

Abstract
The rapid emergence of Gram-negative ‘superbugs’ has become a significant threat to human health globally and polymyxins become a last-line therapy for these very problematic pathogens. Polymyxins exhibit their antibacterial killing by the initial interaction with lipid A in Gram-negative bacteria. Polymyxin resistance can be mediated by phosphoethanolamine (PEA) modification of lipid A that abolishes the initial electrostatic interaction with polymyxins. Both chromosome-encoded (e.g. EptA, EptB and EptC) and plasmid-encoded PEA transferases (e.g. MCR-1 and MCR-2) were reported in Gram-negative bacteria; however, their sequence and functional heterogeneity remain unclear. Here, we report a comparative analysis of PEA transferases across ten clinically relevant Gram-negative bacteria species using multiple sequence alignment and phylogenetic analysis. Our results show that the pairwise identities among chromosome-mediated EptA, EptB and EptC from E. coli are low, and EptA shows the highest similarity with MCR-1/2. Among PEA transferases from representative strains of ten clinically relevant species, the catalytic domain is more conserved compared to the transmembrane domain. Particularly, PEA acceptor sites and zinc binding pockets show high conservation among different species, indicating their potential importance for PEA transferase function. The evolutionary relationship of MCR-1/2 and EptA from ten selected bacterial species was evaluated by phylogenetic analysis. Cluster analysis illustrates that 325 EptA from 275 strains of ten species within each individual species are highly conserved, whereas the inter-species conservation is low. Our comparative analysis provides key bioinformatic information to better understand the mechanism of polymyxin resistance via PEA modification of lipid A.
Keywords: Multi-drug resistance, Polymyxin resistance, Phosphoethanolamine transferase, Multiple sequence alignment, Phylogenetic analysis
1. Introduction
The ‘ESKAPE’ pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter spp.) have presented a significant threat to the global public health [1]. These ‘superbugs’ can cause life-threatening nosocomial infections and most of them are multidrug-resistant (MDR) [2]. The rapid evolution and dissemination of antibiotic resistance among pathogenic bacteria are outpacing the discovery and development of novel antibiotics [3]. Polymyxins (e.g. polymyxin B and colistin) display promising bactericidal activity against the vast majority of Gram-negative bacteria, and are the last-resort antibiotics to treat infections of MDR Gram-negative pathogens [4]. However, along with the extensive use of polymyxins, in particular in the agriculture sector, the concern of potential rapid spread of polymyxin resistance has been raised [5]. Alarmingly, the accumulation of reports on plasmid-borne mcr genes (e.g. mcr-1 and mcr-2 encoding PEA transferases MCR-1/2) indicates the potential for rapid dissemination of polymyxin resistance [6, 7]. Serious infections caused by polymyxin-resistant ‘superbugs’ are very likely untreatable due to resistance to all currently available antibiotics.
The detailed mechanism of antibacterial activity of polymyxins is unknown. Our current understanding involves the initial interaction of polymyxins with lipid A of Gram-negative bacteria, permeabilising the outer and inner membranes, and resulting in cell death [8]. Several other mechanisms have been proposed, such as ribosome binding [9], prevention of cell division [10], and inhibition of bacterial respiration [11]. Bacteria have developed a variety of polymyxin resistance mechanisms, including intrinsic resistance and acquired resistance. For instance, Proteus mirabilis, Serratia marcescens and Burkholderia cepacia are naturally resistant to polymyxins [12]; whereas some strains from P. aeruginosa, A. baumannii and K. pneumoniae can develop resistance in response to cationic antimicrobial peptides (including polymyxins) [13]. The mechanisms of acquired polymyxin resistance include lipopolysaccharide (LPS) modifications to reduce its net negative charge [14], increased drug efflux [15], and loss or functional inactivation of porins [16]. The most well studied resistance mechanism thus far is via modifications of the lipid A moiety of LPS [17]. The initial binding of polymyxins on lipid A mainly depends on the electrostatic interaction between positively charged diaminobutyric acid (Dab) residues of polymyxins and the negatively charged phosphate groups on lipid A in the outer leaflet of the bacterial outer membrane [18]. Addition of positively charged chemical residues (e.g. phosphoethanolamine [PEA] and 4-amino-4-deoxy-L-arabinose [L-Ara4N]) to lipid A abolishes the interaction with polymyxins, and causes the development of resistance.
Recently, mcr-1 and mcr-2 were discovered on plasmids in Escherichia coli, Salmonella enterica and K. pneumoniae [19]. Both mcr genes encode PEA transferases (MCR-1/2) that catalyse the addition of phosphoethanolamine to a phosphate of lipid A in Enterobacteriaceae. In several Gram-negative bacteria such as Neisseria meningitides and A. baumannii, chromosome-encoded EptA (formerly known as PmrC) is responsible for the modification of lipid A with PEA [19]. In addition to lipid A PEA transferases, many Gram-negative bacteria have modifications on different sites of LPS that are catalysed by different PEA transferases [20]. EptB and EptC (CptA) can modify the 3-deoxy-d-manno-oct-2-ulosonate (Kdo II) of the inner core and the O-6 of L-glycero-D-manoheptose I of the LPS, respectively [21]. Among these enzymes, EptA, EptB and EptC are chromosome-mediated PEA transferases; while MCR-1 and MCR-2 are plasmid-mediated, potentially enabling the resistance widespread via horizontal gene transfer. There is lack of conservation and evolutionary investigations on different types of PEA transferases across multiple Gram-negative bacterial species. Here we conducted a computational study of PEA transferases associated with polymyxin resistance across clinically relevant Gram-negative species. This study provides key mechanistic information on polymyxin resistance due to the LPS modification by PEA transferases across multiple Gram-negative bacteria.
2. Material and Methods
2.1. Selection of Gram-negative bacteria and collection of PEA transferase sequences
To cover a wide range of Gram-negative bacteria (including intrinsically polymyxin-resistant and polymyxin-susceptible), ten clinically relevant species with PEA-modified LPS were selected, including N. meningitides, Neisseria gonorrhoeae, E. coli, Shigella flexneri, S. enterica, K. pneumoniae, P. aeruginosa, A. baumannii, Vibrio cholerae and Helicobacter pylori. Table 1 provides additional information on the selected bacteria and their representative strains (collected based on the annotations from NCBI Genome Database) which were ranked based upon their taxonomic order. The noted pathogenicity and polymyxin MICs of the representative strains were extracted from the literature. All types of PEA transferases (i.e. EptA, EptB, EptC, MCR-1 and MCR-2) were retrieved from NCBI RefSeq database [22] and/or KEGG Orthology database [23]. Appendix Text S1 provides the list of selected strains of each species and their full-length EptA sequences in the FASTA format.
Table 1.
Selected bacteria and their representative strains for the bioinformatic analysis.
| Source species | Taxonomy (class and order) | Representative strain | Pathogenicity (Disease) | Polymyxin B MIC (μg/mL)* | Reference |
|---|---|---|---|---|---|
| Neisseria meningitides | Betaproteobacteria, Neisseriales | MC58 | Pathogen (epidemic meningitis) | ≥512 | [20],[38] |
| Neisseria gonorrhoeae | FA 1090 | Pathogen (gonorrhoea) | ≥100 | [20],[38] | |
| Escherichia coli | Gammaprotebacteria, Enterobactriales | K-12 MG1655 | Non-pathogenic | 0.25 | [12],[20],[39] |
| Shigella flexneri | 2a str. 301 | Opportunistic pathogen (diarrhoea) | 0.5 | [20],[39] | |
| Salmonella enterica | subsp. enterica serovar Typhimurium LT2 | Pathogen (gastroenteritis) | 0.5 | [12],[20],[39] | |
| Klebsiella pneumoniae | subsp. pneumoniae HS11286 | Pathogen (pneumonia) | ≤0.5 | [12],[39] | |
| Pseudomonas aeruginosa | Gammaprotebacteria, Pseudomonadales | PAO1 | Opportunistic pathogen (pneumonia) | 0.5 | [12] |
| Acinetobacter baumannii | ATCC 17978 | Pathogen (pneumonia) | 1 | [12] | |
| Vibrio cholerae | Gammaprotebacteria, Vibrionaceae | O1 biovar El Tor N16961 | Pathogen (cholera) | >96 | [20],[35] |
| Helicobacter pylori | Epsilonproteobacteria, Campylobacterales | 26695 | Pathogen (gastric and duodenal ulcers) | >250 | [20],[36] |
PMB: polymyxin B; MIC: Minimum inhibitory concentration. MICs of Pseudomonas aeruginosa PAO1 and Acinetobacter baumannii ATCC 17978 were based on our unpublished data.
2.2. Multiple sequence alignment
We employed Clustal Omega [24] for multiple sequence alignment (MSA) and the conservation among different PEA transferases was examined based on protein sequences. Parameters used for performing MSA were set by default of Clustal Omega. To enhance the visualisation of the MSA, Jalview [25] was utilised to highlight the conserved sequence motifs and key amino acids. To explore the sequence conservation of PEA transferases which have different active sites on LPS, we first performed the MSA of 5 PEA transferases including MCR-1 in E. coli SHP45, MCR-2 in E. coli KP37 (first identified in these two strains [6, 7]), EptA, EptB and EptC in E. coli K-12 MG1655 (a reference strain). Sequence identities were calculated via the SIAS webserver (http://imed.med.ucm.es/Tools/sias.html). Subsequently, the MSA of MCR-1/2 and EptA from ten species was conducted to compare the sequence conservation across polymyxin-susceptible and intrinsically polymyxin-resistant bacteria.
2.3. Phylogenetic analysis
To evaluate the evolutionary relationship and distance of the PEA transferases among different Gram-negative bacteria, MEGA 7 software was employed to construct a phylogenetic tree with the MSA of the representative EptA together with MCR-1/2 based on the maximum likelihood method [26]. Confidence values for branches and nodes of the resulting tree were validated by bootstrap analysis with 1,000 replicates to ensure appreciable reliability [27].
2.4. Cluster analysis of EptA from different species and strains
The original dataset containing ten Gram-negative bacteria was further augmented by adding EptA sequences from all available strains based on the classification of KEGG Orthology database. Pairwise identities among the 325 EptA sequences from different bacteria and strains together with MCR-1/2 sequences were calculated using the Basic Local Alignment Search Tool (BLAST) [28]. Hierarchical cluster analysis based on the pairwise identity values was conducted using R programming package for classifying the PEA transferases and comparing the sequence similarity among the strains from the same and different Gram-negative bacterial species. A heat map was plotted to visualise the hierarchical cluster analysis results.
3. Results and Discussion
3.1. Sequence conservation among chromosome-encoded EptA, EptB, EptC and plasmid-encoded MCR-1/2 in E. coli
MSA was performed to investigate the sequence conservation of PEA transferases in E. coli (Fig. 1A) which have different active sites on LPS. PEA transferases consist of two major domains, the N-terminal transmembrane domain and the C-terminal catalytic domain [29]. Despite the availability of recently published catalytic domain structures of EptA from Neisseria meningitidis [30] and MCR-1 [29, 31–33], the structure of any PEA transferase has not been completely solved. Hence, in the present study we conducted sequence conservation and evolutionary analysis using the protein sequences of PEA transferases. MSA of both chromosome- and plasmid-encoded PEA transferases (Fig. 1A) shows higher conservation in the catalytic domain (mean pairwise sequence identity of ~35%), compared to the transmembrane domain (mean pairwise sequence identity of ~26%). Across both chromosome- and plasmid-encoded PEA transferases, several conserved sites were identified by MSA based on the MCR-1 sequence, including Glu246, Thr285, His395, Asp465, His466 and His478. The phosphorylated site Thr285 is important to the catalytic function of plasmid-encoded MCR-1 and acts as the acceptor for the PEA group during the phosphate transfer reaction [6]. Thr285 is conserved among MCR-1, MCR-2, EptA and EptB (Fig. 1A), which may indicate that these four enzymes bind to PEA in a similar mode. In MCR-1, two zinc-binding pockets have been identified, Glu246-Thr285-Asp465-His466 and His395-His478 [33]. A recent study showed that depletion of zinc ions from culturing media increased the susceptibility of MCR-1-positive E. coli to colistin (MIC decreased from 2 μg/mL to 0.5 μg/mL), indicating zinc-binding is vital for MCR-1 activity [31]. The MSA (Fig. 1A) clearly demonstrates that within the first zinc-binding pocket residues (Glu246-Thr285-Asp465-His466), three residues (i.e. Glu246, Asp465 and His466) are highly conserved across the sequences of all five types of chromosome- and plasmid-encoded PEA transferases from E. coli. In the second zinc-binding pocket, His478 is not conserved in EptC but across all other four types of PEA transferases in E. coli [33].
Fig. 1.

(A) Multiple sequence alignment (MSA) of different PEA transferases selected from representative strains of E. coli, including MCR-1, MCR-2, EptA, EptB and EptC. The residues are coloured by their degree of conservation. The darker the colour is, the more conserved the amino acid is. Black lines underneath the alignment represent conserved motifs among MCR-1, MCR-2 and Ec_EptA but not homologous among Ec_EptB or Ec_EptC; while cyan lines under the alignment represent homologous motifs among MCR-1, MCR-2, Ec_EptA and Ec_EptB but not in Ec_EptC. Abbreviations and relevant accession numbers listed in Fig. 1 are as follows: MCR-1 (from the plasmid in E. coli SHP45, WP_049589868.1); MCR-2 (from the plasmid in E. coli KP37, WP_065419574.1); Ec_EptA (in E. coli K-12 MG1655, NP_418538.2); Ec_EptB (in E. coli K-12 MG1655, NP_418002.2) and Ec_EptC (in E. coli K-12 MG1655, NP_418390.1). (B) Heatmap of pairwise identities among MCR-1, MCR-2, EptA, EptB and EptC based on the sequence BLAST.
The pairwise identities among the three chromosome-encoded PEA transferases EptA, EptB and EptC are low (17–26%; Fig. 1B); while the plasmid-encoded PEA transferases MCR-1 and MCR-2 share 81% identity and the major sequence differences are seen in the N-terminus. MCR-1 and MCR-2 share high sequence identities (both ~33%) with EptA, probably because they all transfer PEA to the same active site, lipid A. In contrast, EptB and EptC show low sequence identities with MCR-1 and MCR-2 (16–24%); this is not unexpected as EptB and EptC transfer PEA to the outer 3-deoxy-D-manno-oct-2-ulosonate (Kdo II) and the heptose residues, respectively, which are different from lipid A PEA transferases [20]. Moreover, EptC shares the lowest similarity with the other PEA transferases, probably due to the fact that EptC catalyses multiple reactions related to resistance to cationic antimicrobial peptides and bacterial motility [34]. For instance, in Campylobacter jejuni, four enzymatic targets of EptC have been identified to date, including heptose I of the core oligosaccharide of LPS, 1- and 4′-phosphate groups of lipid A, N-linked heptasaccharides and the flagellar rod protein FlgG [34].
3.2 Sequence alignment of EptA across strains from ten Gram-negative bacteria and MCR-1/2 from E. coli
Ten clinically relevant Gram-negative bacteria that are able to modify their lipid A with PEA were selected (Table 1). As EptB and EptC cannot be found in most of the species and EptA has been reported to be more relevant to polymyxin resistance [20], in the present study we only focused on the sequence comparison of EptA from the ten species plus MCR-1/2 from E. coli. In total, 325 EptA sequences of all the strains listed in KEGG Orthology Database from the ten Gram-negative bacteria were extracted and the MSA was then constructed with the obtained EptA sequences together with MCR-1/2.
Among the ten clinically relevant Gram-negative bacteria, the C-terminal catalytic domain of EptA among different bacteria was more conserved (mean pairwise sequence identity of 50%) than the N-terminal transmembrane domain (mean pairwise sequence identity of 31%; Fig. 2). This finding is similar to the results shown in Fig. 1A that the catalytic domain is more conserved than the transmembrane domain among EptA, EptB, EptC and MCR-1/2. The higher variability in the transmembrane domain is probably because of different membrane compositions among different Gram-negative bacteria. Regarding the functional sites, the two zinc-binding pockets (i.e. Glu246-Thr285-Asp465-His466 and His395-His478) are both conserved across all the EptA of the ten Gram-negative bacteria species, MCR-1 and MCR-2 (Fig. 2). Such high conservation of zinc-binding pockets among multiple Gram-negative bacteria indicates that the zinc-binding domain plays a crucial role in lipid A modification and polymyxin resistance.
Fig. 2.

MSA of EptA, MCR-1 and MCR-2 from ten Gram-negative bacteria. The residues are coloured by their degree of conservation. The darker the colour is, the more conserved the amino acid is. MCR-1 and MCR-2 sequences are highlighted in light pink, whereas four polymyxin-resistant bacteria (i.e. Vibrio cholerae, Neisseria meningitides, Neisseria gonorrhoeae and Helicobacter pylori) are highlighted in light yellow. The following abbreviations are used to denote the bacteria in the figure, Nm: N. meningitides, Ng: N. gonorrhoeae, Ec: E. coli, Shf: Sh. flexneri, Se: S. enterica, Kp: K. pneumoniae, Pa: P. aeruginosa, Ab: A. baumannii, Vc: V. cholera, and Hp: H. pylori.
The EptA sequences of polymyxin-susceptible (MIC≤2 μg/mL) [5] and intrinsically polymyxin-resistant strains (MIC≥4 μg/mL) together with MCR-1 and MCR-2 were compared. Interestingly, several amino acids (i.e. Ile128, Val240, Val241, Glu310, Asn311, Asp327, Leu522 based on the MCR-1 sequence) are different among PEA transferases of intrinsically polymyxin-resistant species from those of polymyxin-susceptible species (Fig. 2).
There are two PEA transferases annotated as EptA in P. aeruginosa PAO1 in the KEGG Orthology Database, which share the sequence identify of 48%. To analyse the evolutionary relationship among the 11 EptA from ten bacterial species one MCR-1, and one MCR-2, a phylogenetic tree was constructed using the maximum likelihood method with MEGA 7 (Fig. 3). Notably, previous phylogenetic analysis indicated that MCR-1 is closely related to the PEA transferase from Paenibacillus sophorae, a known Gram-positive polymyxin producer. However, such a conclusion is not convincing as Pa. sophorae is a Gram-positive bacterium which lacks LPS and, therefore, transferring PEA to lipid A does not exist. In the present study, we firstly confirmed the existence of lipid A modification in the examined bacteria before the MSA and phylogenetic analysis (Table 1). As shown in Fig. 3, short phylogenetic distance is evident in the EptA from E. coli, Sh. flexneri, S. enterica and K. pneumoniae, indicating their closer evolutionary relationship compared to the other species examined. The representative strain of E. coli used in Fig. 3 is E. coli K-12 MG1655 which has only one EptA, despite that some strains of E. coli have more than one EptA enzymes. The phylogenetic distances in Fig. 3 are consistent with the taxonomical classification of E. coli, Sh. flexneri, S. enterica and K. pneumoniae, as they all belong to the same class (Gammaprotebacteria) and order (Enterobacteriales). In terms of genetic variations, MCR-1 and MCR-2 (i.e. plasmid-encoded PEA transferases conferring resistance to polymyxins) are closer to both types of EptA from P. aeruginosa than from the other examined Gram-negative bacteria. MCR-1 and MCR-2 stay in the same sub-clade within polymyxin-susceptible P. aeruginosa rather than Enterobacteriales where the MCR-1/2 were first identified. Interestingly, the phylogenetic tree shows MCR-1 and MCR-2 have a closer relationship with polymyxin-susceptible Pseudomonadales and Enterobacteriales, than intrinsically polymyxin-resistant Vibrionaceae (e.g. V. cholerae), Neisseriales (e.g. N. meningitides, N. gonorrhoeae) and Campylobacterales (e.g. H. pylori). Polymyxin B MICs of the representative strain of H. pylori 26695 and V. cholerae are >250 μg/mL and ≥512 μg/mL, respectively [35, 36], ranked as two of the most polymyxin-resistant strains in our dataset. EptA in H. pylori (belonging to Epsilonproteobacteria, Campylobacterales) and V. cholerae (belonging to Gammaproteobacteria, Vibrionaceae) have the farthest phylogenetic relationship to EptA in the other Gram-negative bacteria examined.
Fig. 3.

Phylogenetic tree constructed with 11 EptA and MCR-1, MCR-2 sequences by MEGA 7 based on the maximum likelihood method. The scale bar corresponds to proportional length of branch presenting an amount genetic change of 0.10. The percentage bootstrap support (per 1000 replicates) was indicated by the values at each node. Bootstrap support values (%) based on 1,000 replicates are indicated by the values at each node. The number (in percentage) next to each node represents a measure of support for the node. The taxonomy and polymyxin B MICs are listed on the right side. MCR-1 and MCR-2 sequences are highlighted in light pink, whereas four polymyxin-resistant V. cholerae, N. meningitides, N. gonorrhoeae and H. pylori are highlighted in light yellow. The following abbreviations are used to denote the bacteria in the figure, Nm: N. meningitides, Ng: N. gonorrhoeae, Ec: E. coli, Shf: Sh. flexneri, Se: S. enterica, Kp: K. pneumoniae, Pa: P. aeruginosa, Ab: A. baumannii, Vc: V. cholera, and Hp: H. pylori.
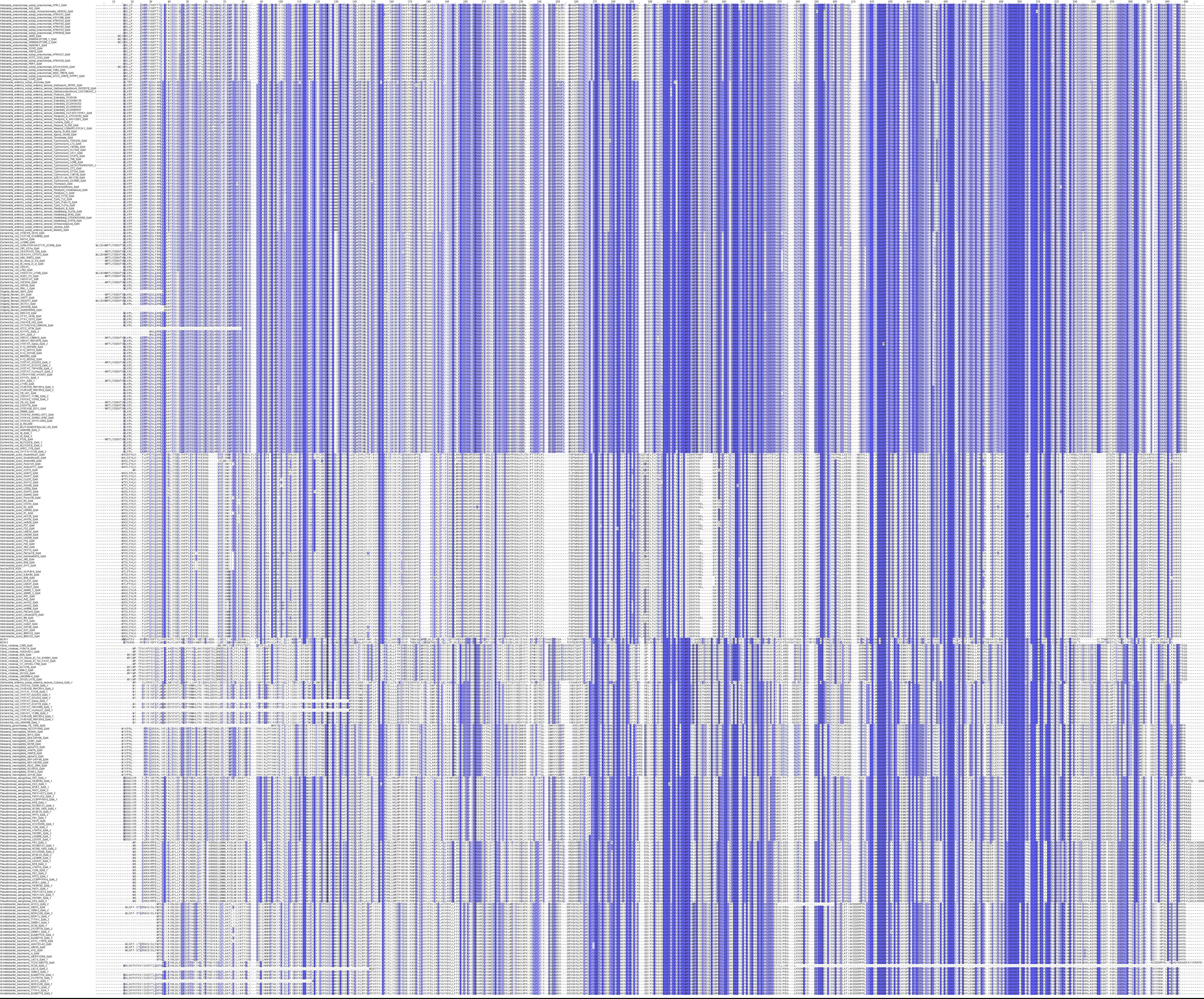
3.3. Cluster analysis based on an augmented dataset with 325 EptA from the selected ten species of Gram-negative bacteria and MCR-1/2 from E. coli
In total, 325 EptA sequences of 275 strains and 10 species from the KEGG Orthology database were employed for cluster analysis. Among them, 230 strains (83.6%) have only one eptA in their genomes while 45 strains (16.4%) have two or three different eptA genes. To examine the sequence diversity of EptA among different species, we performed hierarchical cluster analysis based on the pairwise identities to classify 325 EptA together with MCR-1/2. Overall, the 325 EptA sequences together with MCR-1 and MCR-2 were clustered into 13 groups in a heat map for convenient comparison and visualisation, based on their identity levels (Fig. 4). EptA sequences within different strains in the same species generally have identities >90%, indicating the high conservation within the same species. On the other hand, poorer inter-species conservation was demonstrated by lower pairwise identities of EptA sequences across different species (Fig. 4). EptA sequences from E. coli, S. enterica, K. pneumoniae and Sh. flexneri have higher identity values (> 60%) with each other than with other bacteria species. This is consistent with the generated phylogenetic tree and their taxonomic relationships (Fig. 3). In addition, consistent with the result of the phylogenetic tree (Fig. 3), H. pylori has the lowest similarity with the other Gram-negative bacteria examined in this study. Alignment of 325 EptA together with MCR-1/2 was performed (Appendix Fig. S2) for intuitive visualisation of the conservation among all the strains. By the time of submission of our revised manuscript, we noticed that a very recent study reported that MCR-1 is evolutionarily close (identity of 59–64%) to EptA from Moraxella species [37]. However, rather than the multiple sequence alignment results purely on Moraxella in the recent report [37], our study is featured by three major aspects: (1) large-scale sequence alignment across multiple species and phylogenetic analyses were conducted; (2) the bioinformatic analysis was based on all major PEA transferases, i.e. EptA, EptB, EptC and MCR-1/2; and (3) conservation analysis was performed with the PEA acceptor Thr285 and zinc-binding pockets. In summary, our cluster analysis illustrates that the EptA sequences show higher conservation within the same species but are poorly conserved inter-species.
Fig. 4.

Heatmap of a hierarchical cluster analysis of the pairwise identities among 325 EptA and MCR-1 and MCR-2 sequences. The darker the colour is, the higher the identity is between the two strains. The following abbreviations are used to denote the bacteria in the figure, Nm: N. meningitides, Ng: N. gonorrhoeae, Ec: E. coli, Shf: Sh. flexneri, Se: S. enterica, Kp: K. pneumoniae, Pa: P. aeruginosa, Ab: A. baumannii, Vc: V. cholera, and Hp: H. pylori.
Conclusions
In summary, this is the first comparative study to demonstrate the evolutionary relationship of PEA transferases, including MCR-1, MCR-2 and EptA among 275 strains in 10 major Gram-negative bacteria. Our results reveal that the catalytic domain, particularly PEA acceptor sites and zinc binding pockets, is conserved. EptA within each individual species is highly conserved while the inter-species conservation is low. This study provides key evolutionary insight into PEA transferases and PEA-mediated polymyxin resistance, which may contribute to rescuing the clinical utility of this last-line therapeutic option and the discovery of novel approaches to combat potentially rapid prevalence of resistance.
Supplementary Material
{kind=link}
Highlights.
MCR-1/2 and EptA in 10 bacterial species were examined in the conservation analysis
EptA is highly conserved in each species but not inter-species
PEA acceptor site and zinc binding pocket are highly conserved in all EptA examined
Evolutionary distance of EptA is not related to intrinsic polymyxin susceptibility
Acknowledgments
Funding: J.L., T.V. and J.S. are supported by a research grant from the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (R01 AI111965). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. J.L. is an Australian National Health and Medical Research Council (NHMRC) Senior Research Fellow and T.V. is an NHMRC Career Development Fellow.
Appendix A. Supplementary data
Supplementary data associated with this article can be found in the online version.
Footnotes
Competing Interests: None
Ethical Approval: Not required.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Deslouches B, Steckbeck JD, Craigo JK, Doi Y, Burns JL, Montelaro RC. Engineered cationic antimicrobial peptides to overcome multidrug resistance by ESKAPE pathogens. Antimicrob Agents Chemother. 2015;59:1329–33. doi: 10.1128/AAC.03937-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khan SN, Khan AU. Breaking the spell: combating multidrug resistant ‘superbugs’. Front Microbiol. 2016;7:1–11. doi: 10.3389/fmicb.2016.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dickey SW, Cheung GY, Otto M. Different drugs for bad bugs: antivirulence strategies in the age of antibiotic resistance. Nat Rev Drug Discov. 2017:457–71. doi: 10.1038/nrd.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Velkov T, Roberts KD, Nation RL, Thompson PE, Li J. Pharmacology of polymyxins: new insights into an ‘old’ class of antibiotics. Future Microbiol. 2013;8:711–24. doi: 10.2217/fmb.13.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poirel L, Jayol A, Nordmann P. Polymyxins: antibacterial activity, susceptibility testing, and resistance mechanisms encoded by plasmids or chromosomes. Clin Microbiol Rev. 2017;30:557–96. doi: 10.1128/CMR.00064-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu YY, Wang Y, Walsh TR, Yi LX, Zhang R, Spencer J, et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis. 2016;16:161–8. doi: 10.1016/S1473-3099(15)00424-7. [DOI] [PubMed] [Google Scholar]
- 7.Xavier BB, Lammens C, Ruhal R, Kumar-Singh S, Butaye P, Goossens H, et al. Identification of a novel plasmid-mediated colistin-resistance gene, mcr-2, in Escherichia coli, Belgium, June 2016. Euro Surveill. 2016;21:1–6. doi: 10.2807/1560-7917.ES.2016.21.27.30280. [DOI] [PubMed] [Google Scholar]
- 8.Velkov T, Roberts KD, Nation RL, Wang JP, Thompson PE, Li J. Teaching ‘old’ polymyxins new tricks: new-generation lipopeptides targeting Gram-negative ‘superbugs’. ACS Chem Biol. 2014;9:1172–7. doi: 10.1021/cb500080r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCoy LS, Roberts KD, Nation RL, Thompson PE, Velkov T, Li J, et al. Polymyxins and analogues bind to ribosomal RNA and interfere with eukaryotic translation in vitro. Chembiochem. 2013;14:2083–6. doi: 10.1002/cbic.201300496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mortensen NP, Fowlkes JD, Sullivan CJ, Allison DP, Larsen NB, Molin S, et al. Effects of colistin on surface ultrastructure and nanomechanics of Pseudomonas aeruginosa cells. Langmuir. 2009;25:3728–33. doi: 10.1021/la803898g. [DOI] [PubMed] [Google Scholar]
- 11.Saugar JM, Alarcon T, Lopez-Hernandez S, Lopez-Brea M, Andreu D, Rivas L. Activities of polymyxin B and cecropin A-,melittin peptide CA(1-8)M(1-18) against a multiresistant strain of Acinetobacter baumannii. Antimicrob Agents Chemother. 2002;46:875–8. doi: 10.1128/AAC.46.3.875-878.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olaitan AO, Morand S, Rolain JM. Mechanisms of polymyxin resistance: acquired and intrinsic resistance in bacteria. Front Microbiol. 2014;5:643. doi: 10.3389/fmicb.2014.00643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeannot K, Bolard A, Plesiat P. Resistance to polymyxins in Gram-negative organisms. Int J Antimicrob Agents. 2017;47:526–35. doi: 10.1016/j.ijantimicag.2016.11.029. [DOI] [PubMed] [Google Scholar]
- 14.Trimble MJ, Mlynarcik P, Kolar M, Hancock RE. Polymyxin: alternative mechanisms of action and resistance. Cold Spring Harb Perspect Med. 2016;6:1–22. doi: 10.1101/cshperspect.a025288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsai MH, Wu SR, Lee HY, Chen CL, Lin TY, Huang YC, et al. Recognition of mechanisms involved in bile resistance important to halting antimicrobial resistance in nontyphoidal Salmonella. Int J Antimicrob Agents. 2012;40:151–7. doi: 10.1016/j.ijantimicag.2012.04.016. [DOI] [PubMed] [Google Scholar]
- 16.Muller C, Plesiat P, Jeannot K. A two-component regulatory system interconnects resistance to polymyxins, aminoglycosides, fluoroquinolones, and beta-lactams in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2011;55:1211–21. doi: 10.1128/AAC.01252-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baron S, Hadjadj L, Rolain JM, Olaitan AO. Molecular mechanisms of polymyxin resistance: knowns and unknowns. Int J Antimicrob Agents. 2016;48:583–91. doi: 10.1016/j.ijantimicag.2016.06.023. [DOI] [PubMed] [Google Scholar]
- 18.Velkov T, Thompson PE, Nation RL, Li J. Structure-activity relationships of polymyxin antibiotics. J Med Chem. 2010;53:1898–916. doi: 10.1021/jm900999h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cabello FC, Godfrey HP. Comment on: Transferable resistance to colistin: a new but old threat. J Antimicrob Chemother. 2017;72:636–7. doi: 10.1093/jac/dkw432. [DOI] [PubMed] [Google Scholar]
- 20.Needham BD, Trent MS. Fortifying the barrier: the impact of lipid A remodelling on bacterial pathogenesis. Nat Rev Microbiol. 2013;11:467–81. doi: 10.1038/nrmicro3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salazar J, Alarcon M, Huerta J, Navarro B, Aguayo D. Phosphoethanolamine addition to the heptose I of the pipopolysaccharide modifies the inner core structure and has an impact on the binding of polymyxin B to the Escherichia coli outer membrane. Arch Biochem Biophys. 2017;620:28–34. doi: 10.1016/j.abb.2017.03.008. [DOI] [PubMed] [Google Scholar]
- 22.O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44:D733–45. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40:D109–14. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, et al. Analysis tool web services from the EMBL-EBI. Nucleic Acids Res. 2013;41:W597–600. doi: 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–91. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao R, Hu Y, Li Z, Sun J, Wang Q, Lin J, et al. Dissemination and mechanism for the MCR-1 colistin resistance. PLoS Pathog. 2016;12:e1005957. doi: 10.1371/journal.ppat.1005957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boratyn GM, Camacho C, Cooper PS, Coulouris G, Fong A, Ma N, et al. BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 2013;41:W29–33. doi: 10.1093/nar/gkt282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stojanoski V, Sankaran B, Prasad BV, Poirel L, Nordmann P, Palzkill T. Structure of the catalytic domain of the colistin resistance enzyme MCR-1. BMC Biol. 2016;14:81. doi: 10.1186/s12915-016-0303-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anandan A, Evans GL, Condic-Jurkic K, O’Mara ML, John CM, Phillips NJ, et al. Structure of a lipid A phosphoethanolamine transferase suggests how conformational changes govern substrate binding. Proc Natl Acad Sci U S A. 2017;114:2218–23. doi: 10.1073/pnas.1612927114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hinchliffe P, Yang QE, Portal E, Young T, Li H, Tooke CL, et al. Insights into the mechanistic basis of plasmid-mediated colistin resistance from crystal structures of the catalytic domain of MCR-1. Sci Rep. 2017;7:39392. doi: 10.1038/srep39392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu M, Guo J, Cheng Q, Yang Z, Chan EW, Chen S, et al. Crystal structure of Escherichia coli originated MCR-1, a phosphoethanolamine transferase for colistin resistance. Sci Rep. 2016;6:38793. doi: 10.1038/srep38793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma G, Zhu Y, Yu Z, Ahmad A, Zhang H. High resolution crystal structure of the catalytic domain of MCR-1. Sci Rep. 2016;6:39540. doi: 10.1038/srep39540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cullen TW, O’Brien JP, Hendrixson DR, Giles DK, Hobb RI, Thompson SA, et al. EptC of Campylobacter jejuni mediates phenotypes involved in host interactions and virulence. Infect Immun. 2013;81:430–40. doi: 10.1128/IAI.01046-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hankins JV, Madsen JA, Giles DK, Brodbelt JS, Trent MS. Amino acid addition to Vibrio cholerae LPS establishes a link between surface remodeling in Gram-positive and Gram-negative bacteria. Proc Natl Acad Sci U S A. 2012;109:8722–7. doi: 10.1073/pnas.1201313109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tran AX, Whittimore JD, Wyrick PB, McGrath SC, Cotter RJ, Trent MS. The lipid A 1-phosphatase of Helicobacter pylori is required for resistance to the antimicrobial peptide polymyxin. J Bacteriol. 2006;188:4531–41. doi: 10.1128/JB.00146-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kieffer N, Nordmann P, Poirel L. Moraxella species as potential sources of MCR-like polymyxin resistance determinants. Antimicrob Agents Chemother. 2017;61:1–22. doi: 10.1128/AAC.00129-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lewis LA, Choudhury B, Balthazar JT, Martin LE, Ram S, Rice PA, et al. Phosphoethanolamine substitution of lipid A and resistance of Neisseria gonorrhoeae to cationic antimicrobial peptides and complement-mediated killing by normal human serum. Infect Immun. 2009;77:1112–20. doi: 10.1128/IAI.01280-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Falagas ME, Kasiakou SK. Colistin: the revival of polymyxins for the management of multidrug-resistant Gram-negative bacterial infections. Clin Infect Dis. 2005;40:1333–41. doi: 10.1086/429323. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.