar]
- Franco D. L., Mainez J., Vega S., Sancho P., Murillo M. M., de Frutos C. A., Del Castillo G., Lopez-Blau C., Fabregat I. and Nieto M. A. (2010). Snail1 suppresses TGF-beta-induced apoptosis and is sufficient to trigger EMT in hepatocytes. J. Cell Sci. 123, 3467-3477. 10.1242/jcs.068692 [DOI] [PubMed] [Google Scholar]
- Fuxe J., Vincent T. and Garcia de Herreros A. (2010). Transcriptional crosstalk between TGF-β and stem cell pathways in tumor cell invasion: role of EMT promoting Smad complexes. Cell Cycle 9, 2363-2374. 10.4161/cc.9.12.12050 [DOI] [PubMed] [Google Scholar]
- Grebbin B. M., Hau A.-C., Groß A., Anders-Maurer M., Schramm J., Koss M., Wille C., Mittelbronn M., Selleri L. and Schulte D. (2016). Pbx1 is required for adult subventricular zone neurogenesis. Development 143, 2281-2291. 10.1242/dev.128033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritli-Linde A. (2008). The etiopathogenesis of cleft lip and cleft palate: usefulness and caveats of mouse models. Curr. Top. Dev. Biol. 84, 37-138. 10.1016/S0070-2153(08)00602-9 [DOI] [PubMed] [Google Scholar]
- Grumolato L., Liu G., Haremaki T., Mungamuri S. K., Mong P., Akiri G., Lopez-Bergami P., Arita A., Anouar Y., Mlodzik M. et al. (2013). beta-Catenin-independent activation of TCF1/LEF1 in human hematopoietic tumor cells through interaction with ATF2 transcription factors. PLoS Genet. 9, e1003603 10.1371/journal.pgen.1003603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin C.-H. and Moustakas A. (2012). Role of Smads in TGFbeta signaling. Cell Tissue Res. 347, 21-36. 10.1007/s00441-011-1190-x [DOI] [PubMed] [Google Scholar]
- Heldin C.-H., Landström M. and Moustakas A. (2009). Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr. Opin. Cell Biol. 21, 166-176. 10.1016/j.ceb.2009.01.021 [DOI] [PubMed] [Google Scholar]
- Hurtado R., Zewdu R., Mtui J., Liang C., Aho R., Kurylo C., Selleri L. and Herzlinger D. (2015). Pbx1-dependent control of VMC differentiation kinetics underlies gross renal vascular patterning. Development 142, 2653-2664. 10.1242/dev.124776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang R., Bush J. O. and Lidral A. C. (2006). Development of the upper lip: morphogenetic and molecular mechanisms. Dev. Dyn. 235, 1152-1166. 10.1002/dvdy.20646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J.-Z. and Ding J. (2006). Analysis of cell migration, transdifferentiation and apoptosis during mouse secondary palate fusion. Development 133, 3341-3347. 10.1242/dev.02520 [DOI] [PubMed] [Google Scholar]
- Johansson S., Berland S., Gradek G. A., Bongers E., de Leeuw N., Pfundt R., Fannemel M., Rodningen O., Brendehaug A., Haukanes B. I. et al. (2014). Haploinsufficiency of MEIS2 is associated with orofacial clefting and learning disability. Am. J. Med. Genet. A 164A, 1622-1626. 10.1002/ajmg.a.36498 [DOI] [PubMed] [Google Scholar]
- Kaartinen V., Cui X.-M., Heisterkamp N., Groffen J. and Shuler C. F. (1997). Transforming growth factor-beta3 regulates transdifferentiation of medial edge epithelium during palatal fusion and associated degradation of the basement membrane. Dev. Dyn. 209, 255-260. [DOI] [PubMed] [Google Scholar]
- Kamps M. P., Murre C., Sun X.-H. and Baltimore D. (1990). A new homeobox gene contributes the DNA binding domain of the t(1;19) translocation protein in pre-B ALL. Cell 60, 547-555. 10.1016/0092-8674(90)90658-2 [DOI] [PubMed] [Google Scholar]
- Kim S. K., Selleri L., Lee J. S., Zhang A. Y., Gu X., Jacobs Y. and Cleary M. L. (2002). Pbx1 inactivation disrupts pancreas development and in Ipf1-deficient mice promotes diabetes mellitus. Nat. Genet. 30, 430-435. 10.1038/ng860 [DOI] [PubMed] [Google Scholar]
- Kim S., Lewis A. E., Singh V., Ma X., Adelstein R. and Bush J. O. (2015). Convergence and extrusion are required for normal fusion of the mammalian secondary palate. PLoS Biol. 13, e1002122 10.1371/journal.pbio.1002122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama T., Newberg J. Y., Kodama M., Rangel R., Yoshihara K., Tien J. C., Parsons P. H., Wu H., Finegold M. J., Copeland N. G. et al. (2016). Transposon mutagenesis identifies genes and cellular processes driving epithelial-mesenchymal transition in hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 113, E3384-E3393. 10.1073/pnas.1606876113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koss M., Bolze A., Brendolan A., Saggese M., Capellini T. D., Bojilova E., Boisson B., Prall O. W. J., Elliott D. A., Solloway M. et al. (2012). Congenital asplenia in mice and humans with mutations in a Pbx/Nkx2-5/p15 module. Dev. Cell 22, 913-926. 10.1016/j.devcel.2012.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K., Zheng T. S., Na S., Kuan C.-Y., Yang D., Karasuyama H., Rakic P. and Flavell R. A. (1996). Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature 384, 368-372. 10.1038/384368a0 [DOI] [PubMed] [Google Scholar]
- Lamouille S., Xu J. and Derynck R. (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178-196. 10.1038/nrm3758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y., Xu J. and Jiang R. (2015). Cellular and molecular mechanisms of palatogenesis. Curr. Top. Dev. Biol. 115, 59-84. 10.1016/bs.ctdb.2015.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landin Malt A., Cesario J. M., Tang Z., Brown S. and Jeong J. (2014). Identification of a face enhancer reveals direct regulation of LIM homeobox 8 (Lhx8) by wingless-int (WNT)/beta-catenin signaling. J. Biol. Chem. 289, 30289-30301. 10.1074/jbc.M114.592014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letra A., Maili L., Mulliken J. B., Buchanan E., Blanton S. H. and Hecht J. T. (2014). Further evidence suggesting a role for variation in ARHGAP29 variants in nonsyndromic cleft lip/palate. Birth Defects Res. A Clin. Mol. Teratol 100, 679-685. 10.1002/bdra.23286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long A. B., Kaiser W. J., Mocarski E. S. and Caspary T. (2013). Apaf1 apoptotic function critically limits Sonic hedgehog signaling during craniofacial development. Cell Death Differ. 20, 1510-1520. 10.1038/cdd.2013.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H. K., Prescott S. L. and Wysocka J. (2016). Ever-changing landscapes: transcriptional enhancers in development and evolution. Cell 167, 1170-1187. 10.1016/j.cell.2016.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longobardi E., Penkov D., Mateos D., De Florian G., Torres M. and Blasi F. (2014). Biochemistry of the tale transcription factors PREP, MEIS, and PBX in vertebrates. Dev. Dyn. 243, 59-75. 10.1002/dvdy.24016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losa M., Latorre V., Andrabi M., Ladam F., Sagerström C., Novoa A., Zarrineh P., Bridoux L., Hanley N. A., Mallo M. et al. (2017). A tissue-specific, Gata6-driven transcriptional program instructs remodeling of the mature arterial tree. eLife 6, e31362 10.7554/eLife.31362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louw J. J., Corveleyn A., Jia Y., Hens G., Gewillig M. and Devriendt K. (2015). MEIS2 involvement in cardiac development, cleft palate, and intellectual disability. Am. J. Med. Genet. A 167A, 1142-1146. 10.1002/ajmg.a.36989 [DOI] [PubMed] [Google Scholar]
- Luo D., Wang J., Li J. and Post M. (2011). Mouse snail is a target gene for HIF. Mol. Cancer Res. 9, 234-245. 10.1158/1541-7786.MCR-10-0214 [DOI] [PubMed] [Google Scholar]
- Macias M. J., Martin-Malpartida P. and Massagué J. (2015). Structural determinants of Smad function in TGF-beta signaling. Trends Biochem. Sci. 40, 296-308. 10.1016/j.tibs.2015.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marazita M. L. (2012). The evolution of human genetic studies of cleft lip and cleft palate. Annu. Rev. Genomics Hum. Genet. 13, 263-283. 10.1146/annurev-genom-090711-163729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Álvarez C., Tudela C., Pérez-Miguelsanz J., O'Kane S., Puerta J. and Ferguson M. W. J. (2000). Medial edge epithelial cell fate during palatal fusion. Dev. Biol. 220, 343-357. 10.1006/dbio.2000.9644 [DOI] [PubMed] [Google Scholar]
- Martínez-Álvarez C., Blanco M. J., Pérez R., Rabadán M. A., Aparicio M., Resel E., Martínez T. and Nieto M. A. (2004). Snail family members and cell survival in physiological and pathological cleft palates. Dev. Biol. 265, 207-218. 10.1016/j.ydbio.2003.09.022 [DOI] [PubMed] [Google Scholar]
- McCulley D. J., Wienhold M. D., Hines E. A., Hacker T. A., Rogers A., Pewowaruk R. J., Zewdu R., Chesler N. C., Selleri L. and Sun X. (2018). PBX transcription factors drive pulmonary vascular adaptation to birth. J. Clin. Invest., 128, 655-667. 10.1172/JCI93395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moens C. B. and Selleri L. (2006). Hox cofactors in vertebrate development. Dev. Biol. 291, 193-206. 10.1016/j.ydbio.2005.10.032 [DOI] [PubMed] [Google Scholar]
- Monica K., Galili N., Nourse J., Saltman D. and Cleary M. L. (1991). PBX2 and PBX3, new homeobox genes with extensive homology to the human proto-oncogene PBX1. Mol. Cell. Biol. 11, 6149-6157. 10.1128/MCB.11.12.6149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossey P. A., Little J., Munger R. G., Dixon M. J. and Shaw W. C. (2009). Cleft lip and palate. Lancet 374, 1773-1785. 10.1016/S0140-6736(09)60695-4 [DOI] [PubMed] [Google Scholar]
- Murray S. A., Oram K. F. and Gridley T. (2007). Multiple functions of Snail family genes during palate development in mice. Development 134, 1789-1797. 10.1242/dev.02837 [DOI] [PubMed] [Google Scholar]
- Nakazawa M., Matsunaga K., Asamura S., Kusuhara H., Isogai N. and Muragaki Y. (2008). Molecular mechanisms of cleft lip formation in CL/Fr mice. Scand. J. Plast. Reconstr. Surg. Hand Surg. 42, 225-232. 10.1080/02844310802271188 [DOI] [PubMed] [Google Scholar]
- Netherton S. J. and Bonni S. (2010). Suppression of TGFbeta-induced epithelial-mesenchymal transition like phenotype by a PIAS1 regulated sumoylation pathway in NMuMG epithelial cells. PLoS ONE 5, e13971 10.1371/journal.pone.0013971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto M. A. (2011). The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu. Rev. Cell Dev. Biol. 27, 347-376. 10.1146/annurev-cellbio-092910-154036 [DOI] [PubMed] [Google Scholar]
- Nieto M. A. (2013). Epithelial plasticity: a common theme in embryonic and cancer cells. Science 342, 1234850 10.1126/science.1234850 [DOI] [PubMed] [Google Scholar]
- Nieto M. A. (2017). Context-specific roles of EMT programmes in cancer cell dissemination. Nat. Cell Biol. 19, 416-418. 10.1038/ncb3520 [DOI] [PubMed] [Google Scholar]
- Nieto M. A., Huang R. Y.-J., Jackson R. A. and Thiery J. P. (2016). Emt: 2016. Cell 166, 21-45. 10.1016/j.cell.2016.06.028 [DOI] [PubMed] [Google Scholar]
- Nourse J., Mellentin J. D., Galili N., Wilkinson J., Stanbridge E., Smith S. D. and Cleary M. L. (1990). Chromosomal translocation t(1;19) results in synthesis of a homeobox fusion mRNA that codes for a potential chimeric transcription factor. Cell 60, 535-545. 10.1016/0092-8674(90)90657-Z [DOI] [PubMed] [Google Scholar]
- Packard A., Georgas K., Michos O., Riccio P., Cebrian C., Combes A. N., Ju A., Ferrer-Vaquer A., Hadjantonakis A.-K., Zong H. et al. (2013). Luminal mitosis drives epithelial cell dispersal within the branching ureteric bud. Dev. Cell 27, 319-330. 10.1016/j.devcel.2013.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiró S., Escrivá M., Puig I., Barberá M. J., Dave N., Herranz N., Larriba M. J., Takkunen M., Francí C., Muñoz A. et al. (2006). Snail1 transcriptional repressor binds to its own promoter and controls its expression. Nucleic Acids Res. 34, 2077-2084. 10.1093/nar/gkl141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penkov D., Mateos San Martín D., Fernandez-Díaz L. C., Rosselló C. A., Torroja C., Sánchez-Cabo F., Warnatz H. J., Sultan M., Yaspo M. L., Gabrieli A. et al. (2013). Analysis of the DNA-binding profile and function of TALE homeoproteins reveals their specialization and specific interactions with Hox genes/proteins. Cell Rep. 3, 1321-1333. 10.1016/j.celrep.2013.03.029 [DOI] [PubMed] [Google Scholar]
- Pflüger L. S., Oberzaucher E., Katina S., Holzleitner I. J. and Grammer K. (2012). Cues to fertility: perceived attractiveness and facial shape predict reproductive success. Evol. Hum. Behav. 33, 708-714. 10.1016/j.evolhumbehav.2012.05.005 [DOI] [Google Scholar]
- Ray H. J. and Niswander L. (2012). Mechanisms of tissue fusion during development. Development 139, 1701-1711. 10.1242/dev.068338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid B. S., Yang H., Melvin V. S., Taketo M. M. and Williams T. (2011). Ectodermal Wnt/beta-catenin signaling shapes the mouse face. Dev. Biol. 349, 261-269. 10.1016/j.ydbio.2010.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risolino M., Mandia N., Iavarone F., Dardaei L., Longobardi E., Fernandez S., Talotta F., Bianchi F., Pisati F., Spaggiari L. et al. (2014). Transcription factor PREP1 induces EMT and metastasis by controlling the TGF-beta-SMAD3 pathway in non-small cell lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 111, E3775-E3784. 10.1073/pnas.1407074111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagerström C. G. (2004). PbX marks the spot. Dev. Cell 6, 737-738. 10.1016/j.devcel.2004.05.015 [DOI] [PubMed] [Google Scholar]
- Santamaria P. G., Moreno-Bueno G., Portillo F. and Cano A. (2017). EMT: present and future in clinical oncology. Mol. Oncol. 11, 718-738. 10.1002/1878-0261.12091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabel C. A., Selleri L., Jacobs Y., Warnke R. and Cleary M. L. (2001). Expression of Pbx1b during mammalian organogenesis. Mech. Dev. 100, 131-135. 10.1016/S0925-4773(00)00516-5 [DOI] [PubMed] [Google Scholar]
- Sefton M., Sanchez S. and Nieto M. A. (1998). Conserved and divergent roles for members of the Snail family of transcription factors in the chick and mouse embryo. Development 125, 3111-3121. [DOI] [PubMed] [Google Scholar]
- Selleri L., Depew M. J., Jacobs Y., Chanda S. K., Tsang K. Y., Cheah K. S., Rubenstein J. L., O'Gorman S. and Cleary M. L. (2001). Requirement for Pbx1 in skeletal patterning and programming chondrocyte proliferation and differentiation. Development 128, 3543-3557. [DOI] [PubMed] [Google Scholar]
- Selleri L., DiMartino J., van Deursen J., Brendolan A., Sanyal M., Boon E., Capellini T., Smith K. S., Rhee J., Popperl H. et al. (2004). The TALE homeodomain protein Pbx2 is not essential for development and long-term survival. Mol. Cell. Biol. 24, 5324-5331. 10.1128/MCB.24.12.5324-5331.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuler C. F., Halpern D. E., Guo Y. and Sank A. C. (1992). Medial edge epithelium fate traced by cell lineage analysis during epithelial-mesenchymal transformation in vivo. Dev. Biol. 154, 318-330. 10.1016/0012-1606(92)90071-N [DOI] [PubMed] [Google Scholar]
- Slavotinek A., Risolino M., Losa M., Cho M. T., Monaghan K. G., Schneidman-Duhovny D., Parisotto S., Herkert J. C., Stegmann A. P. A., Miller K. et al. (2017). De novo, deleterious sequence variants that alter the transcriptional activity of the homeoprotein PBX1 are associated with intellectual disability and pleiotropic developmental defects. Hum. Mol. Genet. 26, 4849-4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70-71. 10.1038/5007 [DOI] [PubMed] [Google Scholar]
- Sun D., Baur S. and Hay E. D. (2000). Epithelial-mesenchymal transformation is the mechanism for fusion of the craniofacial primordia involved in morphogenesis of the chicken lip. Dev. Biol. 228, 337-349. 10.1006/dbio.2000.9946 [DOI] [PubMed] [Google Scholar]
- Tamarin A. and Boyde A. (1977). Facial and visceral arch development in the mouse embryo: a study by scanning electron microscopy. J. Anat. 124, 563-580. [PMC free article] [PubMed] [Google Scholar]
- van Dijk E. L., Auger H., Jaszczyszyn Y. and Thermes C. (2014). Ten years of next-generation sequencing technology. Trends Genet. 30, 418-426. 10.1016/j.tig.2014.07.001 [DOI] [PubMed] [Google Scholar]
- Van Otterloo E., Feng W., Jones K. L., Hynes N. E., Clouthier D. E., Niswander L. and Williams T. (2016). MEMO1 drives cranial endochondral ossification and palatogenesis. Dev. Biol. 415, 278-295. 10.1016/j.ydbio.2015.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega S., Morales A. V., Ocaña O. H., Valdés F., Fabregat I. and Nieto M. A. (2004). Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 18, 1131-1143. 10.1101/gad.294104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venable J. H. and Coggeshall R. (1965). A Simplified Lead Citrate Stain for Use in Electron Microscopy. J. Cell Biol. 25, 407-408. 10.1083/jcb.25.2.407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent T., Neve E. P. A., Johnson J. R., Kukalev A., Rojo F., Albanell J., Pietras K., Virtanen I., Philipson L., Leopold P. L. et al. (2009). A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat. Cell Biol. 11, 943-950. 10.1038/ncb1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitobello A., Ferretti E., Lampe X., Vilain N., Ducret S., Ori M., Spetz J.-F., Selleri L. and Rijli F. M. (2011). Hox and Pbx factors control retinoic acid synthesis during hindbrain segmentation. Dev. Cell 20, 469-482. 10.1016/j.devcel.2011.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voon D. C., Huang R. Y., Jackson R. A. and Thiery J. P. (2017). The EMT spectrum and therapeutic opportunities. Mol. Oncol. 11, 878-891. 10.1002/1878-0261.12082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. H., Heike C. L., Clarkson M. D., Mejino J. L., Brinkley J. F., Tse R. W., Birgfeld C. B., Fitzsimons D. A. and Cox T. C. (2014). Evaluation and integration of disparate classification systems for clefts of the lip. Front. Physiol. 5, 163 10.3389/fphys.2014.00163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmer J. B., Germine L. T. and Nakayama K. (2014). Face recognition: a model specific ability. Front. Hum. Neurosci. 8, 769 10.3389/fnhum.2014.00769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X. and Weinberg R. A. (2015). Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol. 25, 675-686. 10.1016/j.tcb.2015.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yook J. I., Li X.-Y., Ota I., Hu C., Kim H. S., Kim N. H., Cha S. Y., Ryu J. K., Choi Y. J., Kim J. et al. (2006). A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat. Cell Biol. 8, 1398-1406. 10.1038/ncb1508 [DOI] [PubMed] [Google Scholar]
- Yue F., Cheng Y., Breschi A., Vierstra J., Wu W., Ryba T., Sandstrom R., Ma Z., Davis C., Pope B. D. et al. (2014). A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355-364. 10.1038/nature13992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeytinoglu S. and Davey M. P. (2012). It's a privilege to smile: impact of cleft lip palate on families. Fam. Syst. Health 30, 265-277. 10.1037/a0028961 [DOI] [PubMed] [Google Scholar]
- Zheng X., Carstens J. L., Kim J., Scheible M., Kaye J., Sugimoto H., Wu C.-C., LeBleu V. S. and Kalluri R. (2015). Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 527, 525-530. 10.1038/nature16064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B. P., Deng J., Xia W., Xu J., Li Y. M., Gunduz M. and Hung M.-C. (2004). Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 6, 931-940. 10.1038/ncb1173 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.