

Abstract
As the most abundant internal modification in eukaryotic messenger RNAs identified, N6-methyladenosine (m6A) has been shown recently to play essential roles in various normal bioprocesses. Evidence is emerging that m6A modification and its regulatory proteins also play critical roles in various cancers including leukemia, brain tumor, breast cancer and lung cancer, etc. For instance, FTO, the first m6A demethylase identified, has been reported recently to play an oncogenic role in leukemia and glioblastoma. ALKBH5 (another m6A demethylase) has been reported to exert a tumor-promoting function in glioblastoma and breast cancer. METTL3 (a major m6A methyltransferase) likely plays distinct roles between glioblastoma and lung cancer. Here we discuss the recent progress and future prospects in study of m6A machinery in cancer.
Introduction
N6-methyladenosine (m6A) is the most abundant internal modification in eukaryotic messenger RNAs (mRNAs) that mainly occur at consensus motif of RRm6ACH ([G/A/U][G>A]m6AC[U>A>C] [1,2]. Although m6A was first discovered in 1970s [3,4], functional characteristics and regulatory mechanisms of m6A modification were largely unknown until recent years [1,2]. The identification of the fat mass and obesity-associated protein (FTO) as a bona fide demethylase of m6A modification [5] and the development of transcriptome-wide approaches for m6A sequencing [6,7] have indicated that m6A is a reversible and dynamic RNA modification that may affect thousands of mRNAs and non-coding RNAs in a given type of cells. The deposition of m6A is catalyzed by the m6A methyltransferase complex (MTC) composed of methyltransferase-like 3 and 14 (METTL3 and METTL14) (i.e., writers) and their cofactor, Wilms tumor 1-associated protein (WTAP) [8–11]. The removal of m6A is facilitated by FTO and ALKBH5, two m6A demethylase (i.e., erasers) that may target distinct sets of target mRNAs [2,5,12]. YTHDF1, YTHDF2, YTHDF3, and YTHDC1, members of the YT521-B homology (YTH) domain family of proteins, have been identified as m6A direct readers that affect the translation, stability, and/or splicing of target mRNAs [13–17] (see Figure 1). Recent studies have shown that m6A modification in mRNAs or non-coding RNAs plays essential roles in virtually all types of normal bioprocesses including tissue development, self-renewal and differentiation of stem cells, heat shock response, circadian clock control, DNA damage response, and maternal-to-zygotic transition, likely through affecting RNA fate/metabolism and functions such as mRNA stability, splicing, transport, localization, translation, primary microRNA processing, and RNA-protein interactions [6,7,10,12–14,18–26]. While still in the beginning stage, efforts have also been made to investigate the biological impacts of m6A modification in cancer. In this review, we summarize the recent advance in our understanding of the biological functions and underlying molecule mechanisms of m6A regulatory proteins (i.e., writers, erasers and readers) in various types of cancers, and also discuss future prospects.
Figure 1. The fates of m6A-modified mRNA transcripts are influenced by different m6A readers.
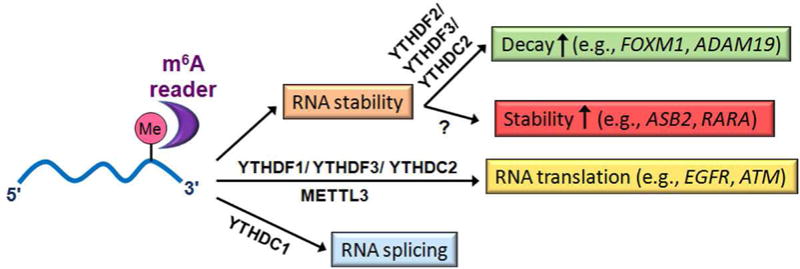
See References [13–17] for more details. The examples of m6A-modification affected target mRNAs shown herein are those that have been reported to be dysregulated in cancer (see Figure 2 and Table 1 for more information). While many m6A-modified mRNA transcripts (e.g., FOXM1 and ADAM19) can be recognized by readers such as YTHDF2, YTHDF3 and/or YTHDC2 that promote mRNA decay, other m6A-modified mRNA transcripts (e.g., ASB2 and RARA) can be recognized by some currently unknown m6A readers that promote mRNA stability. Different m6A readers can also promote translation or affect splicing of m6A-modified target mRNAs. Besides serving as an m6A methyltransferase in nucleus, METTL3 may also serve as an m6A reader in cytoplasm in some scenarios (e.g., Ref. [51]).
FTO plays an oncogenic role in leukemia as an m6A demethylase
FTO was first reported to be associated with increased body mass and obesity in humans [27–30]. In line with a link between the single nucleotide polymorphism (SNP) risk genotype and increased FTO expression in human blood cells and fibroblasts [31,32], transgenic mouse model studies have demonstrated a critical role of FTO in regulating fat mass, adipogenesis and body weight [33,34,35•], though IRX3 has also been suggested to be associated with obesity-associated variants within FTO [36]. The identification of FTO as the first m6A demethylase suggests that FTO involves in m6A-based post-transcriptional regulation of RNA targets. Through analysis of genome-wide gene expression profiles of several large-cohorts of human primary acute myeloid leukemia (AML) patients, Li Z. et al. found that FTO is highly expressed in certain subtypes of AMLs including AMLs carrying t(11q23)/MLL-rearrangements, t(15;17)/PML-RARA, NPM1 mutation (i.e., cytoplasmic localization of NPM1 (NPM1c+)), and/or Fms-like tyrosine kinase 3 with internal tandem duplication (FLT3-ITD) [37••]. More importantly, they provided compelling evidence, based on both in vitro leukemia cell line models and in vivo mouse leukemia models, showing that FTO plays an essential oncogenic role in promoting leukemic cell transformation and AML cell survival/growth and enhancing leukemogenesis, as well as in inhibiting all-trans-retionic acid (ATRA)-induced differentiation of AML cells [37••] (see Figure 2, upper left; Table 1).
Figure 2. The roles of m6A regulatory proteins in AML, breast cancer, GBM and lung cancer.
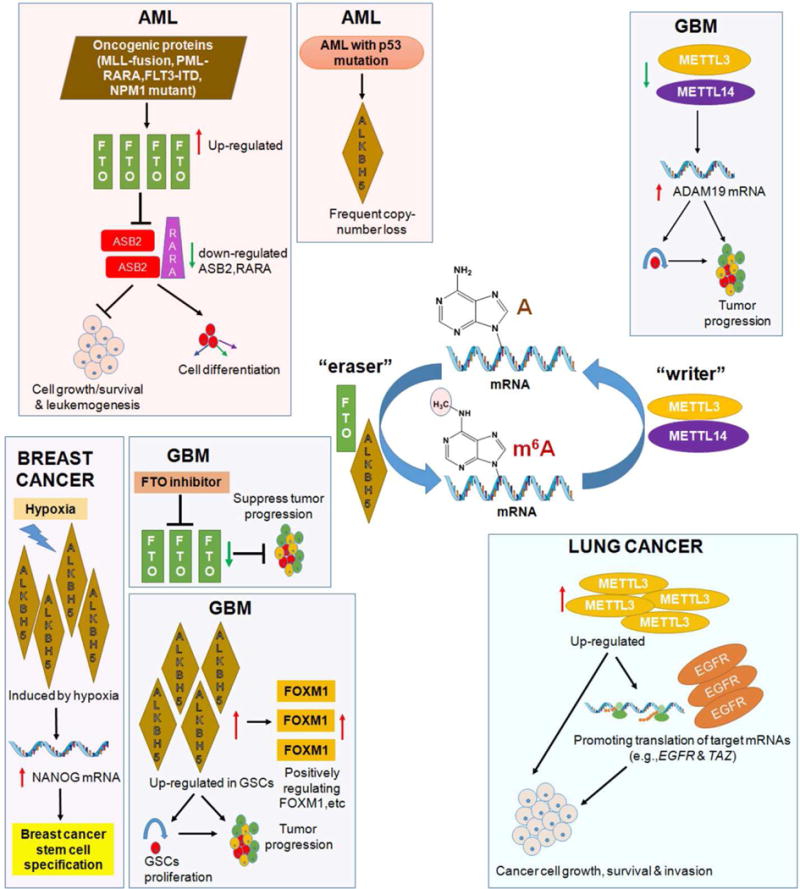
m6A, N6 methyladeosine; AML, acute myeloid leukemia; GBM: glioblastoma; GSCs: glioblastoma(-like) cells.
Table 1.
m6A regulators in cancer
| Protein | Functional classification | m6A-associated function (and underlying mechanism) in cancer | Refs |
|---|---|---|---|
| METTL3 | m6A writer component: catalytic subunit of m6A MTC |
|
[51••] [47••] |
| METTL14 | m6A writer component: core subunit of m6A MTC |
|
[47••] |
| WTAP | m6A writer component: regulatory subunit of m6A MTC |
|
[43] |
| FTO | m6A eraser (demethylase) |
|
[37••] [47••] |
| ALKBH5 | m6A eraser (demethylase) |
|
[46••] [48•] [44•] |
m6A, N6 methyladeosine; m6A MTC: m6A methyltransferase complex; GBM: glioblastoma; GSCs: glioblastoma(−like) cells; AML, acute myeloid leukemia.
Mechanistically, FTO functions as an m6A demethylase that post-transcriptionally regulates expression of its critical target RNAs (such as ASB2 and RARA) in an m6A-dependent manner. ASB2 and RARA have been implicated in leukemia cell growth and drug response, especially in ATRA-induced AML cell differentiation [38–40]. FTO negatively regulates expression of ASB2 and RARA through reducing the m6A abundance of the target RNA transcripts (especially in the 3′ untranslated regions (3′-UTRs)) and thereby decreasing the stability of the RNA transcripts [37••] (see Figure 2, upper left). Notably, in this study, the luciferase-reporter and mutagenesis assays have been introduced into the field of m6A-related research, for the first time, to demonstrate that the putative m6A consensus motif sites on the target RNA transcripts are important for the m6A-based epigenetic regulation of the target mRNA transcripts [37••]. A recent study suggests that FTO also exhibits demethylation activity towards RNA with N6,2′-O-dimethyladenosine (m6Am) in the 5′ cap, as a sub-form of m6A, leading to enhanced mRNA stability [41]. Nonetheless, roughly ¼ of FTO target mRNAs may contain an A as the first encoded nucleotide adjacent to the 7-methylguanosine (m7G) cap and there is a pretty low chance that this A is methylated at both N6 and 2′-O sites; in contrast, on average there are 3–5 internal m6A sites per mRNA transcript [42]. Thus, the overall abundance of internal m6A modifications should be much higher than that of the 5′ cap m6Am modification in cells. Indeed, analysis of the m6A-seq data reported in [37••] showed that over 95% of the m6A peaks with increased abundance upon FTO knockdown in AML cells are located in the internal regions (> 150 nucleotides away from the 5′ ends) and thus are impossible 5′ cap m6Am. In addition, liquid chromatography-tandem mass spectrometry (LC-MS/MS)-based quantification of m6A and m6Am in human AML cells showed that the internal m6A abundance is approximately 20–30 times of the near 5′ cap m6Am abundance, and the internal m6A peaks are the main substrates of FTO and represent the major changes in the N6-methyladenosine abundance when FTO is forced expressed or knocked down in AML cells (Su R, et al. unpublished). Moreover, as demonstrated by the luciferase reporter and mutagenesis assays, the internal m6A sites on ASB2 and RARA transcripts are required for FTO-mediated post-transcriptional regulation of their stability and expression [37••]. Therefore, internal m6A sites, rather than the rare m6Am in the 5′ cap, are responsible for FTO-mediated post-transcription regulation of target mRNAs in cancer cells.
Dysregulation of other m6A regulatory proteins in leukemia
WTAP, which was first identified as a partner of Wilms’ tumor gene 1 (WT1), has been reported recently to play an oncogenic role in AML [43]. WTAP was found to be aberrantly overexpressed in 32% of AML patients, especially in patients carrying NPM1 mutation and/or FTL3-ITD; depletion of expression of WTAP significantly inhibited AML cell growth and promoted AML cell differentiation [43] (see Table 1). Further studies are warranted to determine whether WTAP’s such oncogenic function in AML is related to its role as a cofactor of METTL3 and METTL14 that form m6A MTC. Interestingly, the phenomenon that both WTAP and FTO are highly expressed in AML carrying NPM1 mutation and/or FLT3-ITD [37••,43] suggests that m6A modification in this subtype of AML is tightly and sophistically controlled by both writer and eraser regulators.
Gene mutations and copy number variations (CNVs) of m6A regulatory genes, including METTL3, METTL14, FTO, ALKBH5, YTHDF1 and YTHDF2 have been investigated in leukemia patients through analysis of The Cancer Genome Atlas (TCGA) sequence data [44•]. Around 2.6% (5/191) and 9.7% (18/186) of AML patients were found to carry mutations and CNVs, respectively, in one or more of these genes; the mutations and CNVs are especially enriched in AML patients carrying p53 mutations, but rarely in patients carrying mutations in NPM1, FLT3, IDH1 and IDH2 [44•]. Patients with genetic alterations in these genes are often associated with poor prognosis, largely due to their strong association with p53 mutations [44•]. Notably, among the CNV events of m6A regulatory genes, copy number loss of ALKBH5 is the most frequent one (12/191; 6.3%); depletion of ALKBH5 was significantly associated with poorer cytogenetic risk and the presence of p53 mutations in AML [44•] (see Figure 2, upper left; Table 1). Interestingly, as the second m6A demethylase identified, ALKBH5 was reported to affect mRNA export and RNA metabolism, and Alkbh5 deficiency leads to aberrant spermatogenesis and apoptosis in mouse testes, likely through regulating genes associated with the p53 network [12]. Consistently, according to The cBioPortal for Cancer Genomics database [45], unlike METTL3, METTL14, WTAP, FTO, YTHDF2 and YTHDC1 that are expressed at a relatively higher level in AML than in many other types of cancers, ALKBH5 (as well as YTHDF1) is expressed at a low level in AML. Thus, opposite to the oncogenic role of FTO, ALKBH5 may exert a tumor-suppressor function in AML, which warrants further experimental validation.
m6A RNA modification in solid tumors
In brain tumors, Zhang et al. found that ALKBH5 expression is elevated in glioblastoma (GBM) stem(-like) cells (GSCs) and elevated expression of ALKBH5 predicts poor prognosis in GBM patients [46••]. ALKBH5 enhances GSC self-renewal proliferation and promotes tumorigensis through demethylating FOXM1 nascent transcripts, with the aid of the lncRNA antisense (FOXM1-AS) that promotes the interaction of FOXM1 nascent transcripts with ALKBH5, and thereby enhancing FOXM1 expression [46••] (see Figure 2, lower left; Table 1). Similarly, Cui et al. [47••] reported that m6A levels in GSCs were elevated upon induced differentiation. Accordingly, knockdown of METTL3 or METTL14 significantly enhanced GSC growth and self-renewal and promoted tumor progression, and the opposite is true when METTL3 was overexpressed or FTO function was pharmaceutically inhibited [47••] (see Figure 2, upper right and lower left; Table 1). A number of GSC-associated genes (e.g., ADAM19) are likely the targets of m6A modifications in GSCs [47••] (see Figure 2, upper right).
In breast cancer cells, ALKBH5 expression was found to be up-regulated by hypoxia-stimulated HIF1α and HIF2α, and thereby promoted mRNA stability and expression of NANOG, a gene encoding a pluripotency factor, which in turn enhanced breast cancer stem cell (BCSC) enrichment; knockdown of ALKBH5 inhibited tumor formation and decreased BCSC population in breast tumors [48•] (see Figure 2, lower left; Table 1). ZNF217 may also participate in the hypoxia-induced up-regulation of expression of NANOG and KLF4 (another pluripotency factor gene) in breast cancer cells [49], likely through sequestering METTL3 and thereby inhibiting m6A methylation on NANOG and KLF4 transcripts [50].
METTL3 was observed to be up-regulated in lung adenocarcinoma and played an oncogenic role that promotes growth, survival and invasion of human lung cancer cells [51••]. Notably, METTL3 was shown to promote translation of its target mRNA transcripts (e.g., EGFR and TAZ) by interaction with translation initiation machinery, in a manner independent of its methytransferase activity [51••] (see Figure 2, lower right; Table 1). However, the biological function of METTL3 in lung cancer cells was determined solely by loss-of-function studies through knockdown of METTL3 expression, and it is unclear whether METTL3 mutants that lose its methyltransferase activity can exert the same degree of oncogenic effects as wild-type METTL3 in promoting growth, survival and invasion of human lung cancer cells. Thus, systematic gain-of-function studies with both wild-type and mutant METTL3 constructs, under conditions of with or without depletion of endogenous MELLT3 expression, are necessary to determine the functional importance of METTL3 as an m6A reader in cytoplasm in the pathogenesis of lung cancer and other types of cancers.
Conclusions & Perspectives
While we are just beginning to understand the function of the m6A modification machinery in cancer, recent studies have shown that m6A regulatory proteins likely play essential roles in various types of cancers, including leukemia, brain tumor, breast cancer and lung cancer (see Figure 2 and Table1). Interestingly, some proteins likely play a similar role across different types of cancers, whereas some others may function differently in distinct types of cancers. For instance, FTO has been shown to function as an oncoprotein in both leukemia and GBM [37••,47••]. In contrast, while ALKBH5 functions as an oncoprotein in GBM and breast cancer [46••,48•], it may exert a tumor-suppressor role in AML as implied by its frequent copy number loss [44•] and low expression level in AML. Similarly, METTL3 likely plays an oncogenic role in lung cancer [51••] but a tumor-suppressor role in GBM [47••]. Notably, both FTO and WTAP are highly expressed and play an oncogenic role in certain subtypes of AMLs. In addition, METTL3 and METTL14 are also highly expressed in AML and thus they may also play an oncogenic role in AML. These phenomena suggest that an m6A eraser (e.g., FTO) does not necessarily function oppositely than a component of the m6A methyltransferase (writer) complex (e.g., WTAP, METTL3 or METTL14) in the same cancer type. Similarly to this, it was known that both DNMT3A (a DNA methyltransferase) and TET2 (a DNA demethylase) are frequently associated with loss-of-function mutations and both function as tumor-suppressors in myeloid malignancies [52,53], and they may cooperate in repressing lineage differentiation of hematopietic stem cells [54]. Thus, it is possible that a writer and an eraser of the same epigenetic modification could play similar functions in the same cell context, likely through regulating distinct sets of targets. Future systematic studies are warranted to determine the biological function of each individual m6A regulatory genes in different types of cancers, and to identify their cirtical target genes to reveal the underlying molecule mechansims.
Interestingly, besides its role as the major m6A methyltransferase in nucleus, METTL3 may also exert some function independent of its catalytic activity, such as promoting translation of target transcripts as an m6A reader in cytoplasm in lung cancer cells [51••]. Nevertheless, since METTL3’s translation-promoting function also relies on the m6A modification on its target mRNAs, METTL3’s catalytic activity is still required for its function in promoting translation of the target transcripts. It would be important to determine how critical its role as an m6A reader is in its overall pathological function in each type of cancer. It is possible that in some types of cancers, its role as an m6A reader is dispensible, whereas its m6A methyltransferase role is still required.
Given the critical roles of the m6A regulatory proteins in cancers, they (especially the methylatransferase and demethylase proteins with catalytic activies) appear to be good drug targets for cancer therapy. Several FTO small-molecule inhibitors have been identified [55–58]; among them, meclofenamic acid (MA) [56] and MO-I-500 [57] have been shown to be able to effectively inhibit the survival and growth of GBM and breats cancer cells by inhibition of the catalytic activity of FTO [47••,58]. Development of clinically applicable selective and effective inhibitors for FTO and other m6A regulatory proteins may provide more effetcive novel therapeutic strategies to treat cancers, especially in combination with other therapeutic agents to treat cancers that are resistant to currently available therapies.
Acknowledgments
The authors apologize to colleagues whose work could not be included due to space limitations. This work was supported in part by grants NO.81603149 (X.D.) from National Nature Science Foundation of China, as well as the National Institutes of Health (NIH) R01 Grants CA214965 (J.C.), CA211614 (J.C.), and CA178454 (J.C.). J.C. is a Leukemia & Lymphoma Society (LLS) Scholar.
Abbreviations
- m6A
N6-methyladenosine
- MTC
methyltransferase complex
- FTO
the fat mass and obesity-associated protein
- SNP
single nucleotide polymorphism
- LC-MS/MS
liquid chromatography coupled with tandem mass spectrometry
- METTL3 and METTL14
methyltransferase-like 3 and 14
- WTAP
Wilms tumor 1-associated protein
- AML
acute myeloid leukemia
- FLT3-ITD
Fms-like tyrosine kinase 3 with internal tandem duplication
- NPM1c+
NPM1 mutant that causes aberrant cytoplasmic localization of nucleophosmin
- ATRA
all-trans-retionic acid
- CNV
copy number variation
- GBM
glioblastoma
- GSC
glioblastoma stem(-like) cell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
Nothing declared.
References and recommended reading
Papers of particular interest, published within the period of review (last two years), have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA Modifications in Gene Expression Regulation. Cell. 2017;169:1187–1200. doi: 10.1016/j.cell.2017.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nature reviews Molecular cell biology. 2017;18:31–42. doi: 10.1038/nrm.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perry RP, Kelley DE. Existence of methylated messenger RNA in mouse L cells. Cell. 1974;1:37–42. [Google Scholar]
- 4.Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci USA. 1974;71:3971–3975. doi: 10.1073/pnas.71.10.3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 7.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. Rna. 1997;3:1233–1247. [PMC free article] [PubMed] [Google Scholar]
- 9.Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014;16:191–198. doi: 10.1038/ncb2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–189. doi: 10.1038/cr.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vagbo CB, Shi Y, Wang WL, Song SH, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, He C. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 2017 doi: 10.1038/cr.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, Sun HY, Li A, Ping XL, Lai WY, et al. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell. 2016;61:507–519. doi: 10.1016/j.molcel.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 17.Li A, Chen YS, Ping XL, Yang X, Xiao W, Yang Y, Sun HY, Zhu Q, Baidya P, Wang X, et al. Cytoplasmic m6A reader YTHDF3 promotes mRNA translation. Cell Res. 2017;27:444–447. doi: 10.1038/cr.2017.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao X, Yang Y, Sun BF, Shi Y, Yang X, Xiao W, Hao YJ, Ping XL, Chen YS, Wang WJ, et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014;24:1403–1419. doi: 10.1038/cr.2014.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen T, Hao YJ, Zhang Y, Li MM, Wang M, Han W, Wu Y, Lv Y, Hao J, Wang L, et al. m(6)A RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell. 2015;16:289–301. doi: 10.1016/j.stem.2015.01.016. [DOI] [PubMed] [Google Scholar]
- 20.Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–564. doi: 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alarcon CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015;519:482–485. doi: 10.1038/nature14281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science. 2015;347:1002–1006. doi: 10.1126/science.1261417. [DOI] [PubMed] [Google Scholar]
- 23.Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian SB. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature. 2015;526:591–594. doi: 10.1038/nature15377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, Jaffrey SR. 5′ UTR m(6)A Promotes Cap-Independent Translation. Cell. 2015;163:999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiang Y, Laurent B, Hsu CH, Nachtergaele S, Lu Z, Sheng W, Xu C, Chen H, Ouyang J, Wang S, et al. RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature. 2017;543:573–576. doi: 10.1038/nature21671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao BS, Wang X, Beadell AV, Lu Z, Shi H, Kuuspalu A, Ho RK, He C. m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature. 2017;542:475–478. doi: 10.1038/nature21355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scuteri A, Sanna S, Chen WM, Uda M, Albai G, Strait J, Najjar S, Nagaraja R, Orru M, Usala G, et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 2007;3:e115. doi: 10.1371/journal.pgen.0030115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JR, Elliott KS, Lango H, Rayner NW, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dina C, Meyre D, Gallina S, Durand E, Korner A, Jacobson P, Carlsson LM, Kiess W, Vatin V, Lecoeur C, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. 2007;39:724–726. doi: 10.1038/ng2048. [DOI] [PubMed] [Google Scholar]
- 30.Yang J, Loos RJ, Powell JE, Medland SE, Speliotes EK, Chasman DI, Rose LM, Thorleifsson G, Steinthorsdottir V, Magi R, et al. FTO genotype is associated with phenotypic variability of body mass index. Nature. 2012;490:267–272. doi: 10.1038/nature11401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berulava T, Horsthemke B. The obesity-associated SNPs in intron 1 of the FTO gene affect primary transcript levels. Eur J Hum Genet. 2010;18:1054–1056. doi: 10.1038/ejhg.2010.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karra E, O’Daly OG, Choudhury AI, Yousseif A, Millership S, Neary MT, Scott WR, Chandarana K, Manning S, Hess ME, et al. A link between FTO, ghrelin, and impaired brain food-cue responsivity. J Clin Invest. 2013;123:3539–3551. doi: 10.1172/JCI44403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fischer J, Koch L, Emmerling C, Vierkotten J, Peters T, Bruning JC, Ruther U. Inactivation of the Fto gene protects from obesity. Nature. 2009;458:894–898. doi: 10.1038/nature07848. [DOI] [PubMed] [Google Scholar]
- 34.Church C, Moir L, McMurray F, Girard C, Banks GT, Teboul L, Wells S, Bruning JC, Nolan PM, Ashcroft FM, et al. Overexpression of Fto leads to increased food intake and results in obesity. Nat Genet. 2010;42:1086–1092. doi: 10.1038/ng.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •35.Merkestein M, Laber S, McMurray F, Andrew D, Sachse G, Sanderson J, Li M, Usher S, Sellayah D, Ashcroft FM, et al. FTO influences adipogenesis by regulating mitotic clonal expansion. Nature communications. 2015;6:6792. doi: 10.1038/ncomms7792. This study together with Ref. [18] showed that FTO protein influences adipogenesis directly by regulating RUNX1T1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smemo S, Tena JJ, Kim KH, Gamazon ER, Sakabe NJ, Gomez-Marin C, Aneas I, Credidio FL, Sobreira DR, Wasserman NF, et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507:371–375. doi: 10.1038/nature13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••37.Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, Huang H, Nachtergaele S, Dong L, Hu C, et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N6-Methyladenosine RNA Demethylase. Cancer Cell. 2017;31:127–141. doi: 10.1016/j.ccell.2016.11.017. This study demonstrated the functional importance of m6A methylation in cancer, by providing compelling in vitro and in vivo evidence showing the oncogenic impact of FTO (the first m6A demethylase (i.e., eraser) identified) and its m6A-based post-transcriptional regulation of the targets (e.g., ASB2 and RARA) on leukemogenesis and drug response in leukemia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kohroki J, Fujita S, Itoh N, Yamada Y, Imai H, Yumoto N, Nakanishi T, Tanaka K. ATRA-regulated Asb-2 gene induced in differentiation of HL-60 leukemia cells. FEBS Lett. 2001;505:223–228. doi: 10.1016/s0014-5793(01)02829-0. [DOI] [PubMed] [Google Scholar]
- 39.Guibal FC, Moog-Lutz C, Smolewski P, Di Gioia Y, Darzynkiewicz Z, Lutz PG, Cayre YE. ASB-2 inhibits growth and promotes commitment in myeloid leukemia cells. J Biol Chem. 2002;277:218–224. doi: 10.1074/jbc.M108476200. [DOI] [PubMed] [Google Scholar]
- 40.Glasow A, Prodromou N, Xu K, von Lindern M, Zelent A. Retinoids and myelomonocytic growth factors cooperatively activate RARA and induce human myeloid leukemia cell differentiation via MAP kinase pathways. Blood. 2005;105:341–349. doi: 10.1182/blood-2004-03-1074. [DOI] [PubMed] [Google Scholar]
- 41.Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, Linder B, Pickering BF, Vasseur JJ, Chen Q, et al. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature. 2017;541:371–375. doi: 10.1038/nature21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat Rev Genet. 2014;15:293–306. doi: 10.1038/nrg3724. [DOI] [PubMed] [Google Scholar]
- 43.Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia DA, Penalva LO, Uren PJ, Suresh U, Carew JS, Karnad AB, et al. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia. 2014;28:1171–1174. doi: 10.1038/leu.2014.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •44.Kwok CT, Marshall AD, Rasko JE, Wong JJ. Genetic alterations of m6A regulators predict poorer survival in acute myeloid leukemia. Journal of hematology & oncology. 2017;10:39. doi: 10.1186/s13045-017-0410-6. This study showed that over 10% of m6A regulatory genes are associated with mutations and/or copy number variations in AML, with the copy number loss of ALKBH5 being the most frequent one that is often associated with p53 mutations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••46.Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, Chen Y, Sulman EP, Xie K, Bogler O, et al. m6A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell. 2017;31:591–606 e596. doi: 10.1016/j.ccell.2017.02.013. This study showed that ALKBH5 is up-regulated in glioblastoma stem cells (GSCs) and its expression and function is required for GSC proliferation and tumor progression through positively regulating expression of FOXM1, etc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••47.Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, Lu Z, Huang Y, Yang CG, Riggs AD, et al. m6A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell reports. 2017;18:2622–2634. doi: 10.1016/j.celrep.2017.02.059. This study, together with Ref. [46••], showed the functional importance of m6A mRNA modification in GSC self-renewal and glioblastoma tumor progression, as evidenced by functional studies through manipulating expression of METTL3 or METTL14, or pharmacologically suppressing activity of FTO in GSCs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •48.Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, He X, Semenza GL. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113:E2047–2056. doi: 10.1073/pnas.1602883113. This study showed that ALKBH5 expression is up-regulated by hypoxia in breast cancer cells and its function is required for hypoxic tumor microenvironment through positively regulating expression of NANOG, etc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang C, Zhi WI, Lu H, Samanta D, Chen I, Gabrielson E, Semenza GL. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget. 2016;7:64527–64542. doi: 10.18632/oncotarget.11743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aguilo F, Zhang F, Sancho A, Fidalgo M, Di Cecilia S, Vashisht A, Lee DF, Chen CH, Rengasamy M, Andino B, et al. Coordination of m(6)A mRNA Methylation and Gene Transcription by ZFP217 Regulates Pluripotency and Reprogramming. Cell Stem Cell. 2015;17:689–704. doi: 10.1016/j.stem.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••51.Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol Cell. 2016;62:335–345. doi: 10.1016/j.molcel.2016.03.021. This study showed that METTL3 is overexpressed in lung cancer, in which it plays an oncogenic role presumably through promoting translation of its targets (e.g., EGFR and TAZ) in a manner independent of its catalytic activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, Kosmider O, Le Couedic JP, Robert F, Alberdi A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360:2289–2301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 54.Zhang X, Su J, Jeong M, Ko M, Huang Y, Park HJ, Guzman A, Lei Y, Huang YH, Rao A, et al. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat Genet. 2016;48:1014–1023. doi: 10.1038/ng.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen B, Ye F, Yu L, Jia G, Huang X, Zhang X, Peng S, Chen K, Wang M, Gong S, et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. Journal of the American Chemical Society. 2012;134:17963–17971. doi: 10.1021/ja3064149. [DOI] [PubMed] [Google Scholar]
- 56.Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H, Gan J, Jiang H, Jia GF, Luo C, et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Research. 2015;43:373–384. doi: 10.1093/nar/gku1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zheng G, Cox T, Tribbey L, Wang GZ, Iacoban P, Booher ME, Gabriel GJ, Zhou L, Bae N, Rowles J, et al. Synthesis of a FTO inhibitor with anticonvulsant activity. ACS chemical neuroscience. 2014;5:658–665. doi: 10.1021/cn500042t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Singh B, Kinne HE, Milligan RD, Washburn LJ, Olsen M, Lucci A. Important Role of FTO in the Survival of Rare Panresistant Triple-Negative Inflammatory Breast Cancer Cells Facing a Severe Metabolic Challenge. PLoS ONE. 2016;11:e0159072. doi: 10.1371/journal.pone.0159072. [DOI] [PMC free article] [PubMed] [Google Scholar]