

Abstract
Increasing evidence has pointed to that dysregulation of the endo-lysosomal system is an early cellular phenotype of pathogenesis for Alzheimer’s disease (AD). Rab5, a small GTPase, plays a critical role in mediating these processes. Abnormal overactivation of Rab5 has been observed in post-mortem brain samples of Alzheimer’s patients as well as brain samples of mouse models of AD. Recent genome-wide association studies of Alzheimer’s disease have identified RIN3 (Ras and Rab Interactor 3) as a novel risk factor for the disease. RIN3 that functions as a guanine nucleotide exchange factor for Rab5 may serve as an important activator for Rab5 in AD pathogenesis. In the review, we present recent research highlights on the possible roles of dysregulation of Rab5-mediated endocytic pathways in contributing to early pathogenesis of Alzheimer’s disease.
Keywords: Alzheimer’s disease, Rab5, endocytosis, axonal transport, axonal dysfunction, atrophy, neurodegeneration
Graphical abstract
Introduction
Genetic complexity of Alzheimer’s disease
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder that often results in memory loss and cognitive impairment1–9. The classical neuropathological hallmarks for AD include: 1) Aβ-amyloid-containing neuritic plaques and 2) phosphorylated Tau-containing neurofibrillary tangles (NFT)2,3,10,11. The clinical phase of AD is also marked significantly by synaptic loss, selective neuronal death, neurotransmitter loss, and neuroinflammation1,3,4,6,8,10,11. At present, there are no disease-modifying treatments for this fatal illness. As of today, mutations in three genes: amyloid precursor protein (APP), presenilin (PS)1 and PS2, have all been linked to early onset of AD (EOAD)12–15, which only account for a very small number of AD cases. Extensive investigation of these mutations have helped to formulate the Aβ-amyloid hypothesis1,16, positing that small Aβ-amyloid peptides (Aβ40/42) underlie the pathogenesis of AD. This has led to many clinical trials that use approaches (inhibitors, antibodies etc.) to target and to reduce the buildup of toxic Aβ-amyloid peptides. Unfortunately, the anti-amyloid-based treatments have all failed in clinical trials thus far17,18. Similarly, intensive efforts have also been made to develop inhibitors to prevent aggregation of Tau-containing neurofibrillary tangles. However, these efforts have also failed in recent clinical trials19. Nevertheless, studies of these hereditary mutations in APP, PS1/2 have helped to gain a greater understanding of the molecular pathogenesis of AD.
One of the most important lessons we have learned from these failed clinical efforts is that the cause(s) for AD is far more complex than we originally anticipated11. For example, the incidence of late-onset AD (LOAD) accounts for the majority cases of AD and the cause(s) for LOAD is far from clear in comparison to that for EOAD. In 2009, apolipoprotein E (APOE) that plays an important role in lipid transport, Aβ trafficking, synaptic function, immune regulation, and intracellular signaling20, was identified as an established risk factor for LOAD21. More recently, large scale genome-wide association studies (GWAS) and a meta-analysis have identified significant associations between LOAD with SNPs in ~20 additional loci including CLU, CR1, PICALM, BIN1, ABCA7, MS4A4, EPHA1, CD2AP, CD33, INPP5D, MEF2C, HLA-DRB1/HLA-DRB5, NME8, ZCWPW1, PTK2B, SORL1, CELF1, SLC24A4/RIN3, FERMT2 and CASS422–27. Identification of these additional risk factors highlights the extraordinary genetic complexity of AD.
Dysfunction for the endocytic pathways contributes to early AD pathogenesis
A significant number of the newly identified AD risk loci (PICALM, BIN1, EPHA1, CD2AP, MEF2C, PTK2B, SORL1 and RIN3) that encode products that function predominantly in endocytic trafficking10. For instance, PICALM (phosphatidylinositol binding clathrin assembly protein), whose expression is reduced in AD28–30, plays a central role in internalization, trafficking and clearance of Aβ via the low density lipoprotein receptor related protein-1 and Rab proteins (Rab5, Rab11)29,31,32; Expression of the longest isoform of bridging integrator 1 (BIN1), an adaptor protein that functions in clathrin-mediated endocytosis and endocytic recycling, was significantly increased in the AD brains compared to age-matched controls33,34. Furthermore, BIN1 is significantly correlated with the amount of pTau-containing NFT pathology34 and has been implicated in modulating Tau-associated neuronal toxicity34–36. The sortilin-related receptor 1 (SORL1) regulates intracellular trafficking and processing of APP37–40. In the absence or reduced expression of SORL1, APP is released into late endosomal pathway, where it is subjected to both β- and γ-secretase cleavage, thus driving Aβ production41,42. Mutations in SORL1 have been associated with both EOAD and LOAD43,44. In addition, APOE and CLU (Clusterin) both influence Aβ-aggregation and receptor-mediated Aβ clearance by endocytosis20,45,46. Although each of these genes confers only an incremental risk to AD, together they may play a significant role in altering endocytic processes, such as internalization, endocytic trafficking and signaling, that ultimately contributes to molecular pathogenesis of LOAD47.
Endocytic trafficking and amyloidogenic processing of APP
Neurons with the most extraordinary architecture have elaborate dendrites and axons, so it is not surprising that strong evidence has suggested that these early endocytic processes are dysregulated in AD48–51 and in AD cases of people with Down syndrome52. For instance, amyloidogenic cleavage of APP to yield Aβ likely occurs predominantly in the intracellular compartments53–58. Under normal conditions, the early endosome, marked by Rab5, is a major site of APP processing by β-secretase (BACE1) to yield the β-cleavage C-terminal fragment (β-CTFs)59–62, which is further processed in late endosomes or trans-Golgi network (TGN) to give rise to Aβ. Therefore, APP metabolism that produces toxic β-CTFs and Aβ is intimately regulated by the endocytic pathways48–51,63–68. Dysregulation of Rab5 can thus profoundly impact APP processing and Aβ production. In AD, abnormal enlargement of Rab5-positive early endosomes is not only characteristic, it is early; it was observed in non-demented individuals who were diagnosed postmortem as fulfilling the pathological diagnosis of sporadic AD48. Interestingly, endosomal pathology was detected in brain regions that were free of Aβ or pTau pathology48,69, suggesting that early endosomal abnormalities precede not only the onset of dementia but also the emergence of plaques and tangles.
Axonal toxicity of APP β-CTFs and pTau
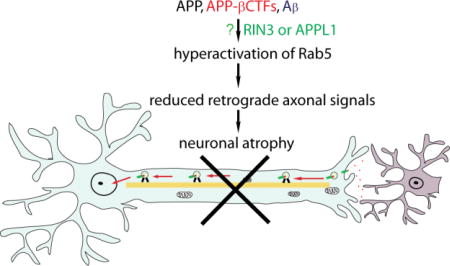
Although neuronal toxicities by β-CTF or C99 have been observed for quite some time70–73, little is known about the underlying cellular mechanism(s). Recent evidence has pointed to a strong adverse impact of β-CTF on synaptic plasticity and neuronal function. The effect appears to be independent of Aβ, but is intimately linked to early cellular pathology in AD55,60,73–80. Intra-neuronal accumulation of excessive β-CTFs likely contributed to worsening cognitive function in AD patients in failed clinical trials of γ-secretase inhibitors17,18,74,78,79. We and others have recently demonstrated that excessive β-CTFs increased the level of activated Rab5, i.e. GTP-Rab5, impaired retrograde axonal delivery of endosomes or trophic factors that resulted in neuronal atrophy79–81.
Moreover, a recent study has demonstrated that pathogenic tau species, phorsphorylated tau (pTau) is also impacted by endocytic trafficking and by APP processing82. Using human-stem-cell-derived forebrain neurons, Moore and colleagues have shown that APP mutations and increased gene dosage of APP caused marked increase in both total Tau and phosphorylated Tau in these AD neurons82. They further demonstrated that γ-secretase inhibition that resulted in accumulation of APP- β-CTF/α-CTF led to an increase in tau, while treatment with inhibitors of β-secretase decreased Tau82. Therefore, intracellular trafficking and processing of APP impacts tau proteostasis as well. Since disruption of tau homeostasis has been implicated in disrupting axonal function19,35,36,83–87, the link between β-CTF and Rab5 dysregulation, and between endosomal dysfunction and pTau thus warrants further investigation. RIN3, the Ras and Rab interactor 3, a stimulator and stabilizer for GTP-Rab588,89 identified in recent GWAS studies22,24,25, may fill an important gap linking β-CTF to Rab5 dysregulation in AD.
Dysregulation of Rab5 in Alzheimer’s disease
Rab5 is a small GTPase that cycles between an inactivate GDP-bound form and an active GTP-bound form for its biological functions90–94. Activation of Rab5 requires guanine-nucleotide exchange factors (GEFs) such as RIN390,94. Inactivation of Rab5 is under the control of GTP-hydrolysis-activating proteins (GAPs)95 due to a low intrinsic rate of GTP hydrolysis by Rab5 itself. It is under the dual control by both GEFs and GAPs, and Rab5 regulates early steps of endocytosis, ensuing endosomal membrane trafficking, sorting and endosomal fusion90–92,94,96.
Through interaction with numerous effectors such as: APPL1/2 [adaptor protein containing pleckstrin homology (PH) domain, phosphotyrosine binding (PTB) domain and leucine zipper motif 1]97, Rabaptin-5/Rabex-5, early endosome antigen 1 (EEA1), phosphatidylinositol 3-kinases98–102, Rab5 contributes importantly to cellular homeostasis. These effector proteins specifically bind to the activated form of Rab5, i.e. the GTP-bound form (GTP-Rab5) and regulates tethering and fusion of the early endosomes99,103–106. Therefore, Rab5 plays a critical role in docking of endosomal membranes, and motility of endosomes and intracellular signal transduction91,92,107.
Sustained activation of Rab5 will result its interaction with the class C VPS/HOPS complex, an established GEF for Rab7108 residing on late endosomes, thus leading to conversion from Rab5+ early endosomes to Rab7+-late endosomes for degradation. Therefore, the timing and duration of Rab5 in GTP-bound form is tightly regulated to ensure a smooth transition from early to late endosomes. This is especially important for axonal neurotrophic signals. Extensive studies have demonstrated that neurotrophic factors such as nerve growth factor (NGF) binds to their Trk receptor to be internalized into Rab5+ signaling endosomes that carry important downstream signaling machineries such as Erk, PI3K, PLCγ for axonal transport to deliver the trophic signals to the soma and nucleus109–114. An intriguing question is how the signaling endosome is retained as Rab5+ early endosomes, but not progress to Rab7+ late endosomes, during their transit within the long axons. Work from the laboratory of Dr. G. Li has shed lights into the potential mechanisms115. As it turns out, following activation by NGF, TrkA recruits a Rab5-GAP to quickly convert GTP-Rab5 to GDP-Rab5 to keep the level of GTP-Rab5 in check, thus preventing the Rab5 to Rab7 conversion. As a result, the NGF/TrkA signaling endosomes is prohibited from premature degradation115.
Given the important roles played by Rab5 in retrograde endosomal sorting and trafficking, it is not surprising that recent studies have demonstrated that the function and activity of Rab5 is compromised in early phases of Alzheimer’s disease49,50,64,80,81,92,93,116, as well as in mouse models of Parkinson’s disease117,118. Persistent hyperactivation of Rab5 upsets other endocytic pathways such as late endosomes, lysosomes and autophagy, processes that have all been found to be altered in AD48,51,63,119,120. An important consequence of these changes under the pathological condition results in premature degradation of the neurotrophic factor signaling, thus effectively preventing the delivery of retrograde trophic signals to the soma/nucleus, leading to neuronal atrophy. This is indeed the case for early degeneration of basal forebrain cholinergic neurons in Down syndrome111,113.
Dysfunction of retromers has also been implicated in AD62,121,122. Retromer is a protein complex that mediates endosome-to-Golgi transport. The levels of Vps35 and Vps26, two key retromer proteins, were significantly reduced in the entorhinal cortex of AD patients62,121. Vps26 binds to a hexapeptide motif (FANSHY) in the cytoplasmic tail of the sorting receptor SorLA (aka SORL1 or LR11)123,124. SorLA is a neuronal sorting protein that directs trafficking of APP also from endosomes to the Golgi123,124. It is thus conceivable that reduced levels of SorLA in conjunction with dysfunctional retromer would exacerbate the deficit of APP trafficking from endosomes to Golgi, resulting in accumulation of APP in endosomes. It is within the endosomal compartments enriched with proteolytic enzymes, APP is processed by β-secretase to drive production of β-CTF and Aβ62,122–124.
In addition to Rab5 containing-early endosomes, the recycling endosomal pathways mediated by Rab4/Rab11 that regulate intracellular sorting, trafficking and processing of APP has been found to be altered as well in AD125. Accumulation of APP β-CTF has been shown to impair a key neuronal specific soma-to-axon transcytosis pathway126,127. Using induced pluripotent stem cell (iPSC)-derived human neurons with familiar AD mutations in PS1 (presenilin 1) and APP, a recent study from Dr. L. Goldstein’s laboratory has demonstrated that subcellular distribution and trafficking of APP was altered in these neurons126; the level of APP was increased in the soma while APP in the axons was significantly reduced, a pattern closely reminiscent the distribution of Rab11 in these neurons126. Furthermore, knocking down Rab11 induced a similar defect in soma-to-axon transcytosis of APP in isogenic control neurons126. These studies have demonstrated that Rab11 plays a key role in the neuronal soma-to-axon transcytosis and dysfunction of Rab11 may also contribute to the early phase of AD pathogenesis.
Increased APP and its β-CTF leads to enlargement of Rab5+ early endosomes
One of the earliest neuronal pathologies of AD is endosomal dysfunction, that was observed in AD patients prior to the deposition of any extracellular β-amyloid (Aβ)128–130. In addition, patients with Down Syndrome (DS), who are known to develop AD pathology after the age of 40, also show these same endosomal abnormalities as early as 28 weeks of gestation111,128. These findings suggest that neurons become compromised intracellularly well before any plaque formation or tau accumulation occurs in the brain. Endosomal alterations reported in these two neurological disorders include increased endocytosis and enlarged early endosomes that contain the early endosomal protein Rab5129–132. Studies have also provided evidence that the cellular level of amyloid precursor protein (APP) is linked to Rab5 containing early endosomal abnormalities111,128–132. APP, which is triplicated in DS and is one of the main proteins implicated in AD, is a transmembrane protein which is cleaved first by β- or α-secretase producing the corresponding APP C-terminal fragments: β-CTF or α-CTF. Cleavage by γ-secretase then produces the APP intracellular domain (AICD) and generates the Aβ peptides. Of these, several APP gene products and processed fragments, the full-length APP and β-CTF, have been shown to play a direct role in the early endosomal pathologies seen in AD and DS110,128,131,132. Recently, we shown that excessive full-length APP, APP mutants and APP β-CTFs, but not APP α-CTFs or AICD, could elevate the Rab5 activity and induce the enlargement of early endosome in both PC12M cell and primary basal forebrain cholinergic neurons (BFCNs)80, which degenerates early in AD113,133–138. Importantly, APP β-CTFs induced neuronal atrophy in cultured rat E18 BFCNs and these effects by APP β-CTFs were rescued by a dominant-negative Rab5 mutant in vitro in cultured BFCNs as well as in a fly model of AD80. Therefore, consistent with studies from Dr. Nixon’s group81, these studies have demonstrated a role played by APP β-CTFs in mediating cellular toxicity by accumulation of toxic APP products.
APP/β-CTF acts through Rab5 to impair retrograde axonal trafficking of nerve growth factor leading to neuronal atrophy
NGF is a target-derived neurotrophic factor that acts through its surface TrkA receptor to support the survival, differentiation, and maintenance of BFCNs139,140. NGF signaling regulates expression of genes and cellular programs important for the BFCN phenotype, including cell size113,140. Following endocytosis, the NGF/TrkA signaling complexes are trafficked to Rab5+ early endosomes109,114. The “signaling endosomes”, as depicted in Figure 1, are then transported in a retrograde direction to the corresponding cell bodies to transmit NGF trophic signals139,140. Thus, axonal trafficking mediated by Rab5+ early endosomes plays a critical role in maintaining the trophic status of BFCNs. Any alteration in these aspects could potentially disrupt axonal transport, resulting in neurodegenerative disorders. Recently, our group together with Dr. Nixon’s laboratory found that APP β-CTF act though increased activation of Rab5 to cause enlargement of early endosomes79–81. Consequently, retrograde axonal trafficking and signaling of NGF in BFCNs or brain-derived neurotrophic factor in cortical neurons is reduced through increasing pauses79,80. Importantly, disruption of retrograde axonal trafficking of NGF signals results in trophic deficits in BFCNs, leading to neuronal atrophy80. Importantly, we have demonstrated that axonal transport deficits and neuronal atrophy can be rescued through the expression of a dominant negative Rab5 mutant, both in BFCN neurons as well as in a fly model of AD80. These studies have further confirmed the contribution of hyperactivated Rab5 to axonal transport deficits and neuronal atrophy in AD79–81.
Figure 1.

A simplified depiction of an NGF signaling endosome showing that NGF binds to TrkA that activates the Erk, PI3K and PLCγ signaling cascades. In addition, APP/APP βCTF may act through RIN3 or APPL1 to activate Rab5. The dynein motor protein complex is also required to drive the retrograde axonal transport of the NGF signaling endosome.
Based on our studies, we propose the following model to explain how NGF-TrkA signaling transport is impacted in the presence of APP β-CTF through increased activation of Rab5. In normal BFCNs (Figure 2A), NGF binds to and activates TrkA at the axonal terminals. The NGF-TrkA signaling complex undergoes endocytosis and is internalized into the cell cytoplasm to form Rab5+ signaling endosomes. The signaling endosomes are transported in a retrograde direction along the microtubule toward the cell body to propagate the growth and differentiation signals to the nucleus. The NGF/TrkA machinery is then attenuated in the cell body in late endosomes and lysosomes.
Figure 2.

Proposed models for retrograde axonal transport function of NGF signaling endosomes in normal neurons (A) and under conditions of excess APP or APP βCTF (B). Please see the text for a detailed description.
However, under pathogenic conditions, BFCNs are loaded with excessive APP and APP β-CTF (Figure 2B), which in turn results in an increase in the level of GTP-Rab5 leading to sustained activation of Rab5. Overactivation of Rab5 induces the enlargement of early endosomes that impair endocytic trafficking of APP, further exacerbating its processing. In addition, abnormally enlarged early endosomes may interfere with retrograde axonal transport of NGF signals. Additionally, increased Rab5 activity may also impact motor proteins to impair axonal transport. The net effect of all these aspects of APP and APP β-CTF will be reduced trophic signals being delivered to the soma, which will lead to neuronal atrophy111,113.
The missing link between APP/β-CTF and Rab5 activation
1: RIN3
Recent GWAS studies have identified RIN3 as a risk factor for AD. RIN3, a guanidine nucleotide exchange factor (GEF) for selective members of the Rab5 family (Rab5, Rab21, Rab22, Rab24 and Rab31)88,89, functions as the stimulator and stabilizer for GTP-Rab588,89. RIN3 has a Src homology 2 (SH2) domain, a proline rich domain (PRD), a RIN-homology (RH) domain, a Vps9 (vacuolar protein sorting-associated protein 9) conserved in the catalytic domains of the Rab5 GEFs (Vps9p, Rabex-5 etc.) and a Ras-association (RA) domain (Figure 3A)88. It is possible that increased activity of RIN3 is responsible for the increase in the level of GTP-Rab5, that results in early endosomal abnormalities and axonopathy, leading to neuronal degeneration in AD. However, it remains to be defined: 1) if and how the function and expression of RIN3 is altered in AD; 2) if and how the activity of RIN3 is impacted by APP and APP β-CTF that results in hyperactivation of Rab5; and 3) whether or not Rab5 GEFs other than RIN3 also play a role in increased Rab5 activation in AD.
Figure 3.

Domain structures of RIN3 (A) and APPL1 (B). A: RIN3 is consisted of a SH2 domain (a Src homology 2 domain), a proline rich domain (PRD), a RIN-homology domain (RH), a Vsp9 domain [vacuolar protein sorting-associated protein 9 domain that is conserved in the catalytic domains of the Rab5 GEFs (Vps9p, Rabex-5 etc.)], and a Ras-association domain (RA)88,89. B: APPLI contains a BAR (Bin/Amphiphysin/Rvs) domain, a PH (pleckstrin homology) domain and a PTB (phosphotyrosine-binding) domain148.
2. APPL1
In addition to RIN3, APPL1 (adaptor protein, phosphotyrosine interacting with pleckstrin homology domain and leucine zipper 1), an Rab5 effector (Figure 3B), has been shown to link APP β-CTF to Rab5 overactivation in AD and Down syndrome81. In a recent study, Dr. Nixon’s laboratory has elegantly demonstrated that both APPL1 and β-CTF are increased in AD and that β-CTF binds to the YENPTY domain to the PTB domain of APPL1 and recruits APPL1 to Rab5 endosomes81. They have demonstrated that the increased presence of APPL1 to Rab5 endosomes leads to stabilization of active GTP-Rab5, which contributes to endocytic dysfunction e.g. pathologically accelerated endocytosis, abnormal enlargement of endosomes and impaired axonal transport of Rab5 endosomes81. Importantly, these endocytic defects in fibroblasts from Down syndrome were rescued by knocking down APPL1. Therefore, APPL1 represents an important adaptor that links APP β-CTF to hyperactivation of Rab5 and to endosomal dysfunction in AD and Down syndrome. However, this novel APPL1-dependent pathogenic pathway in AD will need to be further validated.
Dysregulation of Rab5 in Parkinson’s disease
In addition to AD, recent studies have also pointed to that Rab5-mediated endocytic functions are impacted in Parkinson's disease (PD), for which α-synuclein aggregates, a major component of Lewy bodies (LBs) and associated Lewy neurites (LNs)141,142, play a central role in the pathogenesis of the disease. The presence of α-synuclein aggregates significantly impacts intracellular vesicular trafficking by Rab proteins58,143. Conversely, these Rab proteins appear to modulate the protein level, aggregation, spreading and also toxicity of α-synuclein143. For example, Rab5A-mediated endocytosis of α-synuclein was found to be correlated with the neuronal cell death and with the that subsequently caused the formation of Lewy body-like intracytoplasmic inclusions144. Importantly, expression of a GTPase-deficient Rab5A mutant reduced endocytosis of α-synuclein and abrogated its cytotoxicity144. Using an APP transgenic mouse model of Alzheimer’s disease, a recent study found that reducing endogenous α-synuclein restored the levels of Rab3a and Rab5 proteins58. In our recent study, we have observed increased expression of α-synuclein interacts with dynein motor and induces endosomal dysfunction by enhancing the level of activated Rab proteins (Rab5, Rab7), and hyperactivated Rab5 in a PD mouse model overexpressing wildtype α-synuclein. And all this may contribute to the impairment of retrograde axonal transport of BDNF and neuronal atrophy145.
Possible interplays between Rab5 activation and PICALM, BIN1 in AD pathogenesis
Increasing evidence has pointed to the disturbance of Rab5-mediated endocytic pathways possibly playing a critical role in early cellular pathogenesis of AD and other neurodegenerative disorders such as PD. It remains unclear how these Rab5-mediated pathways interact with the GWAS hits (PICALM, BIN1, CD2AP, EPHA1, and SORL1), that also function at various stages of endocytosis10,11, to impact pathogenesis in LOAD. We have just begun to understand how these GWAS hits impact endocytic pathways, potentially leading to neurodegeneration in AD. For example, PICALM28,29,31,32 and BIN134–36 both function in clathrin-mediated endocytosis. Yet, the level of PICALM was reduced in LOAD patients10,11, while BIN1 expression was increased in the brain of AD patients who carry high-risk polymorphisms upstream of BIN135,45. Although PICALM has been suggested to promote APP processing and increase Aβ production in neurons, it is possible that PICALM plays a more important role in Aβ clearance from the brain by facilitating internalization of Aβ into endothelial cells and consequently releasing it to the bloodstream146. Since expression of PICALM is much higher in endothelial cells than in neurons147, reduced levels of PICALM may therefore impair Aβ clearance more that Aβ production in LOAD patients. Increased accumulation of extracellular Aβ will undoubtedly upset the Rab5 endocytic pathways in neurons to impact their well-being52. This may explain why a minor PICALM allele (rs3851179 SNP), associated with increased expression of PICALM, is protective against AD likely by increasing clearance of extracellular Aβ to reduce its toxicity on neurons28. On the other hand, although the pathogenic mechanism of BIN1 is presently unknown, one can speculate that elevated level of BIN1 in LOAD patients34–36,45 increase endocytosis and promote Aβ production in neurons35, which in turn may induce activation of Rab5 to impair trafficking and signaling of neurotrophic factors52.
Conclusion
AD is an extremely complex disease that involves many different cell types, factors and pathways. These different components may act independently or work in tandem to contribute AD pathogenesis. It has become increasingly clear that homeostasis of endocytic sorting, trafficking and signaling, that is critical for maintaining neuronal function, is disrupted in AD. Those AD risk factors discovered in GWAS studies that target the endocytic pathways may act alone or work in tandem to impair the normal function and process of the endocytic pathways. Delineating the interplays among these factors and pathways will not only enhance our understanding of the mechanisms responsible for neurodegeneration in AD and other diseases, but also will facilitate the discovery of novel target for developing treatment strategies for these disorders.
A brief synopsis statement.
Dysregulation of the endo-lysosomal system has emerged as one of the early cellular pathologies for Alzheimer’s disease (AD). Rab5 is a small GTPase that plays a critical role in mediating internalization and endocytic trafficking. Recent studies have demonstrated Rab5 is hyper-activated and Rab5+ early endosomes are found to be abnormally enlarged in post-mortem brain of AD patients and in mouse models of AD. In the review, we will summarize recent research findings linking Rab5 dysfunction to AD pathogenesis.
Acknowledgments
We would like to thank our colleagues in the Wu laboratory for their assistance. The study is supported by the following grants: NIH (PN2EY016525), NIH UCSD ADRC P50 Pilot grant, Down Syndrome Research and Treatment Foundation, Larry L. Hillblom Foundation (C. Wu). Dr. J. Ding and Dr. W. Xu are supported by the Ministry of Science and Technology of the People´s Republic of China (2014CB965002, 2012BAI10B03), National Natural Science Foundation of China (81171200), Science and Technology Commission of Shanghai Municipality (13JC1401502, 13140904000), Shanghai Municipal Education Commission (12ZZ115).
Footnotes
Conflict of interest: the authors declare no conflict of interest.
References
- 1.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016 Jun 01;8(6):595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holtzman DM, Mandelkow E, Selkoe DJ. Alzheimer disease in 2020. Cold Spring Harb Perspect Med. 2012 Nov;2(11) doi: 10.1101/cshperspect.a011585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Selkoe DJ. Alzheimer's disease. Cold Spring Harb Perspect Biol. 2011 Jul;3(7) doi: 10.1101/cshperspect.a004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selkoe DJ. Aging, amyloid, and Alzheimer's disease: a perspective in honor of Carl Cotman. Neurochem Res. 2003 Nov;28(11):1705–1713. doi: 10.1023/a:1026065122854. [DOI] [PubMed] [Google Scholar]
- 5.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002 Jul 19;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 6.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001 Apr;81(2):741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 7.Selkoe DJ. Toward a comprehensive theory for Alzheimer's disease. Hypothesis: Alzheimer's disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Ann N Y Acad Sci. 2000;924:17–25. doi: 10.1111/j.1749-6632.2000.tb05554.x. [DOI] [PubMed] [Google Scholar]
- 8.Selkoe DJ. The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol. 1998 Nov;8(11):447–453. doi: 10.1016/s0962-8924(98)01363-4. [DOI] [PubMed] [Google Scholar]
- 9.Rasool CG, Svendsen CN, Selkoe DJ. Neurofibrillary degeneration of cholinergic and noncholinergic neurons of the basal forebrain in Alzheimer's disease. Ann Neurol. 1986 Oct;20(4):482–488. doi: 10.1002/ana.410200407. [DOI] [PubMed] [Google Scholar]
- 10.Karch CM, Goate AM. Alzheimer's disease risk genes and mechanisms of disease pathogenesis. Biological psychiatry. 2015;77(1):43–51. doi: 10.1016/j.biopsych.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karch CM, Cruchaga C, Goate A. Alzheimer’s Disease Genetics: From the bench to the clinic. Neuron. 2014;83(1):11–26. doi: 10.1016/j.neuron.2014.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goate A, Chartier-Harlin MC, Mullan M, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991 Feb 21;349(6311):704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 13.Levy-Lahad E, Wasco W, Poorkaj P, et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995 Aug 18;269(5226):973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 14.Rogaev EI, Sherrington R, Rogaeva EA, et al. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature. 1995 Aug 31;376(6543):775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 15.Sherrington R, Rogaev EI, Liang Y, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995 Jun 29;375(6534):754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 16.Hardy J, Selkoe DJ. The Amyloid Hypothesis of Alzheimer's Disease: Progress and Problems on the Road to Therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 17.Doody RS, Aisen PS, Iwatsubo T. Semagacestat for treatment of Alzheimer's disease. N Engl J Med. 2013 Oct 24;369(17):1661. doi: 10.1056/NEJMc1310845. [DOI] [PubMed] [Google Scholar]
- 18.Doody RS, Raman R, Farlow M, et al. A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N Engl J Med. 2013 Jul 25;369(4):341–350. doi: 10.1056/NEJMoa1210951. [DOI] [PubMed] [Google Scholar]
- 19.Gauthier S, Feldman HH, Schneider LS, et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer's disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet. 2016 Dec 10;388(10062):2873–2884. doi: 10.1016/S0140-6736(16)31275-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer's disease to AIDS. Journal of Lipid Research. 2009 Apr;150(Supplement):S183–S188. doi: 10.1194/jlr.R800069-JLR200. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006 Feb;63(2):168–174. doi: 10.1001/archpsyc.63.2.168. [DOI] [PubMed] [Google Scholar]
- 22.Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41(10):1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hollingworth P, Sweet R, Sims R, et al. Genome-wide association study of Alzheimer's disease with psychotic symptoms. Mol Psychiatry. 2012 Dec;17(12):1316–1327. doi: 10.1038/mp.2011.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lambert J-C, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41(10):1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 25.Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013 Dec;45(12):1452–1458. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sherva R, Baldwin CT, Inzelberg R, et al. Identification of novel candidate genes for Alzheimer's disease by autozygosity mapping using genome wide SNP data. J Alzheimers Dis. 2011;23(2):349–359. doi: 10.3233/JAD-2010-100714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seshadri S, Fitzpatrick AL, Ikram M, et al. Genome-wide analysis of genetic loci associated with alzheimer disease. JAMA. 2010;303(18):1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parikh I, Fardo DW, Estus S. Genetics of PICALM Expression and Alzheimer's Disease. PLoS One. 2014;9(3):e91242. doi: 10.1371/journal.pone.0091242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu W, Tan L, Yu JT. The Role of PICALM in Alzheimer's Disease. Mol Neurobiol. 2015 Aug;52(1):399–413. doi: 10.1007/s12035-014-8878-3. [DOI] [PubMed] [Google Scholar]
- 30.Jones EL, Mok K, Hanney M, et al. Evidence that PICALM affects age at onset of Alzheimer's dementia in Down syndrome. Neurobiology of Aging. 2013;34(10):2441.e2441–2441.e2445. doi: 10.1016/j.neurobiolaging.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Z, Sagare AP, Ma Q, et al. Central role for PICALM in amyloid-beta blood-brain barrier transcytosis and clearance. Nat Neurosci. 2015 Jul;18(7):978–987. doi: 10.1038/nn.4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao Q, Gil SC, Yan P, et al. Role of phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia (PICALM) in intracellular amyloid precursor protein (APP) processing and amyloid plaque pathogenesis. J Biol Chem. 2012 Jun 15;287(25):21279–21289. doi: 10.1074/jbc.M111.338376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiao B, Liu X, Zhou L, et al. Polygenic Analysis of Late-Onset Alzheimer’s Disease from Mainland China. PLoS One. 2015;10(12):e0144898. doi: 10.1371/journal.pone.0144898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holler CJ, Davis PR, Beckett TL, et al. Bridging integrator 1 (BIN1) protein expression increases in the Alzheimer's disease brain and correlates with neurofibrillary tangle pathology. J Alzheimers Dis. 2014;42(4):1221–1227. doi: 10.3233/JAD-132450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chapuis J, Hansmannel F, Gistelinck M, et al. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry. 2013 Nov;18(11):1225–1234. doi: 10.1038/mp.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Calafate S, Flavin W, Verstreken P, Moechars D. Loss of Bin1 Promotes the Propagation of Tau Pathology. Cell Rep. 2016 Oct 18;17(4):931–940. doi: 10.1016/j.celrep.2016.09.063. [DOI] [PubMed] [Google Scholar]
- 37.Willnow TE, Andersen OM. Sorting receptor SORLA--a trafficking path to avoid Alzheimer disease. J Cell Sci. 2013 Jul 01;126(Pt 13):2751–2760. doi: 10.1242/jcs.125393. [DOI] [PubMed] [Google Scholar]
- 38.Yin RH, Yu JT, Tan L. The Role of SORL1 in Alzheimer's Disease. Mol Neurobiol. 2015;51(3):909–918. doi: 10.1007/s12035-014-8742-5. [DOI] [PubMed] [Google Scholar]
- 39.Dumanis SB, Burgert T, Caglayan S, et al. Distinct Functions for Anterograde and Retrograde Sorting of SORLA in Amyloidogenic Processes in the Brain. J Neurosci. 2015 Sep 16;35(37):12703–12713. doi: 10.1523/JNEUROSCI.0427-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young JE, Boulanger-Weill J, Williams DA, et al. Elucidating molecular phenotypes caused by the SORL1 Alzheimer's disease genetic risk factor using human induced pluripotent stem cells. Cell Stem Cell. 2015 Apr 02;16(4):373–385. doi: 10.1016/j.stem.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee JH, Barral S, Reitz C. The Neuronal Sortilin-Related Receptor Gene SORL1 and Late-Onset Alzheimer's Disease. Current neurology and neuroscience reports. 2008;8(5):384–391. doi: 10.1007/s11910-008-0060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Felsky D, Szeszko P, Yu L, et al. The SORL1 gene and convergent neural risk for Alzheimer's disease across the human lifespan. Mol Psychiatry. 2014 Oct;19(10):1125–1132. doi: 10.1038/mp.2013.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cuccaro ML, Carney RM, Zhang Y, et al. SORL1 mutations in early- and late-onset Alzheimer disease. Neurol Genet. 2016 Dec;2(6):e116. doi: 10.1212/NXG.0000000000000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meng Y, Lee JH, Cheng R, George-Hyslop PS, Mayeux R, Farrer LA. Association between SORL1 and Alzheimer disease in a genome-wide study. Neuroreport. 2007;18(17):1761–1764. doi: 10.1097/WNR.0b013e3282f13e7a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones L, Harold D, Williams J. Genetic evidence for the involvement of lipid metabolism in Alzheimer's disease. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2010;1801(8):754–761. doi: 10.1016/j.bbalip.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 46.Nuutinen T, Suuronen T, Kauppinen A, Salminen A. Clusterin: A forgotten player in Alzheimer's disease. Brain Research Reviews. 2009;61(2):89–104. doi: 10.1016/j.brainresrev.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 47.Funk KE, Kuret J. Lysosomal Fusion Dysfunction as a Unifying Hypothesis for Alzheimer's Disease Pathology. International Journal of Alzheimer's Disease. 2012;2012:10. doi: 10.1155/2012/752894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000 Jul;157(1):277–286. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ginsberg SD, Mufson EJ, Alldred MJ, et al. Upregulation of select rab GTPases in cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer's disease. Journal of Chemical Neuroanatomy. 2011;42(2):102–110. doi: 10.1016/j.jchemneu.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ginsberg SD, Mufson EJ, Counts SE, et al. Regional selectivity of rab5 and rab7 protein upregulation in mild cognitive impairment and Alzheimer's disease. J Alzheimers Dis. 2010;22(2):631–639. doi: 10.3233/JAD-2010-101080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nixon RA. Endosome function and dysfunction in Alzheimer's disease and other neurodegenerative diseases. Neurobiol Aging. 2005 Mar;26(3):373–382. doi: 10.1016/j.neurobiolaging.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 52.Chen XQ, Sawa M, Mobley WC. Dysregulation of neurotrophin signaling in the pathogenesis of Alzheimer disease and of Alzheimer disease in Down syndrome. Free Radic Biol Med. 2017 Oct 12; doi: 10.1016/j.freeradbiomed.2017.10.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008 Oct 31;283(44):29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Greenberg SM, Koo EH, Selkoe DJ, Qiu WQ, Kosik KS. Secreted beta-amyloid precursor protein stimulates mitogen-activated protein kinase and enhances tau phosphorylation. Proc Natl Acad Sci U S A. 1994 Jul 19;91(15):7104–7108. doi: 10.1073/pnas.91.15.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koo EH, Squazzo SL. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem. 1994 Jul 01;269(26):17386–17389. [PubMed] [Google Scholar]
- 56.Haass C, Koo EH, Teplow DB, Selkoe DJ. Polarized secretion of beta-amyloid precursor protein and amyloid beta-peptide in MDCK cells. Proc Natl Acad Sci U S A. 1994 Feb 15;91(4):1564–1568. doi: 10.1073/pnas.91.4.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Toh Wei H, Gleeson Paul A. Dysregulation of intracellular trafficking and endosomal sorting in Alzheimer's disease: controversies and unanswered questions. Biochemical Journal. 2016;473(14):1977–1993. doi: 10.1042/BCJ20160147. [DOI] [PubMed] [Google Scholar]
- 58.Spencer B, Desplats PA, Overk CR, et al. Reducing Endogenous alpha-Synuclein Mitigates the Degeneration of Selective Neuronal Populations in an Alzheimer's Disease Transgenic Mouse Model. J Neurosci. 2016 Jul 27;36(30):7971–7984. doi: 10.1523/JNEUROSCI.0775-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kinoshita A, Fukumoto H, Shah T, Whelan CM, Irizarry MC, Hyman BT. Demonstration by FRET of BACE interaction with the amyloid precursor protein at the cell surface and in early endosomes. Journal of Cell Science. 2003;116(16):3339–3346. doi: 10.1242/jcs.00643. [DOI] [PubMed] [Google Scholar]
- 60.Koo EH, Squazzo SL, Selkoe DJ, Koo CH. Trafficking of cell-surface amyloid beta-protein precursor. I. Secretion, endocytosis and recycling as detected by labeled monoclonal antibody. Journal of Cell Science. 1996;109(5):991–998. doi: 10.1242/jcs.109.5.991. [DOI] [PubMed] [Google Scholar]
- 61.Rajendran L, Honsho M, Zahn TR, et al. Alzheimer's disease β-amyloid peptides are released in association with exosomes. Proceedings of the National Academy of Sciences. 2006 Jul;25103(30):11172–11177. doi: 10.1073/pnas.0603838103. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Small SA, Gandy S. Sorting through the cell biology of Alzheimer's disease: intracellular pathways to pathogenesis. Neuron. 2006 Oct 05;52(1):15–31. doi: 10.1016/j.neuron.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cataldo AM, Petanceska S, Terio NB, et al. Abeta localization in abnormal endosomes: association with earliest Abeta elevations in AD and Down syndrome. Neurobiol Aging. 2004 Nov-Dec;25(10):1263–1272. doi: 10.1016/j.neurobiolaging.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 64.Ginsberg SD, Alldred MJ, Counts SE, et al. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer's disease progression. Biol Psychiatry. 2010 Nov 15;68(10):885–893. doi: 10.1016/j.biopsych.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haass C, Kaether C, Thinakaran G, Sisodia S. Trafficking and Proteolytic Processing of APP. Cold Spring Harb Perspect Med. 2012;2(5):a006270. doi: 10.1101/cshperspect.a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang TY, Zhao Y, Li X, et al. SNX27 and SORLA Interact to Reduce Amyloidogenic Subcellular Distribution and Processing of Amyloid Precursor Protein. J Neurosci. 2016 Jul 27;36(30):7996–8011. doi: 10.1523/JNEUROSCI.0206-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang X, Huang T, Zhao Y, et al. Sorting nexin 27 regulates Abeta production through modulating gamma-secretase activity. Cell Rep. 2014 Nov 06;9(3):1023–1033. doi: 10.1016/j.celrep.2014.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang X, Zhao Y, Zhang X, et al. Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down's syndrome. Nat Med. 2013 Apr;19(4):473–480. doi: 10.1038/nm.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ihara Y, Morishima-Kawashima M, Nixon R. The ubiquitin-proteasome system and the autophagic-lysosomal system in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(8) doi: 10.1101/cshperspect.a006361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Berger-Sweeney J, McPhie DL, Arters JA, Greenan J, Oster-Granite ML, Neve RL. Impairments in learning and memory accompanied by neurodegeneration in mice transgenic for the carboxyl-terminus of the amyloid precursor protein. Brain Res Mol Brain Res. 1999 Mar 20;66(1–2):150–162. doi: 10.1016/s0169-328x(99)00014-5. [DOI] [PubMed] [Google Scholar]
- 71.Neve RL, Boyce FM, McPhie DL, Greenan J, Oster-Granite ML. Transgenic mice expressing APP-C100 in the brain. Neurobiol Aging. 1996 Mar-Apr;17(2):191–203. doi: 10.1016/0197-4580(95)02074-8. [DOI] [PubMed] [Google Scholar]
- 72.Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science. 1989 Jul 28;245(4916):417–420. doi: 10.1126/science.2474201. [DOI] [PubMed] [Google Scholar]
- 73.Rockenstein E, Mante M, Alford M, et al. High beta-secretase activity elicits neurodegeneration in transgenic mice despite reductions in amyloid-beta levels: implications for the treatment of Alzheimer disease. J Biol Chem. 2005 Sep 23;280(38):32957–32967. doi: 10.1074/jbc.M507016200. [DOI] [PubMed] [Google Scholar]
- 74.Bittner T, Fuhrmann M, Burgold S, et al. Gamma-secretase inhibition reduces spine density in vivo via an amyloid precursor protein-dependent pathway. J Neurosci. 2009 Aug 19;29(33):10405–10409. doi: 10.1523/JNEUROSCI.2288-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kozlowski MR, Spanoyannis A, Manly SP, Fidel SA, Neve RL. The neurotoxic carboxy-terminal fragment of the Alzheimer amyloid precursor binds specifically to a neuronal cell surface molecule: pH dependence of the neurotoxicity and the binding. J Neurosci. 1992 May;12(5):1679–1687. doi: 10.1523/JNEUROSCI.12-05-01679.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lauritzen I, Pardossi-Piquard R, Bauer C, et al. The beta-secretase-derived C-terminal fragment of betaAPP, C99, but not Abeta, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J Neurosci. 2012 Nov 14;32(46):16243–16255a. doi: 10.1523/JNEUROSCI.2775-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lauritzen I, Pardossi-Piquard R, Bourgeois A, et al. Intraneuronal aggregation of the beta-CTF fragment of APP (C99) induces Abeta-independent lysosomal-autophagic pathology. Acta Neuropathol. 2016 Aug;132(2):257–276. doi: 10.1007/s00401-016-1577-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mitani Y, Yarimizu J, Saita K, et al. Differential effects between gamma-secretase inhibitors and modulators on cognitive function in amyloid precursor protein-transgenic and nontransgenic mice. J Neurosci. 2012 Feb 08;32(6):2037–2050. doi: 10.1523/JNEUROSCI.4264-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Weissmiller AM, Natera-Naranjo O, Reyna SM, et al. A gamma-secretase inhibitor, but not a gamma-secretase modulator, induced defects in BDNF axonal trafficking and signaling: evidence for a role for APP. PLoS One. 2015;10(2):e0118379. doi: 10.1371/journal.pone.0118379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Xu W, Weissmiller AM, White JA, 2nd, et al. Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J Clin Invest. 2016 May 02;126(5):1815–1833. doi: 10.1172/JCI82409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim S, Sato Y, Mohan PS, et al. Evidence that the rab5 effector APPL1 mediates APP-betaCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer's disease. Mol Psychiatry. 2015 Jul 21; doi: 10.1038/mp.2015.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Moore S, Evans LD, Andersson T, et al. APP metabolism regulates tau proteostasis in human cerebral cortex neurons. Cell Rep. 2015 May 05;11(5):689–696. doi: 10.1016/j.celrep.2015.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of Neurofibrillary Tangles in P301L Tau Transgenic Mice Induced by Aβ42 Fibrils. Science. 2001;293(5534):1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 84.Gotz J, Ittner LM, Kins S. Do axonal defects in tau and amyloid precursor protein transgenic animals model axonopathy in Alzheimer's disease? J Neurochem. 2006 Aug;98(4):993–1006. doi: 10.1111/j.1471-4159.2006.03955.x. [DOI] [PubMed] [Google Scholar]
- 85.Le MH, Weissmiller AM, Monte L, et al. Functional Impact of Corticotropin-Releasing Factor Exposure on Tau Phosphorylation and Axon Transport. PLoS One. 2016;11(1):e0147250. doi: 10.1371/journal.pone.0147250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mandelkow EM, Stamer K, Vogel R, Thies E, Mandelkow E. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiology of Aging. 2003;24(8):1079–1085. doi: 10.1016/j.neurobiolaging.2003.04.007. [DOI] [PubMed] [Google Scholar]
- 87.Yetman MJ, Fowler SW, Jankowsky JL. Humanized Tau Mice with Regionalized Amyloid Exhibit Behavioral Deficits but No Pathological Interaction. PLoS One. 2016;11(4):e0153724. doi: 10.1371/journal.pone.0153724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kajiho H, Saito K, Tsujita K, et al. RIN3: a novel Rab5 GEF interacting with amphiphysin II involved in the early endocytic pathway. J Cell Sci. 2003 Oct 15;116(Pt 20):4159–4168. doi: 10.1242/jcs.00718. [DOI] [PubMed] [Google Scholar]
- 89.Kajiho H, Sakurai K, Minoda T, et al. Characterization of RIN3 as a guanine nucleotide exchange factor for the Rab5 subfamily GTPase Rab31. J Biol Chem. 2011 Jul 8;286(27):24364–24373. doi: 10.1074/jbc.M110.172445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Barbieri MA, Li G, Colombo MI, Stahl PD. Rab5, an early acting endosomal GTPase, supports in vitro endosome fusion without GTP hydrolysis. The Journal of biological chemistry. 1994 Jul 22;269(29):18720–18722. [PubMed] [Google Scholar]
- 91.Bucci C, Parton RG, Mather IH, et al. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell. 1992 Sep 4;70(5):715–728. doi: 10.1016/0092-8674(92)90306-w. [DOI] [PubMed] [Google Scholar]
- 92.Gorvel JP, Chavrier P, Zerial M, Gruenberg J. rab5 controls early endosome fusion in vitro. Cell. 1991 Mar 8;64(5):915–925. doi: 10.1016/0092-8674(91)90316-q. [DOI] [PubMed] [Google Scholar]
- 93.Grbovic OM, Mathews PM, Jiang Y, et al. Rab5-stimulated up-regulation of the endocytic pathway increases intracellular beta-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Abeta production. J Biol Chem. 2003 Aug 15;278(33):31261–31268. doi: 10.1074/jbc.M304122200. [DOI] [PubMed] [Google Scholar]
- 94.Li G. Rab5 GTPase and endocytosis. Biocell. 1996 Dec;20(3):325–330. [PubMed] [Google Scholar]
- 95.Haas AK, Fuchs E, Kopajtich R, Barr FA. A GTPase-activating protein controls Rab5 function in endocytic trafficking. Nat Cell Biol. 2005 Sep;7(9):887–893. doi: 10.1038/ncb1290. [DOI] [PubMed] [Google Scholar]
- 96.Liang Z, Mather T, Li G. GTPase mechanism and function: new insights from systematic mutational analysis of the phosphate-binding loop residue Ala30 of Rab5. Biochem J. 2000 Mar 1;346(Pt 2):501–508. [PMC free article] [PubMed] [Google Scholar]
- 97.Miaczynska M, Christoforidis S, Giner A, et al. APPL proteins link Rab5 to nuclear signal transduction via an endosomal compartment. Cell. 2004 Feb 06;116(3):445–456. doi: 10.1016/s0092-8674(04)00117-5. [DOI] [PubMed] [Google Scholar]
- 98.Horiuchi H, Lippe R, McBride HM, et al. A novel Rab5 GDP/GTP exchange factor complexed to Rabaptin-5 links nucleotide exchange to effector recruitment and function. Cell. 1997 Sep 19;90(6):1149–1159. doi: 10.1016/s0092-8674(00)80380-3. [DOI] [PubMed] [Google Scholar]
- 99.Simonsen A, Lippe R, Christoforidis S, et al. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature. 1998 Jul 30;394(6692):494–498. doi: 10.1038/28879. [DOI] [PubMed] [Google Scholar]
- 100.Christoforidis S, McBride HM, Burgoyne RD, Zerial M. The Rab5 effector EEA1 is a core component of endosome docking. Nature. 1999 Feb 18;397(6720):621–625. doi: 10.1038/17618. [DOI] [PubMed] [Google Scholar]
- 101.Simonsen A, Gaullier JM, D'Arrigo A, Stenmark H. The Rab5 effector EEA1 interacts directly with syntaxin-6. The Journal of biological chemistry. 1999 Oct 8;274(41):28857–28860. doi: 10.1074/jbc.274.41.28857. [DOI] [PubMed] [Google Scholar]
- 102.Lippe R, Miaczynska M, Rybin V, Runge A, Zerial M. Functional synergy between Rab5 effector Rabaptin-5 and exchange factor Rabex-5 when physically associated in a complex. Mol Biol Cell. 2001 Jul;12(7):2219–2228. doi: 10.1091/mbc.12.7.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mu FT, Callaghan JM, Steele-Mortimer O, et al. EEA1, an early endosome-associated protein. EEA1 is a conserved alpha-helical peripheral membrane protein flanked by cysteine "fingers" and contains a calmodulin-binding IQ motif. The Journal of biological chemistry. 1995 Jun 02;270(22):13503–13511. doi: 10.1074/jbc.270.22.13503. [DOI] [PubMed] [Google Scholar]
- 104.Stenmark H, Parton RG, Steele-Mortimer O, Lutcke A, Gruenberg J, Zerial M. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. Embo J. 1994 Mar 15;13(6):1287–1296. doi: 10.1002/j.1460-2075.1994.tb06381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Stenmark H, Vitale G, Ullrich O, Zerial M. Rabaptin-5 is a direct effector of the small GTPase Rab5 in endocytic membrane fusion. Cell. 1995 Nov 03;83(3):423–432. doi: 10.1016/0092-8674(95)90120-5. [DOI] [PubMed] [Google Scholar]
- 106.Vitale G, Alexandrov K, Ullrich O, et al. The GDP/GTP cycle of Rab5 in the regulation of endocytotic membrane traffic. Cold Spring Harb Symp Quant Biol. 1995;60:211–220. doi: 10.1101/sqb.1995.060.01.024. [DOI] [PubMed] [Google Scholar]
- 107.Li G, Liang Z. Phosphate-binding loop and Rab GTPase function: mutations at Ser29 and Ala30 of Rab5 lead to loss-of-function as well as gain-of-function phenotype. Biochem J. 2001 May 1;355(Pt 3):681–689. doi: 10.1042/bj3550681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell. 2005 Sep 9;122(5):735–749. doi: 10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 109.Wu C, Ramirez A, Cui B, et al. A functional dynein-microtubule network is required for NGF signaling through the Rap1/MAPK pathway. Traffic. 2007 Nov;8(11):1503–1520. doi: 10.1111/j.1600-0854.2007.00636.x. [DOI] [PubMed] [Google Scholar]
- 110.Cui B, Wu C, Chen L, et al. One at a time, live tracking of NGF axonal transport using quantum dots. Proceedings of the National Academy of Sciences of the United States of America. 2007 Aug 21;104(34):13666–13671. doi: 10.1073/pnas.0706192104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Salehi A, Delcroix JD, Belichenko PV, et al. Increased App expression in a mouse model of Down's syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006 Jul 6;51(1):29–42. doi: 10.1016/j.neuron.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 112.Delcroix JD, Valletta JS, Wu C, Hunt SJ, Kowal AS, Mobley WC. NGF signaling in sensory neurons: evidence that early endosomes carry NGF retrograde signals. Neuron. 2003 Jul 3;39(1):69–84. doi: 10.1016/s0896-6273(03)00397-0. [DOI] [PubMed] [Google Scholar]
- 113.Cooper JD, Salehi A, Delcroix JD, et al. Failed retrograde transport of NGF in a mouse model of Down's syndrome: reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proceedings of the National Academy of Sciences of the United States of America. 2001 Aug 28;98(18):10439–10444. doi: 10.1073/pnas.181219298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Beattie EC, Zhou J, Grimes ML, Bunnett NW, Howe CL, Mobley WC. A signaling endosome hypothesis to explain NGF actions: potential implications for neurodegeneration. Cold Spring Harb Symp Quant Biol. 1996;61:389–406. [PubMed] [Google Scholar]
- 115.Liu J, Lamb D, Chou MM, Liu YJ, Li G. Nerve growth factor-mediated neurite outgrowth via regulation of Rab5. Mol Biol Cell. 2007 Apr;18(4):1375–1384. doi: 10.1091/mbc.E06-08-0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Laifenfeld D, Patzek LJ, McPhie DL, et al. Rab5 mediates an amyloid precursor protein signaling pathway that leads to apoptosis. J Neurosci. 2007 Jul 4;27(27):7141–7153. doi: 10.1523/JNEUROSCI.4599-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fang FYW, Florio JB, Rockenstein E, Spencer B, Orain XM, Dong SX, Li H, Chen X, Sung K, Rissman RA, Masliah E, Ding J, Wu C. Synuclein impairs trafficking and signaling of BDNF in a mouse model of Parkinson’s disease. Scientific Reports. 2017 doi: 10.1038/s41598-017-04232-4. accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Spencer B, Kim C, Gonzalez T, et al. alpha-Synuclein interferes with the ESCRT-III complex contributing to the pathogenesis of Lewy body disease. Hum Mol Genet. 2016 Mar 15;25(6):1100–1115. doi: 10.1093/hmg/ddv633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lee S, Sato Y, Nixon RA. Primary lysosomal dysfunction causes cargo-specific deficits of axonal transport leading to Alzheimer-like neuritic dystrophy. Autophagy. 2011 Dec;7(12):1562–1563. doi: 10.4161/auto.7.12.17956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lee S, Sato Y, Nixon RA. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer's-like axonal dystrophy. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011 May 25;31(21):7817–7830. doi: 10.1523/JNEUROSCI.6412-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Small SA, Kent K, Pierce A, et al. Model-guided microarray implicates the retromer complex in Alzheimer's disease. Ann Neurol. 2005 Dec;58(6):909–919. doi: 10.1002/ana.20667. [DOI] [PubMed] [Google Scholar]
- 122.Small SA, Petsko GA. Retromer in Alzheimer disease, Parkinson disease and other neurological disorders. Nat Rev Neurosci. 2015 Mar;16(3):126–132. doi: 10.1038/nrn3896. [DOI] [PubMed] [Google Scholar]
- 123.Fjorback AW, Andersen OM. SorLA is a molecular link for retromer-dependent sorting of the Amyloid precursor protein. Commun Integr Biol. 2012 Nov 1;5(6):616–619. doi: 10.4161/cib.21433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fjorback AW, Seaman M, Gustafsen C, et al. Retromer binds the FANSHY sorting motif in SorLA to regulate amyloid precursor protein sorting and processing. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012 Jan 25;32(4):1467–1480. doi: 10.1523/JNEUROSCI.2272-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mufson EJ, Ikonomovic MD, Counts SE, et al. Molecular and cellular pathophysiology of preclinical Alzheimer’s disease. Behavioural Brain Research. 2016 Sep 15;311(Supplement C):54–69. doi: 10.1016/j.bbr.2016.05.030. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Woodruff G, Reyna SM, Dunlap M, et al. Defective Transcytosis of APP and Lipoproteins in Human iPSC-Derived Neurons with Familial Alzheimer's Disease Mutations. Cell reports. 2016 Oct 11;17(3):759–773. doi: 10.1016/j.celrep.2016.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.von Bartheld CS. Axonal transport and neuronal transcytosis of trophic factors, tracers, and pathogens. J Neurobiol. 2004 Feb 5;58(2):295–314. doi: 10.1002/neu.10315. [DOI] [PubMed] [Google Scholar]
- 128.Cataldo AM, Petanceska S, Peterhoff CM, et al. App gene dosage modulates endosomal abnormalities of Alzheimer's disease in a segmental trisomy 16 mouse model of down syndrome. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2003 Jul 30;23(17):6788–6792. doi: 10.1523/JNEUROSCI.23-17-06788.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ginsberg SD, Alldred MJ, Counts SE, et al. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer's disease progression. Biol Psychiatry. 2010 Nov 15;68(10):885–893. doi: 10.1016/j.biopsych.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ginsberg SD, Mufson EJ, Counts SE, et al. Regional selectivity of rab5 and rab7 protein upregulation in mild cognitive impairment and Alzheimer's disease. J Alzheimers Dis. 2010;22(2):631–639. doi: 10.3233/JAD-2010-101080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Laifenfeld D, Patzek LJ, McPhie DL, et al. Rab5 mediates an amyloid precursor protein signaling pathway that leads to apoptosis. J Neurosci. 2007 Jul 4;27(27):7141–7153. doi: 10.1523/JNEUROSCI.4599-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jiang Y, Mullaney KA, Peterhoff CM, et al. Alzheimer's-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proceedings of the National Academy of Sciences of the United States of America. 2010 Jan 26;107(4):1630–1635. doi: 10.1073/pnas.0908953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Aloe L, Rocco ML, Bianchi P, Manni L. Nerve growth factor: from the early discoveries to the potential clinical use. Journal of Translational Medicine. 2012;10(1):239. doi: 10.1186/1479-5876-10-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Capsoni S, Ugolini G, Comparini A, Ruberti F, Berardi N, Cattaneo A. Alzheimer-like neurodegeneration in aged antinerve growth factor transgenic mice. Proc Natl Acad Sci U S A. 2000 Jun 06;97(12):6826–6831. doi: 10.1073/pnas.97.12.6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Counts SE, Mufson EJ. The role of nerve growth factor receptors in cholinergic basal forebrain degeneration in prodromal Alzheimer disease. J Neuropathol Exp Neurol. 2005 Apr;64(4):263–272. doi: 10.1093/jnen/64.4.263. [DOI] [PubMed] [Google Scholar]
- 136.Everitt BJ, Robbins TW. Central cholinergic systems and cognition. Annu Rev Psychol. 1997;48:649–684. doi: 10.1146/annurev.psych.48.1.649. [DOI] [PubMed] [Google Scholar]
- 137.Eyjolfsdottir H, Eriksdotter M, Linderoth B, et al. Targeted delivery of nerve growth factor to the cholinergic basal forebrain of Alzheimer’s disease patients: application of a second-generation encapsulated cell biodelivery device. Alzheimer's Research & Therapy. 2016;8:30. doi: 10.1186/s13195-016-0195-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Grothe M, Heinsen H, Teipel SJ. Atrophy of the cholinergic Basal forebrain over the adult age range and in early stages of Alzheimer's disease. Biol Psychiatry. 2012 May 01;71(9):805–813. doi: 10.1016/j.biopsych.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sofroniew MV, Howe CL, Mobley WC. Nerve growth factor signaling, neuroprotection, and neural repair. Annu Rev Neurosci. 2001;24:1217–1281. doi: 10.1146/annurev.neuro.24.1.1217. [DOI] [PubMed] [Google Scholar]
- 140.Li Y, Holtzman DM, Kromer LF, et al. Regulation of TrkA and ChAT expression in developing rat basal forebrain: evidence that both exogenous and endogenous NGF regulate differentiation of cholinergic neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1995 Apr;15(4):2888–2905. doi: 10.1523/JNEUROSCI.15-04-02888.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Braak H, Sandmann-Keil D, Gai W, Braak E. Extensive axonal Lewy neurites in Parkinson's disease: a novel pathological feature revealed by alpha-synuclein immunocytochemistry. Neuroscience letters. 1999 Apr 09;265(1):67–69. doi: 10.1016/s0304-3940(99)00208-6. [DOI] [PubMed] [Google Scholar]
- 142.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997 Aug 28;388(6645):839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 143.Eisbach SE, Outeiro TF. Alpha-synuclein and intracellular trafficking: impact on the spreading of Parkinson's disease pathology. J Mol Med (Berl) 2013 Jun;91(6):693–703. doi: 10.1007/s00109-013-1038-9. [DOI] [PubMed] [Google Scholar]
- 144.Sung JY, Kim J, Paik SR, Park JH, Ahn YS, Chung KC. Induction of neuronal cell death by Rab5A-dependent endocytosis of alpha-synuclein. The Journal of biological chemistry. 2001 Jul 20;276(29):27441–27448. doi: 10.1074/jbc.M101318200. [DOI] [PubMed] [Google Scholar]
- 145.Fang F, Yang W, Florio JB, et al. Synuclein impairs trafficking and signaling of BDNF in a mouse model of Parkinson's disease. Sci Rep. 2017 Jun 20;7(1):3868. doi: 10.1038/s41598-017-04232-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhao Z, Sagare AP, Ma Q, et al. Central role for PICALM in amyloid-beta blood-brain barrier transcytosis and clearance. Nature neuroscience. 2015 Jul;18(7):978–987. doi: 10.1038/nn.4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Baig S, Joseph SA, Tayler H, et al. Distribution and expression of picalm in Alzheimer disease. Journal of neuropathology and experimental neurology. 2010 Oct;69(10):1071–1077. doi: 10.1097/NEN.0b013e3181f52e01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Diggins NL, Webb DJ. APPL1 is a multifunctional endosomal signaling adaptor protein. Biochem Soc Trans. 2017 Jun 15;45(3):771–779. doi: 10.1042/BST20160191. [DOI] [PMC free article] [PubMed] [Google Scholar]