

Abstract
β– actin plays key roles in cell migration. Our previous work demonstrated that β– actin in migratory non-muscle cells is N-terminally arginylated and that this arginylation is required for normal lamellipodia extension. Here we examined the function of β– actin arginylation in cell migration. We found that arginylated β– actin is concentrated at the leading edge of lamellipodia and that this enrichment is abolished after serum starvation as well as in contact-inhibited cells in confluent cultures, suggesting that arginylated β– actin at the cell leading edge is coupled to active migration. Arginylated actin levels exhibit dynamic changes in response to cell stimuli, lowered after serum starvation and dramatically elevating within minutes after cell stimulation by re-addition of serum or lysophosphatidic acid (LPA). These dynamic changes require active translation and are not seen in confluent contact-inhibited cell cultures. Microinjection of arginylated actin antibodies into cells severely and specifically inhibits their migration rates. Together, these data strongly suggest that arginylation of β– actin is a tightly regulated dynamic process that occurs at the leading edge of locomoting cells in response to stimuli and is integral to the signaling network that regulates cell migration.
Keywords: arginylation, actin, cell migration, posttranslational modifications, lamellipodia dynamics
Graphical abstract
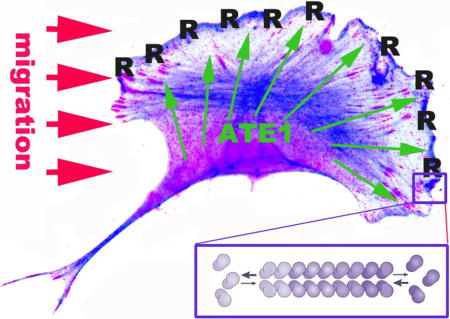
Introduction
β– actin is an essential, ubiquitously expressed protein and the major constituent of the actin cytoskeleton in migratory cell types. During cell migration, actin interacts with multiple proteins 1 and undergoes a number of posttranslational modifications 2, however the underlying regulatory mechanisms that drive cell migration and control actin dynamics at the cell leading edge are not fully understood.
In our previous work we found that β– actin, unlike other actin isoforms, undergoes N-terminal arginylation mediated by arginyltransferase Ate1 3. This arginylation is tightly co-translationally regulated via unique features of β– actin coding sequence that set it apart from the closely related equally abundant non-muscle γ– actin, which is not N-terminally arginylated 4. Lack of arginylation leads to impairments in lamellipodia formation and cell migration 3,5, the effects that can be at least partially rescued by expression of arginylated actin in cells, however the exact contribution of actin arginylation to the arginylation-dependent impairments in cell migration are unknown. Moreover, it has never been studied whether actin arginylation in cells is segregated into a specific cell area or a subset of the total actin pool, and whether it represents a stationary mark or a dynamic modification that can mediate cell responses to stimuli.
Here we used a newly developed arginylated β– actin antibody to address these questions and analyze the distribution and dynamics of actin arginylation during cell migration. Our results indicate that arginylation represents a dynamic actin modification that likely mediates cellular responses to migratory stimuli at the cell leading edge.
Results
Arginylated β- actin localizes to the leading edge of migrating cells
Previous studies of actin arginylation in cells have been difficult, due to the lack of the available tools that would enable reliable detection of endogenous arginylated actin in cells. Development of specific arginylation antibodies has been generally challenging due to the fact that the posttranslationally added Arg is too similar to the Arg natively present in multiple proteins in vivo. However, recently, EMD Millipore has successfully raised new polyclonal peptide antibodies against N-terminally arginylated β– actin (ABT264). We used these antibodies to characterize β– actin arginylation in cells. The specific batch of antibodies used for this project selectively recognize baculovirus-expressed N-terminally arginylated actin and do not cross-react with β– actin lacking N-terminal arginylation (R–and M-actin, respectively, Fig. 1A), even though they still retain some residual cross-reactivity with an actin-sized band in arginylation deficient cell extracts (prepared from mouse embryonic fibroblasts with knockout of arginyltransferase Ate1 as described in 3) (Fig. 1B). Moreover, these antibodies pull down a residual band from Ate1 knockout cell extracts that shows cross-reactivity with pan-actin antibodies but not with the β– actin antibodies (Fig. 1C).
Figure 1. Characterization of arginylated actin (R-actin) antibodies.

A. Baculovirus-expressed non-arginylated and N-terminally arginylated actin (M-and R-actin, respectively) fractionated on the SDS gel next to wild type cell lysate (WT lysate), Coomassie-stained (left) and probed with antibodies to R-actin (middle) and β– actin (right). B. Diluted lysates from wild type (WT) and Ate1 knockout (KO) cells probed with the antibodies to arginyltransferase Ate1, R-actin, and pan-actin. R-actin antibodies recognize a residual actin-sized band in KO cells, which can be titered out by dilution. C. Immunoprecipitates from WT and KO cells using R-actin antibodies. A ~43 kDa actin band is prominent in WT precipitates and also faintly visible in the KO. This band is faintly recognized by the R-actin and pan-actin, but not the beta actin antibodies. Arrow indicates the level at which the membrane was cut to eliminate the IgG heavy chain band and enable the detection of the actin bands without the secondary antibody background.
We first tested the overall distribution of arginylated β– actin (R-actin) in migratory mouse embryonic fibroblasts using non-detergent cell extraction with acetone (as described, e.g., in 6). This staining revealed a narrow zone at the very edge of the lamellipodia (Fig. 2A, top right and 2B, top left), in addition to the diffuse perinuclear staining that did not appear to coincide with the majority of the actin filament distribution in that area. Notably, this leading edge R-actin staining disappeared after cell extraction with Triton X-100 (Fig. 2A, bottom right), while being retained after methanol fixation (Fig 2B, bottom left) suggesting that R-actin localization at the cell leading edge is detergent labile. Notably, residual staining in these regions, as well as faint leading edge staining, could also be seen in some of the Ate1 knockout cells Fig. 2B, right panels). While this staining was considerably weaker in these cells (as seen by comparison left and right panels in Fig. 2B, which have been scaled to the same intensity levels), in combination with the western blot results shown in Fig. 1 this result suggest that residual level of arginylated β– actin may still be retained in these cells.
Figure 2. R-actin antibodies show prominent reactivity within a narrow detergent-labile zone at the edge of the lamellipodia.
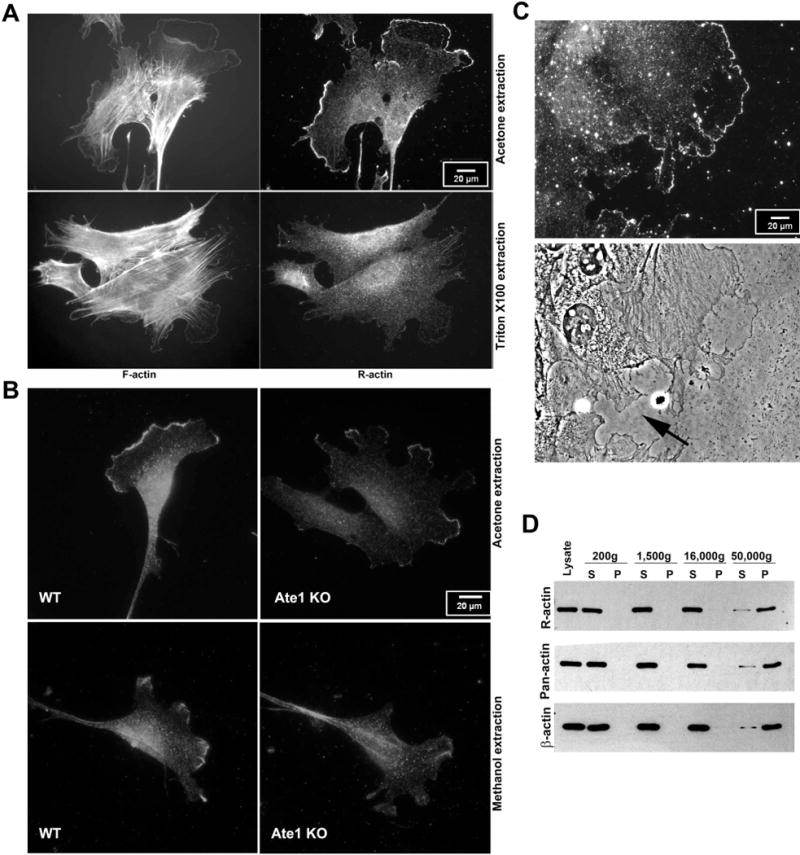
A. Immunofluorescence staining of mouse embryonic fibroblasts grown in normal serum conditions using phalloidin (F-actin) and R-actin antibodies with acetone (top) or detergent (bottom) extraction. Acetone-extracted cells show prominent antibody reactivity with a narrow zone at the leading edge, absent after Triton X100 extraction. B. Immunofluorescence staining of acetone (top) and methanol (bottom) extracted wild type (WT) and Ate1 knockout (KO) cells using R-actin antibodies. Images are scaled to equal gray levels to enable direct comparison of the staining in WT and KO cells. C. R-actin staining (top) and phase contrast (bottom) images of WT cells migrating into the scratch wound. The cell edges facing the wound exhibit prominent leading edge staining, while the cell edges facing away from the wound and partially contact-inhibited (indicated by arrow in the bottom image) show no such leading edge enrichment. A-C scale bars, 20 μm. D. western blots of whole cell lysates fractionated by differential centrifugation steps (identified by g-force on the top) to reveal actin distribution between monomeric and polymeric fraction. S, supernatant, P, pellet.
To test whether the staining at the edge of the lamellipodia region correlates with active migration, we stained cells at the edge of the scratch wound, where the lamellipodia facing outward into the wound areas are formed by the cells that are actively locomoting, while other lamellipodia-like structures inward to the edge of the monolayer can be transiently contact-inhibited and not undergoing active migration (Fig. 2C). R-actin staining was prominently seen at the edge of the lamellipodia facing the wound, while being absent in the “inward” regions where cells were facing each other and thus were unlikely to migrate in that direction (an example is indicated by arrow in Fig. 2C), suggesting that R-actin leading edge localization likely coincides with active migration.
Given that this leading edge R-actin staining is detergent-labile, it is possible that it preferentially targets actin monomers or short oligomers that can be washed out after Triton X-100 extraction. To test this, we performed differential fractionation of cell extracts by sequential centrifugation and tested the presence of R-actin in the supernatant and pellet from each step by western blotting (Fig. 2D). In this experiment, R-actin appeared to be uniformly distributed in the G– and F-actin pool, similar to the distribution of β– actin and total actin. Thus, the overall R-actin reactivity does not appear to be biased to either actin polymer or monomer. However, it is still possible that the detergent-labile zone at the edge of the cells represents a pool of monomeric/oligomeric R-actin.
Arginylated β– actin enrichment at the leading edge is diminished after serum starvation
Our previous work showed that arginylated actin facilitates cell leading edge formation during migration 3. To test if β– actin arginylation correlates with the cells’ active migratory state, we used serum starvation, a treatment that inhibits cell migration and places cells into quiescent state without strongly affecting their overall morphology or lamellipodial activity (supplemental videos 1–4). We then compared the R-actin staining in cells before and after serum starvation.
While virtually every cell grown in normal serum at low density contained at least one area of its well-spread edge prominently highlighted with arginylated actin antibodies (Fig. 3A, left panel), after serum starvation virtually none of the cells showed such arginylated actin enrichment, even though, consistent with published studies 7, they did not undergo any apparent lamellipodia retraction and retained residual R-actin staining in other areas of the cell (Fig. 3A, middle panel and supplemental videos 1–4). Moreover, the total level of arginylated actin in cell extracts, grown to ~30% confluency to maintain their migratory ability and prevent growth-dependent contact inhibition, also prominently dropped, compared to pre-starvation state, as seen in total quantifications by western blots (Fig. 3A, right panel). Thus, arginylated actin levels depend on cells’ migratory and metabolic state, and inhibition of cell migration by serum starvation abolishes this leading edge arginylated actin staining and appears to lower the overall levels of arginylated actin in the cell.
Figure 3. Arginylated actin levels dynamically respond to stimuli.
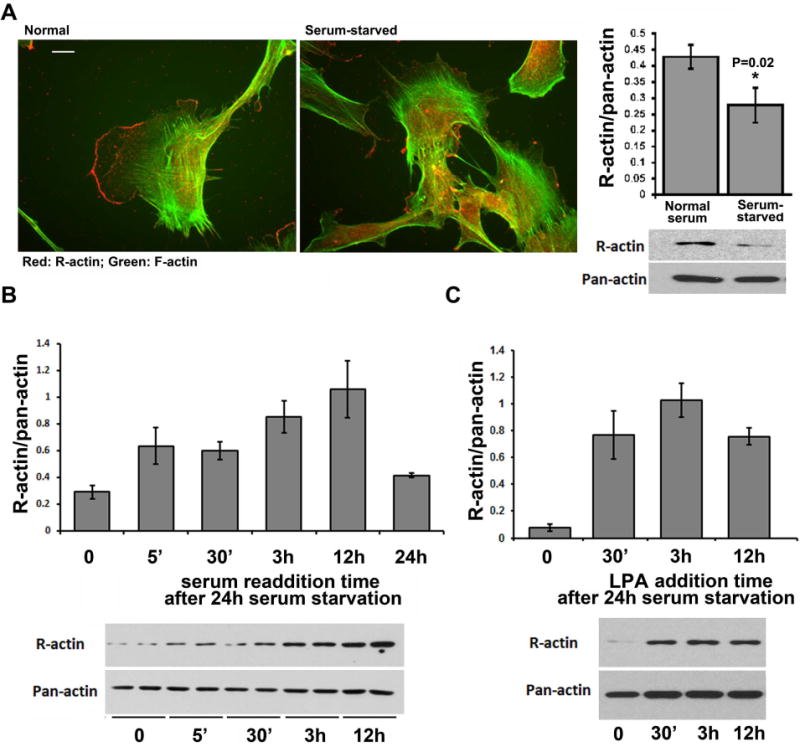
A, immunofluorescence staining of cultured mouse embryonic fibroblasts using R-actin antibodies (red) and fluorescent phalloidin (green). Scale bar, 10 μm. B, Quantifications (top) and images of representative immunoblots (bottom) of R-actin levels in cells grown under normal serum conditions, and after 24 hr serum starvation. Error bars represent SEM from 15 different experiments. P-value was determined by 2-tailed T-test. C, Quantifications (top) and images of representative immunoblots (bottom) of R-actin levels in serum-starved cells stimulated by serum readdition for different intervals of time. R-actin levels are shown normalized to pan-actin signal in the same cells, which was used as a loading control. Error bars represent SEM from 6 different experiments analyzed in duplicates. D, Quantifications (top) and images of representative immunoblots (bottom) of R-actin levels in serum-starved cells stimulated by LPA for different intervals of time. R-actin levels are shown normalized to pan-actin signal in the same cells, which was used as a loading control. Error bars represent SEM from 3 different experiments.
Arginylated β– actin levels are dynamically regulated in response to stimuli
The disappearance of arginylated actin staining at the cell leading edge and its overall reduction in cells after serum starvation suggests that the levels and the leading edge localization of arginylated β– actin are regulated events that can respond to intracellular stimuli. Such regulation could in principle be majorly achieved by redistribution of arginylated actin between different areas of the cell during serum starvation and stimulation, or by changes in arginylated actin levels due to its de novo arginylation and recycling. To test this possibility and to determine the dynamics and speed of actin arginylation-dependent response to the serum stimulation, we used western blot to measure the overall levels of arginylated actin in a ~30% confluent cell culture grown in low-serum media overnight, then stimulated by serum re-addition. We measured the overall changes of the levels of arginylated actin in total lysates from these cells at different time intervals after stimulation (Fig. 3B). Remarkably, 5 min after serum re-addition the total levels of arginylated actin in cells dramatically increased, suggesting that these cells responded to stimulation by a burst of rapid de novo actin arginylation. Arginylated actin levels in these cells continued to further increase over the next few hours, before dropping to approximate pre-starvation levels after 24 hours.
As a control experiment, we used similarly treated 100% confluent cells that undergo quiescence upon serum starvation, but cannot initiate migration after serum stimulation, due to confluency-dependent contact inhibition. In these cells, arginylated actin levels did not reduce after serum starvation and did not significantly increase after stimulation, confirming that the dynamics of arginylated actin levels in response to stimulation likely depends on active migration rather than on other metabolic changes in response to serum treatment (Fig. S1).
To confirm that this R-actin response is mediated via cell migration stimuli and not any other serum components, we performed a similar experiment using cell stimulation in low serum media by addition of lysophosphatidic acid (LPA), which stimulates cell migration via modulation of G-protein signaling 8–10 but does not alter the balance of nutrients or overall composition of the cell culture media. Addition of LPA to serum-starved cells under low serum conditions induced a rapid increase in R-actin levels that changed over time similarly to the serum stimulation experiment (Fig. 3C), suggesting that this R-actin response was indeed linked to cell migratory stimuli.
Thus, actin arginylation is a dynamic event that exhibits a rapid response to cell stimulation to migrate and is regulated over time.
Dynamic changes in actin arginylation require active protein synthesis
We have previously found that β– actin arginylation is coupled to its translation rate and co-translational arginylation-dependent degradation 4. Our prior work also showed that a subset of Ate1 in the cell is associated with the ribosomes, potentially making it available to participate in co-translational arginylation 11. It has been previously found that β- actin mRNA is transported to the cell leading edge, where it is likely translated on the spot to facilitate cell migration 12. Notably, sequence analysis of Ate1 mRNA also reveals a putative zipcode-binding leading edge targeting signal, suggesting that β– actin and Ate1 mRNA can potentially co-localize at the cell leading edge where their simultaneous translation may facilitate β- actin arginylation. To test this hypothesis, we used fluorescence in situ hybridization (FISH) to localize the β– actin and Ate1 mRNA in migratory wild type fibroblasts (Fig. 4A). These tests revealed that a subset of both β– actin and Ate1 mRNA localized to the cell periphery, consistent with the idea that both of these mRNAs are targeted there by specific mechanisms involving long-range mRNA transport. However, the FISH signals for these two mRNAs failed to co-localize in the same spots, suggesting that the majority of β– actin and Ate1 are unlikely to undergo simultaneous translation. It is still possible, however that leading edge-synthesized Ate1 associates with the ribosomes and aginylates the newly synthesized actin.
Figure 4. Arginylated β– actin increase after serum stimulation depends on active translation.
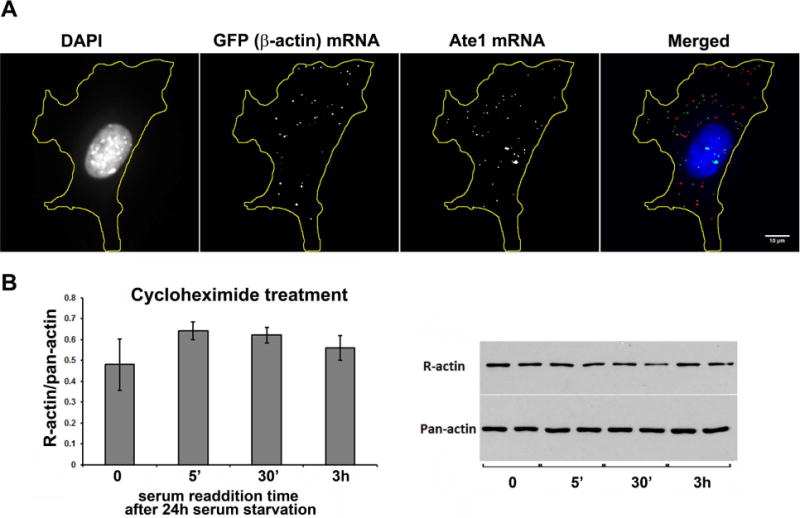
A. Fluorescence in situ hybridization (FISH) to detect the distribution of beta actin and Ate1 mRNA in cells cultured under normal serum conditions. Scale bar, 10 μm. B. Quantifications (left) and images of representative immunoblots (right) of R-actin levels in serum starved cells (0) and cells stimulated by serum readdition for different intervals of time in the presence of translation inhibitor cycloheximide (added 1 hour prior to serum readdition). R-actin levels are shown normalized to pan-actin signal in the same cells, which was used as a loading control. Error bars represent SEM from 3 different experiments analyzed in duplicates.
To test this possibility and determine whether the changes in R-actin seen after serum/LPA stimulation require active protein synthesis, we performed serum stimulation experiments similar to those shown in Fig. 3 in the presence of translation inhibitor cycloheximide (Fig. 4B). This treatment completely abolished the dynamic changes in actin arginylation levels seen in serum-stimulated cells, confirming that these changes require active translation.
Inhibition of actin arginylation inhibits cell migration
To directly test the effect of R-actin inhibition on cell migration, we used scarce cells, grown in normal serum to exhibit random migration in culture, and microinjected them with either control IgG or the antibodies to arginylated actin used in the experiments shown above. As a control, we also microinjected these antibodies into similarly prepared Ate1 knockout cells, which have been previously shown to exhibit severe leading edge defects that can be restored by reintroduction of arginylated β– actin 3. These cells migrate at much slower rates than wild type 5. We then compared the speed and distance of spontaneous cell migration in control and antibody-injected wild type and Ate1 knockout cultures (Fig. 5A).
Figure 5. Inhibition of R-actin suppresses cell migration.
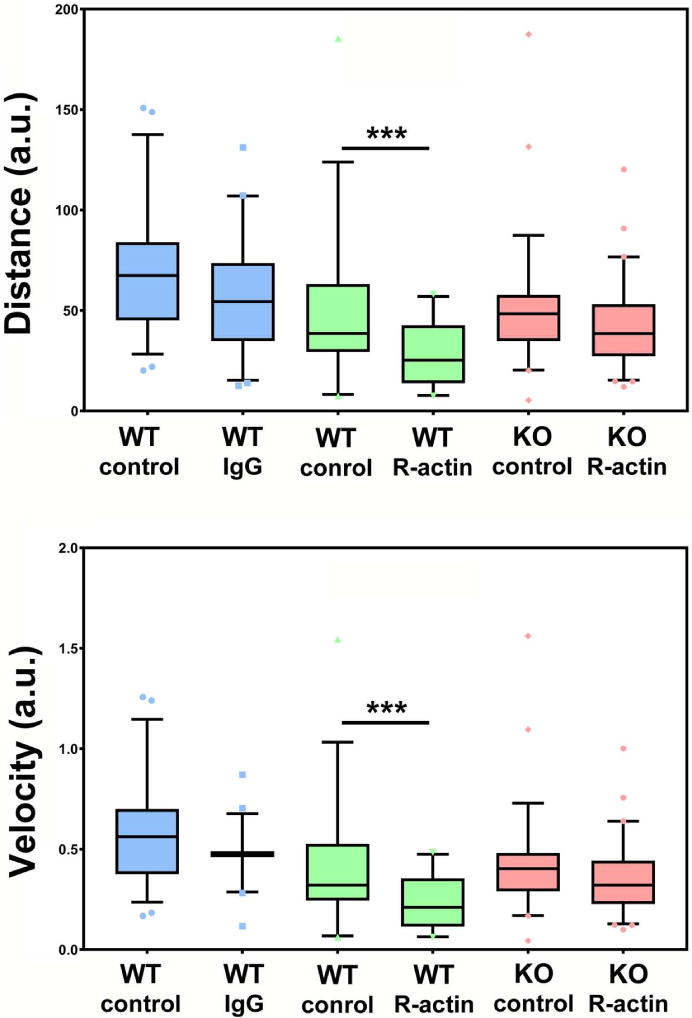
Quantifications of the migration of randomly migrating cells (bottom) over 130 min of observation, before and after injection with R-actin antibodies (Anti-R-actin) or control IgG. WT, wild type. KO, knockout. Comparisons are presented pairwise between uninjected (control) and injected (IgG or R-actin) cells migrating in the same fields of view. See also Supplemental Videos 1–3.
The migration speed of wild type cells in our spontaneous cell migration assay reduced only slightly after the injections with control antibodies (Fig. 5A, left (blue) blue set of bars). In contrast, injecting these cells with R-actin antibodies resulted in a dramatic decrease in speed (Fig. 5A, middle (green) set of bars). In Ate1 knockout cells, microinjection of arginylated actin antibodies did not lead to a significant change in their migration speeds (Fig. 5A, right (pink) set of bars). These changes were seen both in total migration distance over 130 min of observation (Fig. 5A, top), and in the overall migration velocity over this time (Fig. 5A, bottom), confirming that arginylated actin antibodies severely inhibit cell migration in wild type, but not Ate1 knockout cells.
As a control, we injected both cells with antibodies to β– actin, taken at the same concentration. In wild type cells, this injection inhibited cell migration (Fig. S2), consistent with prior findings that β– actin is essential for cell migration 13. Notably, however, the inhibition seen after this injection was not as severe as that seen after injection of R-actin antibodies, consistent with the fact that β– actin antibody likely acts by somewhat reducing the overall β– actin pool rather than by specifically targeting an essential cell migration component. Injection of Ate1 knockout cells with β– actin antibodies produced a negligible effect (Fig. S2). This is likely due to the fact that these cells have a dramatic reduction in the actin polymer:monomer ratio 14, and thus sequestering some of this extra actin monomer pool does not have a strong effect on the overall actin polymerization, which is required for cell migration. Thus, R-actin inhibition produces a more dramatic effect on cell migration.
To look closer at the changes that occur at the cell leading edge during inhibition of R-actin, we studied the effect of R-actin antibody injection on cell leading edge dynamics. For this experiment, we used a wild type mouse embryonic fibroblast cell line stably expressing GFP- β– actin migrating into the wound (introduced by scraping off an area of a dense cell monolayer), microinjected with either control or R-actin antibodies. We found that injection of R-actin antibody, unlike control, greatly suppresses the overall activity of the cell leading edge (data not shown), an effect that likely underlies these cells’ reduced migration speed.
Thus, arginylated actin is required for maintaining normal cell migration.
Discussion
This study demonstrates for the first time that arginylation of β– actin is a dynamic posttranslational modification that specifically targets a subset of actin at the cell leading edge and responds to stimuli to maintain cell migration rate and the dynamics of the cell leading edge. We have previously shown that β– actin is arginylated, and linked this arginylation to the overall morphology of the lamellipodia 3, however it has been unclear whether actin arginylation is regulated, whether it is confined to any particular area of the cell, and whether it is required for rapid responses to environment during cell movement. Our current study resolves these questions and demonstrates that actin arginylation can directly and dynamically modulate its in vivo function in cell migration.
We find that arginylated actin enrichment at the cell leading edge is confined to a detergent-labile actin pool. While our fractionation studies suggest that arginylation shows no overall preference for actin monomer or polymer, it is possible that in the leading edge zone arginylated actin exists as monomers and/or oligomers. If so, R-actin enrichment in the lamellipodia may directly or indirectly mediate actin turnover and disassembly, or modulate monomer sequestering and the maintenance of the higher G-actin concentration that would in turn promote polymerization of the non-arginylated actin pool to facilitate leading edge protrusion. We find that inhibition of arginylated β– actin by antibody microinjection suppresses cell migration and reduces the overall leading edge activity. Thus arginylated β– actin is required for the normal leading edge protrusion and dynamics, likely through a combination of mechanisms that also involve additional actin binding proteins and partners.
Our data show that arginylation constitutes a rapid and dynamic response to cell migration stimuli, such as LPA, or serum re-addition after starvation, previously shown to stimulate actin polymerization and leading edge activity in the cell 15. In fact, prominent increase in arginylated actin occurs within minutes of serum addition, suggesting that this modification is highly dynamic. Moreover, R-actin localization to the narrow leading edge zone suggests a specialized mechanism that maintains this localization over time. Such mechanism could involve R-actin-mediated targeting of the growing actin ends, specialized actin-binding proteins, and direct or indirect binding of R-actin to the leading edge plasma membrane.
The R-actin antibody shows partial cross-reactivity with Ate1 knockout cells, suggesting that a residual amount of arginylated actin may still form in these cells in the absence of Ate1. It has been consistently proposed that additional arginylation enzyme(s), likely encoded by different genes, may exist in cells. Despite extensive studies, this possibility has never been either supported or disproven. It appears likely, however, that even if such an activity exists, β- actin is an off-target substrate in this case, arginylated only at residual levels insufficient to support normal cell migration. It is also possible that another actin variant in these cells exhibits partial cross-reactivity with the antibodies.
Our data show that stimulation of actin arginylation upon serum addition requires active protein synthesis and does not occur in the presence of translation inhibitor cycloheximide. It has been previously found that arginyltransferase can partially colocalize with the ribosomes, where it presumably has an advantage in utilizing the locally synthesized Arg-tRNA. It has also been shown that β– actin mRNA is partially targeted to the cell leading edge in migrating cells 16–18 and that serum stimulation leads to an increase in actin synthesis 15,19. Our study for the first time combines the results of these findings into a general model, which predicts that cell migration stimuli lead to localized actin arginylation via a mechanism coupled to its de novo synthesis at the cell leading edge. It seems likely that this combined mechanism at least in part underlies the known dependence of cell migration on actin mRNA localization at the leading edge, which may enable the cell to produce a differentiated subset of arginylated actin on the spot. Dissecting this regulatory mechanism, and the underlying mechanics that links actin arginylation to cell movement, constitutes an exciting direction of future studies.
Materials and Methods
R-actin antibody
Rabbit polyclonal antibody to arginylated β-actin used in this study was developed by EMD Millipore (ABT264). The current work utilizes mostly Batch 13 of the antibody, different from the bulk of the commercially available Batch 15, due to the fact that the former antibody showed better R-actin specificity in the initial tests. We confirmed, however, that both antibodies show similar staining in cultured cells and have nearly equal reactivity on western blots with wild type cell lysates.
Cell culture
Wild-type and arginyltransferase 1 (Ate1) KO mouse embryonic fibroblasts (MEF) cell lines were obtained by immortalization of primary cells as described previously (Kwon et al., 2002; Karakozova et al., 2006). Cell lines stably expressing GFP-β-actin fusion for the Fluorescence in situ hybridization experiment were generated by transfecting immortalized wild type MEF with the plasmid containing full length β-actin plasmid backbone 20, in which the human β-actin promoter and the TC-GFP on the N-terminus were preserved, but the human β-actin coding sequence was replaced with mouse full length β-actin cDNA.
Cells were grown in in a 1:1 mixture of DMEM (high glucose with GlutaMAX, Gibco Life Technology) and F10 nutrient mix (Invitrogen) supplemented with 10% fetal bovine serum and antibiotics (Antibiotic-Antimycotic solution; Life Technology). Cells were passaged at the ratio of 1:4 every 3 days.
Preparation of cell lysates and actin fractionation experiments
For cell lysates, treated cells were harvested by scraping and centrifugation at 1,000 rpm in 1.5-ml tubes in a tabletop centrifuge for 5 min at 4°C. The cell pellet was promptly resuspended in dPBS, transferred to a new tube, and spin down twice to thoroughly remove the supernatant. This procedure was generally finished within 10 min after the cells were harvested. The pellet was then weighed and lysed in 5 vol of 2× SDS loading buffer by boiling for 5 min.
Baculovirus expression and purification of arginylated and nonarginylated β– actin for the western blot shown in Fig. 1A was performed as described in 21 using mouse M-β and R-β ubiquitin-actin fusion constructs made as described in Karakazowa et al., 2006.
Biochemical fractionation of actin and estimation of G–and F-actin content was performed as described in 14. MEFs were harvested and lysed in F-actin stabilization buffer (50 mM PIPES, pH 6.9, 50 mM NaCl, 5 mM MgCl2, 5 mM EGTA, 5% glycerol, 0.1% NP40, 0.1% Triton X-100, 0.1% Tween 20, 0.1% 2-mercaptoethanol, 1 mM ATP, and protease inhibitor cocktail; Sigma-Aldrich, St. Louis, MO; P8340). The resulting lysates (considered as totals in the course of the analysis) were sequentially sedimented at 37°C at 200 × g for 5 min, 1,500 × g for 15 min, 16,000 × g for 15 min, and 66,000 × g for 60 min. The amount of actin isoforms in the resulted supernatant and pellet fractions, and in totals was detected using monoclonal antibodies against β-actin (Sigma-Aldrich, A1978), total actin (Cytoskeleton, AAN01) and R-actin (EMD Millipore, ABT264) by means of western blot analysis.
Cycloheximide and LPA treatment
For cycloheximide treatment, cells were plated at 30% confluency followed by 24 h of serum starvation (0.5 % serum) and 1 h pretreatment with 50 µg/ml of cycloheximide. Next, culture media supplemented with 10% of serum, containing 100 µg/ml cycloheximide was added to each plate and cells were incubated for 5, 30 min and 3 h and harvested at time points as indicated for analysis of the protein levels.
For LPA stimulation, 30% confluent cells starved for 24 h in 0.5% serum were treated by addition of 10 μM LPA (Sigma-Aldrich, L7260) into the 0.5% serum media and harvested at 0, 30 min, 3 hr, and 12 hr time points for western blotting.
Immunoprecipitation
Cells were washed by dPBS twice, then lysed with 25-gauge syringe needle in a lysis buffer containing 10 mM Tris-Cl pH 8, 0.5 mM CaCl2, 100 mM KCI, 20 mM EDTA, 0.5 mM ATP, 1% Triton X100, 0.5% NP-40, mammalian protease inhibitor cocktail (Sigma-Aldrich), and 0.1 mM PMSF. The mixture was incubated at 4°C for 30 min with occasional vortexing. R-actin was immunoprecipitated using a polyclonal anti-β- actin arginylated (N-terminal) antibody that reacts with the N-terminal region of mouse R-actin (catalog no. ABT 264, Millipore). The antibody–protein complexes were pulled down using protein A beads (catalog no. 15918-014, Invitrogen) and washed three times with lysis buffer. The beads were resuspended in 2× SDS-loading buffer and boiled for 10 min. The resulting protein samples were analyzed by SDS-PAGE.
Western blotting
Cell lysates samples were separated by electrophoresis in SDS-PAGE. The electrophoresed proteins were transferred onto a 0.45 μm pore size nitrocellulose membrane (Bio-Rad). Membranes were incubated with primary antibodies for 2 h in room temperature. Monoclonal antibodies against β-actin, clone AC 15 (Sigma-Aldrich, cat # A5441) were used at 1:5000 dilution, anti-Pan actin at 1:2000 dilution (Cytoskeleton, Inc., cat. # AAN01), anti-R-β–actin at 1:1000 dilution (EMD Millipore, cat # ABT 264), anti-Ate1 at 1:2000 dilution. Chemifluorescence visualization was performed with secondary antibodies conjugated with HRP and reagents provided in the BM Chemifluorescence western Blotting kit Mouse/Rabbit (Roche) or the SuperSignal West Femto Chemiluminescence kit (Thermo Fisher Scientific). In most cases at least three different exposures with time ranging from 5 s to 1 min on x-ray films were performed and the films showing moderate intensity of chemifluorescence signals were chosen for quantification. The corresponding films were scanned by an Epson 4490 Perfection scanner into grayscale digital files with 1,200 dpi. The densitometry of the protein bands was analyzed with ImageJ (NIH).
Immunofluorescence staining
Cells cultured on coverslips were fixed by incubation with 4% paraformaldehyde (PFA), washed twice with PBS, and permeabilized for 5 min with ice cold acetone or 0.5% Triton X100, followed by incubation for 30 min in a blocking solution (5% horse serum in PBS). Cells were incubated overnight at 4°C or for 2 h at room temperature with primary antibodies against polyclonal anti-R-actin, washed with PBS, and incubated for 1 h at room temperature with Alexa Fluor 488-conjugated anti-rabbit antibody (Life Technologies, cat. #A21207) at 1:1000 dilution. After the staining, cover slips with cells were washed in PBS and mounted into ProLong Diamond anti-fade mounting media (Life Technologies). To visualize filamentous actin, the cells were additionally stained for 20 min with Alexa 488-conjugated phalloidin (diluted 1:40 in PBS) (Life Technologies, cat. #12379), and then washed three times with PBS before mounting.
Microinjection
Cells were seeded on MatTek coverglass tissue culture dishes (20,000 cells per dish). Before microinjection, the medium was overlaid with mineral oil to prevent evaporation and retard pH changes. Culture dishes were placed on the stage of a Nikon Eclipse microscope and microinjected with anti-R-actin of β– actin antibody loaded at 1 mg/ml into a glass microneedle, mixed with an equal volume of mouse FITC–or TRITC-conjugated IgG to produce the fluorescent signal that was then used to identify the injected cells. Control cells were injected with equal volume of mouse FITC–or TRITC-conjugated IgG alone.
Random motility assays
Random motility assay was assessed by a time-lapse microscopy as previously described 22. Cells were seeded onto a MatTek dish, and imaged using inverted Leica microscope with 20× dry objective, equipped with an environmental chamber. The photographs were taken every 10 min for up to 2.5 h by DFC 9000 GT camera. Analysis was done using Metamorph software (Molecular Devices). At least 20 cells of each of experimental condition were tracked; cells undergoing division or apoptosis were excluded from the analysis. Following parameters: cell track (reoriented to zero in migration traces) and velocity were calculated using Microsoft Excel software 23.
Kymography
WT MEFs cell line stably expressing GFP-β– actin were allowed to form a confluent monolayer in a 35 mm MatTek dish, and then the layer was scratched using 20-μL tip and photographed with 100× objective of Nikon Eclipse Ti microscope equipped with environmental chamber. Movies were 20 min long with frames taken every 10 s. Kymographs were produced and analyzed using Metamorph software (Molecular Devices). Kymographs were generated along 1-pixel-wide line regions oriented in the direction of individual protrusions.
Fluorescence in situ hybridization
eGFP mRNA probes (conjugated to Quasar 670 dye, VSMF 1015-5), and ATE1 mRNA probes (conjugated to Quasar 570 dye, custom designed) were purchased from LGC Biosearch Technologies and fluorescence in situ hybridization was carried out as per manufacturers’ protocol. Briefly, cells were seeded onto coverslips in six well plate at 20, 000 cells/well overnight and fixed in 4% (w/v) PFA at room temperature for 30 minutes followed by treatment with 70% alcohol at 4°C for 1 hour. Cells were incubated with 125nM probes at 37°C overnight. Cells were stained with DAPI (5ng/mL) and mounted using Prolong Diamond (Life Technologies). Images were acquired using Nikon Ti inverted microscope equipped with Andor iXon Ultra 897 camera at 100X. FISH images were then processed using Fiji to remove background (rolling ball background subtraction with a a radius of 6 pixels) and dilated by two pixels to enable better visualization.
Statistics
Data are presented as mean ± SEM. All p values were calculated by two-tailed Student's t-test. The difference was considered to be statistically significant at the level of at least p <0.05.
Supplementary Material
Figure S1. Dynamic response of R-actin to serum stimulation does not occur in confluent cultures with suppressed cell migration Quantifications (top) and images of representative immunoblots (bottom) of R-actin levels in cells grown at 100% confluency under normal serum conditions (C), or serum starved (O) and stimulated by serum re-addition for different intervals of time. R-actin levels are shown normalized to pan-actin signal in the same cells, which was used as a loading control. Error bars represent SEM from 3 different experiments analyzed in duplicates.
Figure S2. Injection of anti-beta-actin suppresses cell migration. A. quantifications of the migration of randomly migrating cells (bottom) over 130 min of observation, before and after injection with b-actin antibodies.
Supplemental video 1. A representative time lapse video of wild type mouse embryonic fibroblasts, grown under normal serum conditions, migrating into the scratch wound. Images were obtained at 20× magnification using phase contrast.
Supplemental video 2. A representative time lapse video of wild type mouse embryonic fibroblasts after 12 hours of serum deprivation (in media containing 0.5% serum), migrating into the scratch wound. Images were obtained at 20× magnification using phase contrast.
Supplemental video 3. A representative time lapse video of Ate1 knockout mouse embryonic fibroblasts, grown under normal serum conditions, migrating into the scratch wound. Images were obtained at 20× magnification using phase contrast.
Supplemental video 4. A representative time lapse video of Ate1 knockout mouse embryonic fibroblasts after 12 hours of serum deprivation (in media containing 0.5% serum), migrating into the scratch wound. Images were obtained at 20× magnification using phase contrast.
Supplemental Video 5. A representative time lapse video of wild type mouse embryonic fibroblasts injected with fluorescent control IgG. Images represent an overlay of the fluorescence channel (green) and phase contrast, to simultaneously detect injected and non-injected cells.
Supplemental Video 6. A representative time lapse video of wild type mouse embryonic fibroblasts injected with anti-R-actin, mixed with fluorescent control IgG for detection of injected cells. Images represent an overlay of the fluorescence channel (green) and phase contrast, to simultaneously detect injected and non-injected cells.
Supplemental Video 7. A representative time lapse video of Ate1 knockout mouse embryonic fibroblasts injected with anti-R-actin, mixed with fluorescent control IgG for detection of injected cells. Images represent an overlay of the fluorescence channel (green) and phase contrast, to simultaneously detect injected and non-injected cells.
Summary.
This study demonstrates that β– actin arginylation occurs primarily at the very edge of migrating cells, that it responds rapidly to cell stimulation inducing migratory behavior, and that its inhibition severely impairs cell migration. These data strongly suggest that arginylation is a dynamic modification of the leading edge actin that is integral to the migration-dependent signaling network.
Acknowledgments
We thank Dr. John Dawson for providing baculovirus-expressed arginylated and non-arginylated β- actin for the westerns shown in Fig. 1A, and Drs. Junling Wang and Dawei Dong for helpful discussions throughout the project. This work was supported by NIH grants GM104003, GM117984, and GM122505 to A.K.
Footnotes
The authors declare no competing financial interests.
Author contributions.
Iuliia Pavlyk: performed and analyzed the experiments; wrote the paper; Nicolae A. Leu, Pavan Vedula: performed the experiments; Satoshi Kurosaka: generated stably transfected cell lines; Anna Kashina: analyzed data, wrote the manuscript.
References
- 1.Pollard TD, Borisy GG. Cellular Motility Driven by Assembly and Disassembly of Actin Filaments. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 2.Terman JR, Kashina A. Post-translational modification and regulation of actin. Current opinion in cell biology. 2013;25:30–38. doi: 10.1016/j.ceb.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karakozova M, et al. Arginylation of beta-actin regulates actin cytoskeleton and cell motility. Science. 2006;313:192–196. doi: 10.1126/science.1129344. [DOI] [PubMed] [Google Scholar]
- 4.Zhang F, Saha S, Shabalina SA, Kashina A. Differential arginylation of actin isoforms is regulated by coding sequence-dependent degradation. Science. 2010;329:1534–1537. doi: 10.1126/science.1191701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kurosaka S, et al. Arginylation-dependent neural crest cell migration is essential for mouse development. PLoS genetics. 2010;6:e1000878. doi: 10.1371/journal.pgen.1000878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vitriol EA, et al. Two functionally distinct sources of actin monomers supply the leading edge of lamellipodia. Cell reports. 2015;11:433–445. doi: 10.1016/j.celrep.2015.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Helfand BT, et al. Vimentin organization modulates the formation of lamellipodia. Molecular biology of the cell. 2011;22:1274–1289. doi: 10.1091/mbc.E10-08-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jans R, et al. Lysophosphatidic acid promotes cell migration through STIM1- and Orai1-mediated Ca2+(i) mobilization and NFAT2 activation. The Journal of investigative dermatology. 2013;133:793–802. doi: 10.1038/jid.2012.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim EK, et al. Lysophosphatidic acid induces cell migration through the selective activation of Akt1. Experimental & molecular medicine. 2008;40:445–452. doi: 10.3858/emm.2008.40.4.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pietruck F, Busch S, Virchow S, Brockmeyer N, Siffert W. Signalling properties of lysophosphatidic acid in primary human skin fibroblasts: role of pertussis toxin-sensitive GTP-binding proteins. Naunyn-Schmiedeberg's archives of pharmacology. 1997;355:1–7. doi: 10.1007/pl00004906. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, et al. Arginyltransferase is an ATP-independent self-regulating enzyme that forms distinct functional complexes in vivo. Chemistry & biology. 2011;18:121–130. doi: 10.1016/j.chembiol.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Condeelis J, Singer RH. How and why does beta-actin mRNA target? Biology of the cell/under the auspices of the European Cell Biology Organization. 2005;97:97–110. doi: 10.1042/BC20040063. [DOI] [PubMed] [Google Scholar]
- 13.Bunnell TM, Burbach BJ, Shimizu Y, Ervasti JM. beta-Actin specifically controls cell growth, migration, and the G-actin pool. Molecular biology of the cell. 2011;22:4047–4058. doi: 10.1091/mbc.E11-06-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saha S, et al. Arginylation regulates intracellular actin polymer level by modulating actin properties and binding of capping and severing proteins. Molecular biology of the cell. 2010;21:1350–1361. doi: 10.1091/mbc.E09-09-0829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–399. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- 16.Shestakova EA, Singer RH, Condeelis J. The physiological significance of beta -actin mRNA localization in determining cell polarity and directional motility. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:7045–7050. doi: 10.1073/pnas.121146098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ross AF, Oleynikov Y, Kislauskis EH, Taneja KL, Singer RH. Characterization of a beta-actin mRNA zipcode-binding protein. Molecular and cellular biology. 1997;17:2158–2165. doi: 10.1128/mcb.17.4.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taneja KL, Singer RH. Detection and localization of actin mRNA isoforms in chicken muscle cells by in situ hybridization using biotinated oligonucleotide probes. Journal of cellular biochemistry. 1990;44:241–252. doi: 10.1002/jcb.240440406. [DOI] [PubMed] [Google Scholar]
- 19.Riddle VG, Dubrow R, Pardee AB. Changes in the synthesis of actin and other cell proteins after stimulation of serum-arrested cells. Proceedings of the National Academy of Sciences of the United States of America. 1979;76:1298–1302. doi: 10.1073/pnas.76.3.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodriguez AJ, Shenoy SM, Singer RH, Condeelis J. Visualization of mRNA translation in living cells. The Journal of cell biology. 2006;175:67–76. doi: 10.1083/jcb.200512137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yates SP, Otley MD, Dawson JF. Overexpression of cardiac actin with baculovirus is promoter dependent. Archives of biochemistry and biophysics. 2007;466:58–65. doi: 10.1016/j.abb.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 22.Pavlyk I, et al. Arginine deprivation affects glioblastoma cell adhesion, invasiveness and actin cytoskeleton organization by impairment of beta-actin arginylation. Amino acids. 2015;47:199–212. doi: 10.1007/s00726-014-1857-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shum MS, et al. gamma-Actin regulates cell migration and modulates the ROCK signaling pathway. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2011;25:4423–4433. doi: 10.1096/fj.11-185447. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Dynamic response of R-actin to serum stimulation does not occur in confluent cultures with suppressed cell migration Quantifications (top) and images of representative immunoblots (bottom) of R-actin levels in cells grown at 100% confluency under normal serum conditions (C), or serum starved (O) and stimulated by serum re-addition for different intervals of time. R-actin levels are shown normalized to pan-actin signal in the same cells, which was used as a loading control. Error bars represent SEM from 3 different experiments analyzed in duplicates.
Figure S2. Injection of anti-beta-actin suppresses cell migration. A. quantifications of the migration of randomly migrating cells (bottom) over 130 min of observation, before and after injection with b-actin antibodies.
Supplemental video 1. A representative time lapse video of wild type mouse embryonic fibroblasts, grown under normal serum conditions, migrating into the scratch wound. Images were obtained at 20× magnification using phase contrast.
Supplemental video 2. A representative time lapse video of wild type mouse embryonic fibroblasts after 12 hours of serum deprivation (in media containing 0.5% serum), migrating into the scratch wound. Images were obtained at 20× magnification using phase contrast.
Supplemental video 3. A representative time lapse video of Ate1 knockout mouse embryonic fibroblasts, grown under normal serum conditions, migrating into the scratch wound. Images were obtained at 20× magnification using phase contrast.
Supplemental video 4. A representative time lapse video of Ate1 knockout mouse embryonic fibroblasts after 12 hours of serum deprivation (in media containing 0.5% serum), migrating into the scratch wound. Images were obtained at 20× magnification using phase contrast.
Supplemental Video 5. A representative time lapse video of wild type mouse embryonic fibroblasts injected with fluorescent control IgG. Images represent an overlay of the fluorescence channel (green) and phase contrast, to simultaneously detect injected and non-injected cells.
Supplemental Video 6. A representative time lapse video of wild type mouse embryonic fibroblasts injected with anti-R-actin, mixed with fluorescent control IgG for detection of injected cells. Images represent an overlay of the fluorescence channel (green) and phase contrast, to simultaneously detect injected and non-injected cells.
Supplemental Video 7. A representative time lapse video of Ate1 knockout mouse embryonic fibroblasts injected with anti-R-actin, mixed with fluorescent control IgG for detection of injected cells. Images represent an overlay of the fluorescence channel (green) and phase contrast, to simultaneously detect injected and non-injected cells.