

Abstract
The field of ion mobility-based omics studies requires high-quality collision cross section (CCS) libraries to effectively utilize CCS as a molecular descriptor. Absolute CCS values with the highest precision are obtained on drift tube instruments by measuring the drift time of ions at multiple drift voltages, commonly referred to as a ‘stepped field’ experiment. However, generating large scale absolute CCS libraries from drift tube instruments is time consuming due to the current lack of high-throughput methods. This communication reports a fully automated stepped-field method to acquire absolute CCS on commercially available equipment. Using a drift tube ion mobility-mass spectrometer (DTIM-MS) coupled to a minimally modified liquid chromatography (LC) system, CCS values can be measured online with a carefully timed flow injection analysis (FIA) experiment. Results demonstrate that the FIA stepped-field method yields CCS values which are of high analytical precision (< 0.4% relative standard deviation, RSD) and accuracy (≤ 0.4% difference) comparable to CCS values obtained using traditional direct-infusion stepped-field experiments. This high-throughput CCS method consumes very little sample volume (20 μL) and will expedite the generation of large-scale CCS libraries to support molecular identification within global untargeted studies.
GRAPHICAL ABSTRACT
This work presents a fully automated method to generate absolute Collision Cross Sections for annotated libraries.
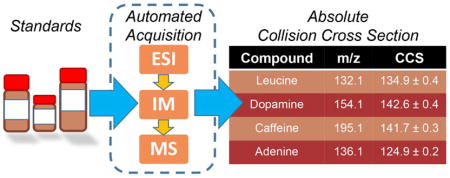
MAIN TEXT
Drift tube IM-mass spectrometers (DTIM-MS) are best suited for generating CCS libraries because unlike most ion mobility techniques in use today, DTIM-MS can determine absolute CCS values from a fundamental ion mobility relationship derived from first principles of the kinetic theory of gases. Specifically, DTIM-MS techniques utilize a low magnitude, uniform electric field sustained under constant pressure and temperature conditions, allowing the ions to traverse the drift tube at a constant velocity with minimal ion heating thus satisfying the ‘low field’ assumptions of the Mason-Schamp relationship.1, 2 A recent interlaboratory study evaluated a wide range of experimental parameters for absolute CCS determination on a DTIM-MS platform to produce a standardized stepped-field method consisting of seven drift potentials from 10.9 to 18.5 V/cm with a dwell time of 30 seconds for each field, resulting in a 3.5 minute long direct infusion method. The results demonstrate that a commercially-available DTIM-MS instrument is capable of obtaining absolute, nitrogen-based CCS measurements (DTCCSN2) with a precision of better than 0.3% RSD and were reproducible to within 0.5% RSD across four participating international laboratories.3 Considering that only three years ago, a similar interlaboratory evaluation of three traveling wave IM-MS instruments reported a %RSD of ca. 2 to 3% for CCS measurements,4 this is a significant improvement in the precision of commercial IM platforms.
In addition to the stepped field experiments, the DTIM interlaboratory study reported a faster semi-empirical method for generating calibrated, relative CCS values based on a single electric field, which is amendable to the timescale of front-end separations such as liquid chromatography.3 However, the single-field method demonstrated an empirical variation of ca. 0.5% RSD when compared to the stepped-field CCS values, and thus is not preferred when the highest obtainable CCS precision is desired (i.e. when generating CCS libraries). Furthermore, the importance of high precision CCS measurements for small molecule characterization is underscored by a recent study of eleven leucine/isoleucine isomers (131 Da) which demonstrated that the majority of these small molecule isomers (ca. 60%) exhibit CCS differences that are within 0.5% of one another.5 Thus, the 0.5% interlaboratory reproducibility is important for isomeric differentiation, although it should be noted that the limitations of current IM technology cannot resolve these small differences in CCS,6 which can lead to ambiguous identifications for mixed isomer systems.
Absolute CCS libraries which support the use of CCS as a molecular descriptor are necessary to improve the confidence level of identifications. These libraries are increasingly important as IM is becoming commonplace in untargeted metabolomics workflows. CCS values provided by IM are orthogonal to other descriptors such as retention time, m/z, and fragmentation (MS/MS) data. When these descriptors are combined, the number of confident identifications in global untargeted experiments increases.4, 7, 8 While metabolomics has several reliable MS and MS/MS libraries to support identification in global experiments, there is currently a lack of comprehensive, validated CCS libraries.9 Furthermore, while ion mobility measurements have been around for half a century, small biological molecules are still largely underrepresented.10 To overcome these challenges, many research laboratories worldwide are compiling large-scale, empirically-derived CCS libraries that support contemporary IM-MS instrumentation. Two recent reports include the compiling of absolute DTCCSN2 values for over 500 small molecules,8 and calibration-based TWCCSN2 measurements for over 1,400 drug and drug-like compounds.11 The latter is particularly noteworthy in that a similar FIA-based method as described in this study was used to analyze over 1,400 analytes in less than 24 hours, although the relative CCS measurements obtained in this study were reported with a ca. 3% calibration error. Despite these important contributions, tens of thousands of additional CCS measurements will need to be generated if good coverage of the metabolome and lipidome is to be achieved.
This communication reports a high-throughput, fully automated DTIM-MS method for collecting stepped-field data and determining absolute CCS values with high analytical. The method utilizes a commercial LC system (1290, Agilent) operated without a chromatography column in flow injection analysis (FIA) mode and coupled directly to a commercial DTIM-MS (6560A, Agilent) via electrospray ionization (Jet Stream ion source, Agilent).
Figure 1 contains the total ion chromatogram (arbitrary intensity) versus time (minutes) for the analyte subjected to variation of the sample loop volume and solvent flow dynamics. Here, the overlaid chromatograms are not normalized to facilitate direct comparisons between the different flow rates evaluated. To measure absolute CCS with the standardized method,3 the ion intensity must be sufficiently high (but not necessarily constant) for at least 3.5 minutes. In order to achieve this using FIA via an LC system, several conditions were optimized. First, the sample loop volume was increased. Figure 1A shows the analyte elution profile of a 20 μL injection using four sample loops with a static flow rate of 30 μL/min. Using the default 20 μL sample loop, the analyte completely eluted in ~1.2 min (FWHM; Figure 1A, black trace). By increasing the sample loop volume to 100 μL, the analyte elution time was extended to ~3.2 min (FWHM, Figure 1A, blue trace). Further increasing the sample loop volume to 200 and 300 μL provided marginal increases in elution peak width (Figure 1A, red and blue trace). Second, a dynamic carrier solvent flow rate was tested to extend the analyte peak width, Figure 1B. For each total ion chromatogram in Figure 1B, 20 μL of sample was injected into a 100 μL loop. The dynamic flow rate was changed from 800 to 30 μL/min (t = 0.19 min), 500 to 30 μL/min (t = 0.50 min), and 200 to 30 μL/min (t=0.70 min) (Figure 1B; green, black, and red trace, respectively). For comparison, the elution peak for a static flow rate of 30 μL/min is included (Figure 1B, blue trace). It is observed that when the flow rate is decreased, the elution peak width increases. Decreasing the solvent flow from either 800, 500, or 200 to a final flow rate of 30 μL/min all provided elution peak widths (~5.5 min, FWHM) sufficient for absolute CCS measurements.
Figure 1.

Total ion chromatogram peak elution profiles. A) Four sample loop volumes were tested with a 20 μL injection and a flow rate of 30 μL/minute. B) One static (blue) and three dynamic (green, black, red) solvent flow rates were tested using a 20 μL injection into a 100 μL sample loop. Note that these chromatograms are extended to the full elution profile, whereas the final FIA-DTIM-MS analysis method is performed in ca. 5 min using a purge step at ca. 4.5 min.
Figure 2 outlines the final experimental setup and time scales for this experiment. For the final method, 20 μL of analyte is injected into a 100 μL loop. Under-filling the sample loop was chosen to minimize sample waste, and thus preserves the maximal amount of sample for subsequent analyses. To create a 100 μL loop, an 80 μL loop (large volume injection kit, Agilent) was added to extend the default 20 μL loop (Figure 2A). The carrier solvent begins pushing the analyte towards the ESI (t = 0 min) at 800 μL/min, and upon analyte elution (t = 0.19 min), the solvent flow rate is decreased to 30 μL/min (Figure 2B, top). While the sample elutes, the IM dispersion field is stepped by increasing the voltage across the drift tube in 100 V increments every 30 seconds (Figure 2B, middle), which are equivalent to the fields and timeframes used in the standardized stepped-field method.3 Because the total ion intensity decreases throughout the measurement and because ion transmission efficiency marginally increases at the larger drift potentials, this method is best performed in order of increasing drift voltage, which also follows the standardized method. Data is acquired from t = 0 to 4.0 min (Figure 2B, bottom). To minimize carryover, the solvent flow rate is increased back to 800 μL/min at the end of the experiment to purge any remaining analyte. To test and optimize this method, CCS values for the nine exogenous compounds (acetaminophen, caffeine, sulfaguanidine, sulfadimethoxine, val-tyr-val, verapamil, terfenadine, leucine-enkephalin, and reserpine, Table S1) were measured using both the FIA method and the traditional DI method. The CCS results from both methods are contained in Table 1. Here, the nine chemical standards were concurrently injected at the concentrations indicated in Table S1. To facilitate sample infusion with a wide temporal peak shape for each analyte, the solvent composition was optimized. Using 100% water as the mobile phase resulted in the less-polar analyte (i.e., verapamil, terfenadine, and reserpine) to elute too rapidly (~1.6 min, FWHM), but increasing the organic composition of the mobile phase to 30% permitted the less-polar analyte to elute with a broader temporal distribution (~5 min, FWHM; Figure S2), which is interpreted as a result of sample-solvent compatibility as the sample plug mixes with the carrier solvent. Otherwise, a 20 μL injection would elute in ~0.67 minutes using the 30 μL/min. flow rate. Both the DI and FIA CCS values measured in Table 1 were obtained using a carrier solvent composition of 7:3 (water:acetonitrile). For the experiments herein, the sample solvent is matched to the carrier solvents, so solvent hysteresis effects should be minimal, although it should be noted that a detailed study of the effects of mismatching the solvents on the resulting CCS measurements was not conducted. The CCS values measured with the FIA stepped-field method in both positive and negative ion modes are reported in Table 1. For both polarities, the CCS values exhibited a percent RSD of 0.34% or less for N = 18 measurements per analyte across multiple days (N = 6 per day) using the FIA method. Additionally, the CCS values from the FIA method were compared to CCS values acquired using the DI stepped-field method, and the values agree within the precision of the instrument (percent difference of less than 0.4%). A table of experimental drift tube settings and timing (Table S2), the pump flow rate timing (Table S3), and source conditions (Table S4) can be found in the supplemental information. While the total FIA stepped-field method is 1 min longer than the DI method (4.5 vs. 3.5 minutes), it comes with several important analytical benefits. First, the FIA method consumes less than half the sample volume (20 μL per injection) that the DI method uses (~40–100 μL). Additionally, the FIA stepped-field method is self-cleaning as the analyte is purged from the system after each measurement. Finally, and perhaps most importantly, the FIA stepped-field method can be fully automated for high-throughput absolute CCS measurements. The current method can analyze approximately 12 samples per hour unsupervised (12 samples in 60.8 minutes). This includes sample injection, cleanup, and switching, all while mirroring the standardized DI stepped-field experiment in accuracy and precision. This method will assist rapid generation of large scale absolute CCS libraries.
Figure 2.

A) Schematic for flow injection analysis CCS experiments. B) Method timing for flow injection analysis CCS measurements. Top, Solvent flow rate; middle, drift tube entrance potential; bottom, total ion counts for analyte in Table 1. Data acquisition completes at 4.3 minutes, and a one minute purge begins.
Table 1.
Positive and Negative ion CCS values measured with FIA and DI stepped-field methods
| Analyte | Positive Ions | Negative Ions | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||||||||||
| [M+H]+ (Da)a | FIA DTCCSN2 (Å2)b | DI DTCCSN2(Å2)c | % Diffd | [M-H]− (Da)a | FIA DTCCSN2 (Å2)b | DI DTCCSN2(Å2)c | % Diffd | |||||||||
|
|
|
|
|
|||||||||||||
| Avg. | σ | %RSD | Avg. | σ | %RSD | Avg. | σ | %RSD | Avg. | σ | %RSD | |||||
|
|
|
|
|
|
|
|
|
|
||||||||
| Acetaminophen | 152.07 | 131.84 | 0.45 | 0.34 | 132.01 | 0.35 | 0.26 | 0.13 | 150.06 | 134.48 | 0.07 | 0.05 | 134.16 | 0.11 | 0.08 | −0.24 |
|
| ||||||||||||||||
| Caffeine | 195.09 | 141.68 | 0.16 | 0.11 | 141.27 | 0.25 | 0.18 | −0.29 | – | – | – | – | – | – | – | – |
|
| ||||||||||||||||
| Sulfaguanidine | 215.06 | 150.18 | 0.18 | 0.12 | 149.60 | 0.26 | 0.17 | −0.39 | 213.04 | 149.51 | 0.10 | 0.07 | 149.41 | 0.14 | 0.09 | −0.07 |
|
| ||||||||||||||||
| Sulfadimethoxine | 311.08 | 170.57 | 0.21 | 0.12 | 170.16 | 0.20 | 0.12 | −0.24 | 309.07 | 172.61 | 0.09 | 0.05 | 172.38 | 0.18 | 0.11 | −0.13 |
|
| ||||||||||||||||
| Val-Tyr-Val | 380.22 | 192.97 | 0.34 | 0.18 | 192.34 | 0.20 | 0.11 | −0.33 | 378.20 | 194.52 | 0.12 | 0.06 | 194.64 | 0.15 | 0.08 | 0.06 |
|
| ||||||||||||||||
| Verapamil | 455.29 | 208.66 | 0.46 | 0.22 | 208.74 | 0.38 | 0.18 | 0.04 | – | – | – | – | – | – | – | – |
|
| ||||||||||||||||
| Terfenadine | 472.32 | 227.37 | 0.46 | 0.20 | 227.93 | 0.43 | 0.19 | 0.25 | – | – | – | – | – | – | – | – |
|
| ||||||||||||||||
| Leucine-Enkephalin | 556.28 | 230.02 | 0.40 | 0.18 | 229.86 | 0.38 | 0.16 | −0.07 | 554.26 | 223.81 | 0.12 | 0.06 | 224.26 | 0.16 | 0.07 | 0.20 |
|
| ||||||||||||||||
| Reserpine | 609.28 | 251.69 | 0.39 | 0.15 | 252.18 | 0.54 | 0.22 | 0.20 | 607.27 | 266.22 | 0.20 | 0.07 | 266.49 | 0.25 | 0.09 | 0.10 |
The average (Avg.), standard deviation of the mean (σ), and percent relative standard deviation (%RSD) of CCS values reported for N = 18 measurements.
Exact mass of the protonated or deprotonated ion.
Flow injection analysis (FIA) CCS values.
Direct infusion (DI) CCS values.
Percent difference between average FIA and DI CCS values.
CONCLUSION
This work reports an automated stepped-field method using FIA for determining CCS on a drift tube instrument using the fundamental low-field ion mobility relationship (i.e., Mason-Schamp theory). With this method, high-precision CCS measurements (RSD < 0.4%) using low volumes (20 μL) of pure analytical standards can be rapidly acquired (12 samples/hr), which will enable the development of large-scale libraries of absolute CCS measurements in support of untargeted workflows that incorporate IM measurements.
Supplementary Material
Acknowledgments
This work was supported in part using the resources of the Center for Innovative Technology at Vanderbilt University. C.M.N. acknowledges a postdoctoral fellowship from Agilent Technologies. Financial support for this work was provided by the National Institutes of Health (NIH NIGMS R01GM092218) and the U.S. Environmental Protection Agency under Assistance Agreement No. 83573601. The EPA does not endorse any products or commercial services mentioned in this publication. This work has not been formally reviewed by the EPA and the views expressed in this document are solely those of the authors and do not necessarily reflect those of the funding agencies and organizations.
Footnotes
Concentrations and commercial vendors for the analyte in this study. Experimental conditions including source parameters, acquisition settings, advanced parameters, and binary pump flow rates. Extracted ion chromatograms for each analyte under various solvent compositions. Composite chromatograms for multiple sample injections.
References
- 1.Mason EA, McDaniel EW. Transport Properties of Ions in Gases. John Wiley & Sons; New York: 1988. [Google Scholar]
- 2.Siems WF, Viehland LA, Hill HH. Anal Chem. 2012;84:9782–9791. doi: 10.1021/ac301779s. [DOI] [PubMed] [Google Scholar]
- 3.Stow SM, Causon TJ, Zheng X, Kurulugama RT, Mairinger T, May JC, Rennie EE, Baker ES, Smith RD, McLean JA, Hann S, Fjeldsted JC. Anal Chem. 2017;89:9048–9055. doi: 10.1021/acs.analchem.7b01729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paglia G, Williams JP, Menikarachchi L, Thompson JW, Tyldesley-Worster R, Halldórsson S, Rolfsson O, Moseley A, Grant D, Langridge J, Palsson BO, Astarita G. Anal Chem. 2014;86:3985–3993. doi: 10.1021/ac500405x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dodds JN, May JC, McLean JA. Anal Chem. 2016;89:952–959. doi: 10.1021/acs.analchem.6b04171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dodds JN, May JC, McLean JA. Anal Chem. 2017;89:12176–12184. doi: 10.1021/acs.analchem.7b02827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.May JC, Gant-Branum RL, McLean JA. Current Opinion in Biotechnology. 2016;39:192–197. doi: 10.1016/j.copbio.2016.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng X, Aly NA, Zhou Y, Dupuis KT, Bilbao A, Paurus VL, Orton DJ, Wilson R, Payne SH, Smith RD. Chemical Science. 2017;8:7724–7736. doi: 10.1039/c7sc03464d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schrimpe-Rutledge AC, Codreanu SG, Sherrod SD, McLean JA. J Am Soc Mass Spectrom. 2016;27:1897–1905. doi: 10.1007/s13361-016-1469-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.May JC, Morris CB, McLean JA. Anal Chem. 2017;89:1032–1044. doi: 10.1021/acs.analchem.6b04905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hines KM, Ross DH, Davidson KL, Bush MF, Xu L. Anal Chem. 2017;89:9023–9030. doi: 10.1021/acs.analchem.7b01709. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.