

Abstract
It is over 20 years since the first fragment-based discovery projects were disclosed. The methods are now mature for most ‘conventional’ targets in drug discovery such as enzymes (kinases and proteases) but there has also been growing success on more challenging targets, such as disruption of protein–protein interactions. The main application is to identify tractable chemical startpoints that non-covalently modulate the activity of a biological molecule. In this essay, we overview current practice in the methods and discuss how they have had an impact in lead discovery – generating a large number of fragment-derived compounds that are in clinical trials and two medicines treating patients. In addition, we discuss some of the more recent applications of the methods in chemical biology – providing chemical tools to investigate biological molecules, mechanisms and systems.
Keywords: chemical biology, drug discovery and design, fragment based lead discovery
Introduction
The key feature of fragment methods is that discovery begins by identification of low molecular weight (MW) compounds or fragments (typically approximately 200 Da) that bind to the biological molecule of interest. Because they are small, the fragments are less likely to contain portions that prevent binding and so are more likely to identify functional motifs that match the requirements of the target. The fragment hits can then be optimized, usually through structure-based design, into higher affinity compounds that can be used to probe the biology of the target or as a starting point (lead) for drug discovery.
Typically, fragments bind to most target binding sites with an equilibrium dissociation constant (KD) in the 100s µM to low mM range. This places constraints on the assays that can reliably detect such weak binding (usually biophysical methods) and the design of the fragment library (solubility for the high concentrations of fragments needed in assays). Although there are some reports of using fragment methods against nucleic acids [1], the overwhelming majority of the published work is on proteins – which will be the type of target covered in this essay.
The first report of fragment-based discovery was in 1996 [2] (discussed later) and there was rapid development of the methods and their application in the late 1990s and early 2000s, particularly in small, structure-based discovery companies such as Astex [3], Sunesis [4] and Vernalis [5]. What was vital was that progress could be made with a small (approximately 1000 compounds) fragment library providing novel chemical matter to start a discovery project, and the companies were able to exploit high-throughput crystallographic methods to generate candidate compounds. The successes of the mid-2000s led to the widespread adoption of the approach across pharmaceutical companies and academia. There are now many compounds in the clinic (http://practicalfragments.blogspot.co.uk/2016/07/fragments-in-clinic-2016-edition.html) and two fragment-derived compounds treating patients [6,7].
There have been many reviews and articles summarizing the methods in more detail than will be covered here. Some representative ones are a review by some of the leading practitioners summarizing the developments of the last 20 years [8], a review of the application of biophysical methods in drug discovery [9]; a recent summary of fragment to lead campaigns [10]; an excellent book with chapters from leading figures in fragment-based discovery [11]; and a chapter written by one of the authors of this essay in the RSC Handbook of Medicinal Chemistry [12]. In addition, there is an excellent blog that contains précis and comments on recent developments, conferences and publications on topics in fragment-based lead discovery (FBLD) (http://practicalfragments.blogspot.co.uk/).
There are variations in the phrase and acronym used to describe the field. We prefer FBLD – fragment-based lead discovery – because the key step in either a drug discovery or a chemical biology project is identifying a compound good enough to answer questions about the target (a lead). Discovery of a drug does depend on the properties introduced as that lead is optimized, and the fragment discovery process can have an impact. However, it is also heavily dependent on whether binding to the target has the required effect on the disease or condition – a reason why some projects fail in clinical trials – which often has little to do with the compound itself.
Here, we will provide a brief overview of the key features of FBLD and give a perspective on some of the more recent developments – in particular the use of fragments in chemical biology, exploring features of protein structure and activity.
Development of the key ideas and methods
In most areas of science, the development of a field is built on a set of ideas established by many researchers. In the case of FBLD, there are a number of contributions that should be highlighted.
The first is from the work of Jencks in the early 1970s [13]. He pointed out that for a molecule made up of two interacting parts, there are contributions to free energy from the interactions made by each of the two parts and additionally from the rotational and translational entropy of the whole molecule. This highlights that although the binding of an individual part (as in the case of a fragment) may have quite low affinity as it overcomes the entropy of binding, all the interaction energy of another binding part is added when it is incorporated (or linked). He also reminded us that ΔG = −RTlnKD – i.e. doubling free energy, squares the binding affinity.
The second set of ideas came from computational methods. Goodford [14] developed the GRID method that visualizes the calculated energy of interaction between a probe atom and a protein surface, highlighting where particular types of interactions could be made. This approach was extended to mapping where functional groups preferentially bind in the multiple copy simultaneous search (MCSS) method of Miranker and Karplus [15]. This idea of functional groups (i.e. very small fragments) binding was explored experimentally by Ringe and Mattos [16] and English et al. [17] through the determination of the structure of protein crystals soaked in high concentrations of various solvents (so-called multiple solvent crystal structures, MSCS). This combination of studies inspired the ideas that small molecules binding to a protein surface can be constructed from small pieces.
It was the work of Fesik and co-workers at Abbott that first reduced these ideas to practice for lead discovery, in a set of papers using a method they termed structure–activity relationship (SAR) by nuclear magnetic resonance (NMR) [2], reviewed in [18]. They used protein-observed NMR (see later) to identify fragments binding to two independent binding sites and linked these fragments together to make potent inhibitors. The approach was extended, first by Abbott, to use X-ray crystallography to identify the fragments [19], an approach that was rapidly exploited and developed by Astex [3] during the early 2000s, along with developments at Sunesis [4] and Vernalis [5]. Although a few larger companies did experiment with the approaches [20], it was the small companies that primarily developed the methods and demonstrated success, which by the mid-2000s led to the wider take-up of the methods across the pharmaceutical industry.
Two other developments made a contribution to the thinking in FBLD. The first was from Hann et al. [21], which developed models around molecular complexity – i.e., that a molecule needs to have sufficient features to make interactions with a binding site, but not so many features (or size) that prevent binding. This provided a framework for thinking about the size of compounds in a fragment library. The second was the idea of ligand efficiency [22] – which in its simplest form is the amount of free energy per non-hydrogen atom in a compound. This had a major impact on medicinal chemistry thinking, providing a guide as to whether atoms added to a compound during a cycle of optimization really are making optimal interactions with the protein.
Current practice in FBLD
Figure 1 summarizes the main features of an FBLD platform; its five main components – fragment libraries, target enablement, fragment screening, generating a model for fragment binding and fragment to lead optimization – are described in the sections that follow.
Figure 1. The FBLD process.

See text for a description of the five main components of a fragment-based discovery platform.
Fragment libraries
There are three main considerations in assembling a fragment library. Firstly, the properties of the fragments – they should not contain groups known to be reactive to proteins, should be soluble at the high concentrations used for screening and should be as diverse as possible (see [23] for one of the first descriptions of these properties). Secondly, the MW is a major consideration for the number of fragments in a library. Work by the group of Reymond [23,24] has estimated that the size of chemical space increases by approximately 8-fold for each heavy atom in a molecule. There are many approximations, but this means that a truly diverse library of 1000 fragments with MW of 190 Da covers the available chemical space in an equivalent way to 108 molecules of MW 280 Da or 1018 molecules of MW 440 Da. Hence, many practitioners work successfully with libraries of 1–2000 compounds of average MW 200 Da. Finally, it is important that the library is regularly checked for stability or precipitation/aggregation of the fragments to avoid false positives in the high concentration assays. Some of the other issues to be considered are discussed in a recent review of fragment library design [24] and the development of a new fragment library at Pfizer [25].
It is important to remember that this is screening and that small differences in the structure of the compounds can have a big impact on binding affinity. The hope is that if the library is diverse enough then some fragments will bind to the target – they will not be the best fragments but may give a starting point for identifying fragments more suitable for optimization.
Target enablement
In thinking about the issues and opportunities for FBLD, it is possible to define two classes of target: (a) a conventional target, where a crystal structure and a robust binding or functional assay is readily available and there is precedence for obtaining drug-like compounds (such as a protein kinase) and (b) a challenging target, such as disruption of a protein–protein interaction or inhibition of the activity of a multi-protein complex, where there is no precedence for lead discovery and where there are many problems to solve in establishing a platform for lead discovery – such as establishing a robust assay or generation of a model of compound binding. The difficulty and time taken for target enablement will clearly be different for these two classes of target.
Fragment and structure-based lead discovery relies upon the production of sufficient quantities of pure, functional, homogeneous protein suitable for screening and structure determination. Although this has usually been relatively routine for conventional targets, it can be an issue for challenging targets, particularly knowing which post-translational modifications affect the target binding site or which additional proteins are involved in affecting activity in the cell. Finally, it is vital that robust assays (both binding and functional) are available – this can be a real bottleneck for challenging targets as a compound is usually required to validate activity assays – and the assays are required to identify the compound. These issues should be self-evident, but they can be a major barrier to initiating a successful fragment-based discovery project.
Fragment screening
The main methods used to identify fragments that bind to a protein are listed in Figure 2, where the figure legend summarizes the main features of each technique. Taking into account the experimental conditions and nature of the assay method, all of the techniques should give the same hits when screening a library against a target [26,27]. What is sometimes forgotten is the relative sensitivity of each method, and whether the fragments (and the target) are still in solution and not aggregated under the conditions of the assay (concentration required, pH, buffer etc). Some practitioners describe screening protocols using a number of different techniques and then taking the intersection of the hits as the true hits. Although such hits will probably be valid, there is a danger that good fragment hits are discarded because of the least robust assay.
Figure 2. Fragment screening methods.
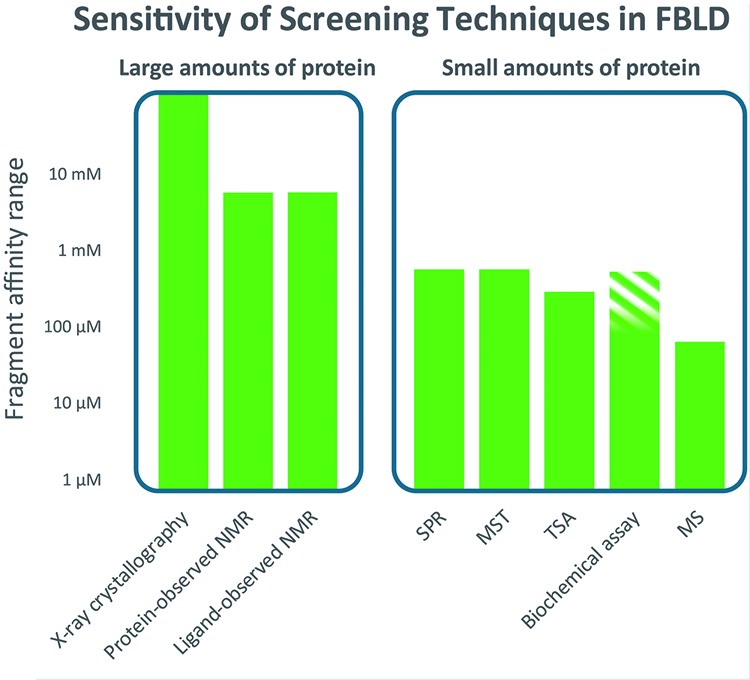
The figure emphasizes that there are two main categories of fragment screening methods – those that require many mg (10s of mg for NMR) of protein and those that require small amounts (typically less than 1 mg can be sufficient). The following is a brief description of the main characteristics and limitations of each of the methods. For all methods, it is important to appreciate the sensitivity of detection (indicated on the vertical axis of the diagram) and the solubility required of the fragment. For some methods, such as NMR, a robust indication of binding can be observed at fragment concentrations around or below the KD; for others, such as X-ray, surface plasmon resonance (SPR) and microscale thermophoresis (MST), it is necessary to achieve a concentration 5–10 times the KD for a robust binding signal. X-ray crystallography – the main requirement is for the protein to form crystals suitable for soaking with high concentrations of fragment (robust crystals with an open binding site not occluded by crystal packing and where any conformational changes on binding can be accommodated within the crystal). Typically, fragments are soaked at 10s mM concentration, usually in mixtures chosen on shape diversity. Advances in synchrotron facilities (in particular Pilates detectors) and streamlining of structure solution software [28] allow many hundreds of crystal structures to be determined in a few weeks – with recent automation (at synchrotrons such as the Diamond facility in Harwell, U.K.) increasing throughput to thousands of structures in a week [29]. The main advantage is that the structure of the fragment bound to the protein is available immediately; as well as the need for a suitable crystal system, the main disadvantage is that it can sometimes take a number of attempts to obtain crystal structures of compounds bound to a protein [26], so there will be false negatives. Protein-observed NMR – the main requirement is for large quantities of labelled protein (usually with 15N, sometimes with 13C), which can only routinely be produced with bacterial overexpression and which is soluble at approximately 100 µM in a suitable buffer (low-salt). The predominant experiment for screening is heteronuclear single quantum correlation spectroscopy (HSQC), which generates a 2D NMR spectrum where each peak (for 15N-labelled) represents each amide – where the chemical shift reports on the local environment that changes on binding of a fragment. It is possible to titrate the fragment to estimate KD and the pattern of peaks can be used to check for same site binding. If the spectrum is assigned then the binding site can be mapped. The method can measure from very strong binding down to mM when the ligands are sufficiently soluble. The main limitations are the time it can take to measure HSQC spectra (10 s of minutes each) for a large fragment library and a size limit on the protein (usually 30 kDa as higher MW gives slower tumbling and broadening of NMR peaks). Ligand-observed NMR – the main requirement is for large quantities of protein (usually 10s of mg, labelling not required, protein can be of any size) that is soluble at approximately 10 µM in a suitable buffer and stable for the time required for screening (with automation, this can be a few days for a full library screen). Practices vary, but what is recommended [5] is to use three different 1D experiments – saturation transfer difference (STD), water-ligand-observed gradient spectroscopy (LOGSY) and Carr–Purcell–Meiboom–Gill sequence (CPMG), and to include a competition step. The NMR experiments report on different properties of the fragments when they bind. For STD, this is transfer of resonance energy from the protein to the fragment when it binds; for water-LOGSY, this is the differential transfer of energy from solvent to the fragment and for CPMG, measuring the difference in relaxation time for the fragment free in solution or binding to the protein. Typically, fragments are screened in mixtures of 4–10 fragments – with a check that there is at least one distinctive peak for each fragment in the combined 1D spectra. As long as none of the fragments is very potent (thus blocking the binding site), then the experiment will report if multiple fragments from a mixture bind. It is beneficial to include a competition step where a known ligand is added to the protein fragment sample – to differentiate between specific and non-specific binding (which can be an issue at the high concentrations, typically 500 µM, used). The main limitation is the amount of protein required – typically 40–50 mg to screen a library of 1200 fragments. The sensitivity is from 100 nM to 5mM, although at stronger binding affinities the requirement for fast binding kinetics is not always fulfilled, leading to false negatives. BU – in this method, the protein is attached to a surface (a variety of methods – either non-specifically e.g. via free amines, or specifically – through a terminal his-tag [30] or introduced biotin tag). The refractive index of the surface varies with the MW of what is attached (see the lower left panel in ‘part 4’ of Figure 1) – there is a rise in MW as the fragment binds and a fall when the fragment is no longer flowing (for larger compounds, SPR can provide very useful kinetics of binding – kon and koff). The sensitivity is to approximately 500 µM affinity – limited by solubility. The main advantage of the method is the small amount of protein required – the main disadvantage is that it can be a challenge to immobilize the protein on the surface and retain binding integrity – some experience is required (and a tool compound) to assess for correct behaviour. In SPR, the protein sample is immobilized and the surface must be washed after each binding experiment. Thus, SPR is more sensitive to compounds inducing aggregation or having slow kinetics. MST – this method works on the principle that the speed at which a molecule moves in a temperature gradient is related to its hydration sphere, electrostatic surface and weight, which are affected by compound binding. The temperature gradient is induced by an infrared laser; movement is detected from a fluorophore (usually added) on the protein. This is a relatively new technique and experience (and suitable controls) is still growing in the community outside the equipment manufacturers [31]. The challenge is to introduce a suitable fluorophore that does not perturb the interaction being measured and success varies with different targets. If successful, the method is rapid and uses small amounts of protein. Thermal shift analysis – it is also known as differential scanning fluorimetry. In this method, fragment binding is detected from changes to the thermal stability (melting temperature or Tm) of the protein – measured by detecting binding of a fluorescent dye to the protein as it unfolds when heated. The advantage is that quite small amounts of materials are needed and that inexpensive thermal cycling instruments (such as for PCR) can be used. The challenge is that small fragments, at best, introduce only a small change to the stability of the protein at the usual Tm values (50–70°C) and so there are a lot of false negatives [32]. However, it is very attractive for academic groups and will usually find some fragments that bind. Biochemical assay – some assay formats (both binding and functional) will withstand the high concentrations that are used in fragment screening, but there can be challenges with interference if detection is spectrophotometric or fluorometric. Sometimes, the assay will successfully report on fragment binding or activity, sometimes not [26]. This is the reason the higher concentration section is marked shaded. Mass spectrometry (MS) – the main challenge with MS for detection of protein–ligand binding is finding suitable conditions to maintain binding in the gas phase – this is particularly challenging for the weak binding of fragments and although a few reports have appeared [33], the method is a challenge for fragments with 100 µM or so the upper limit of sensitivity. One imaginative recent idea is to mix the fragments with the protein, remove the free fragments, liberate the bound fragments and identify them with MS [34].
The three most widely used methods (http://practicalfragments.blogspot.co.uk/) for fragment screening are ligand-observed NMR (reviewed in [35]), SPR (for example, [36]) and X-ray crystallography (for example, [37]). The different characteristics of the techniques are discussed in more detail in the legend of Figure 2, but the distinctive features can be summarized as: (a) the main advantage of ligand-observed NMR is that it is label free and a measurement in free solution. Furthermore, the solubility and stability of both the protein and the fragment can be evaluated for each measurement. This is particularly important for the more challenging target proteins where stability can be an issue – aggregation or precipitation of the protein is the most usual cause of false positives or false negatives in many assay formats; (b) SPR is chosen by many practitioners as the central screening platform as it uses small amounts of protein and can be used to screen relatively rapidly – the main issue is whether the protein can be immobilized to a surface and retain its binding integrity. What can also be important for both SPR and NMR is to assess the effect of a competitive ligand on putative hit fragments to check for non-specific binding; (c) X-ray crystallography provides an immediate model of the fragment binding to the protein. Because it is usually performed with very high concentration of fragment (10s mM), it usually gives a higher hit rate than other techniques [38]. However, it requires a crystal system suitable for soaking of fragments that can be a major issue for some targets.
Generating a model for fragment binding
It is relatively straightforward to find fragments that bind to most binding sites on most proteins [39,40]. What is a challenge is knowing what to do with them. For most targets with only weak binding fragments, it is very difficult to generate useful SAR for a fragment by trial and error synthesis. Most of the modifications result in a loss or a change of binding affinity that is difficult to differentiate across compounds. All successful fragment optimization reported to date has relied on some model of how the fragment binds to the target. The most robust model comes from X-ray crystallography, and for well-behaved conventional targets it can be possible to determine many hundreds of crystal structures in the early stages of a project to enable the medicinal chemist to make informed decisions about optimization. Where crystallography is not possible, then NMR methods can provide sufficient insight to generate a model, although it can take a number of weeks to generate each model.
Fragment to lead optimization
A schematic of the three main approaches in fragment to lead optimization is shown in Figure 3. The following discussion includes some examples of each approach with structures summarized in Figure 4.
Figure 3. Strategies for fragment optimization.

Linking, growing and merging – see text for details.
Figure 4. Fragment to lead (to drug) optimization.

(A) Fragment linking – the discovery of venetoclax – a selective Bcl-2 inhibitor; two-site screening by protein observed NMR followed by structure determination and optimization identified fragments ‘1’ and ‘2’ as inhibitors of Bcl-xL. The initial combination of these to ‘3’ and then ‘4’ gave a potent inhibitor that was subsequently optimized to ‘5’ that as ABT-737 entered clinical trials as a dual Bcl-xL/Bcl-2 inhibitor [41] that established proof of concept that inhibition of these anti-apoptotic proteins generates a therapeutic effect. Subsequent design of ‘6’ (ABT-263) gave a compound with improved properties that highlighted that Bcl-xL inhibition led to thrombocytopenia, further design led to the selective Bcl-2 inhibitor ‘7’ that as venetoclax was approved for chronic lymphocytic leukaemia (CLL) [6]. (B) Fragment growing – the discovery of vemurafenib – a selective inhibitor of the B-Raf V600E mutant kinase; fragment ‘8’ was identified from a biochemical screen against the kinase Pim-1 and early structure-guided design used structures bound to Pim-1 and FGFR to identify fragment ‘9’ and the compound ‘10’ (PLX4720). This was then optimized to ‘11’ (vemurafenib) that is now used for the treatment of mutant B-Raf driven melanoma [7]. (C) Fragment merging – a ligand-observed NMR screen against the protein kinase, PDPK1, identified fragments ‘12’ and ‘13’ and compound ‘14’ was identified by SAR by catalogue and subsequent optimization. All these information were combined with the core of the promiscuous literature inhibitor ‘15’ to give the optimized compound ‘16’ that was relatively selective over other kinases, active on cells and gave the expected pharmacodynamics marker responses when administered in vivo [42].
‘Fragment linking’ is the initial SAR by NMR approach from Fesik and co-workers [2] and consists of five steps – identification of the first fragment, optimization of the first fragment (following structure determination), screening a library of smaller fragments in the presence of the first fragment, optimization of the second fragment and then linking of the two fragments. This was successful for a number of targets for this group [43] but has not been repeated by many others. There are two challenges: the first is whether the binding site has appropriate pockets to accommodate separate fragments; the second is the challenge of the chemistry to link without affecting the detail of the binding orientation and position of the two fragments. The combinatorics of this type of approach is attractive – if each screen is of 1000 fragments and chemistry is possible for 100 ways of linking, then some 1000 × 1000 × 100 (100 million) possible molecules have been assessed in just 1000 + 1000 + 100 (2100) experiments. Figure 4A shows the most advanced example from the SAR by NMR approach – the discovery of the compound ‘7’, venetoclax [6], now treating patients with certain forms of CLL.
‘Fragment growing’ is the most widely applied strategy in FBLD. Figure 4B summarizes the discovery of ‘11’, vemurafenib – a selective inhibitor of V600E mutant B-Raf kinase, a strong driver of melanoma. The initial fragment was optimized by careful structure-based design to introduce the desired selectivity and drug-like properties. There are many similar examples of such structure-guided fragment optimization, such as the Aurora A inhibitor from Astex [44]. A variant of this approach is to search available chemicals for compounds containing the central binding motif of the initial fragment hit – so-called SAR by catalogue. An example of this is contribution to the the rapid discovery by Vernalis and the Institute of Cancer Research of AUY-922, an inhibitor of the molecular chaperone Hsp90, which entered Phase II trials [45].
‘Fragment merging’ is an approach that combines information from multiple chemical hits together. It relies on multiple crystal structures and careful structure-based design. Figure 4C summarizes the steps in the discovery of a cell-active selective inhibitor of the kinase PDPK1 at Vernalis [42], generating a compound that suggested that inhibition of this kinase would not have the desired therapeutic effect. There are fewer examples of this type of approach – as it relies on multiple crystal structures of many chemotypes and a more ambitious approach to chemical design. Another striking example from Vernalis is the identification of an orally active Hsp90 inhibitor, BEP-800, which entered pre-clinical trials [45]. An extension of the merging approach is particularly powerful – i.e. combining the information from the fragments with information about inhibitors identified from the literature, from high-throughput screening (HTS) or natural ligands. One very nice example is work on the enzyme BCATm by a team from GSK, where the fragment was combined with an HTS hit to give a potent lead [46].
One of the common mistakes in fragments to hits to leads chemistry is beginning the process of fragment growth before the core of the fragment itself is explored and optimized or before the full set of fragment hits has been characterized. There are two aspects to this. Firstly, as discussed earlier, screening is a numbers game and it is unlikely that the optimal fragment for a particular binding site will be in the fragment library. It is therefore important to explore closely related chemotypes to a fragment – by compound purchase (SAR by catalogue) or by limited synthesis, if at all possible probing how small changes affect binding (methyl walk, moving nitrogens around a heterocycle etc). Secondly, exploratory evolution around fragments is a powerful way of mapping a binding site for particular features that add to the affinity and selectivity of ligands – having a marked impact on the success of optimization.
Recent developments
So far in this essay, we have summarized current practice in using the now mature fragment-based methods for lead discovery. Here, we briefly summarize some of the recent developments and applications of fragment-based approaches and thinking.
Fragments and chemical biology
As already mentioned, some of the early research used either computational [14,15] or experimental [16,17] methods to explore binding of small functional groups to a protein. Early analyses by a number of groups [39,40] demonstrated how fragments can be found for essentially all binding sites on all proteins – and the number of fragments gives some indication of the ligandability of the binding site, i.e. how chemically attractive it is for a small molecule. An extension of this has been the realization that fragments can reveal potential binding sites on proteins [47] although binding to many of these secondary sites might not affect function.
There have been some recent publications highlighting how such fragment binding can explore a protein or a biological system.
Mutations in the protein K-Ras have long been identified as a key driver of many different cancers. It is a relatively small G-protein, where a switch from binding of GDP to GTP induces a conformational change which triggers multiple signalling cascades. It has proven intractable to drug discovery efforts over the decades. Various groups have recently used fragments to identify novel binding sites on the protein and this has reignited interest in the target [48–50].
There has been considerable interest in covalent fragments – i.e. fragments that contain a reactive group that will form a covalent bond with the protein. The origins of these ideas were in the tethering work of the company Sunesis, who developed a library of fragments which would react with surface exposed cysteines [4]. An extension of this approach is to introduce a cysteine mutation close to the functional site [51] – an interesting example of this type of approach was also demonstrated for K-Ras where a particular mutation seen in some tumours (G12C) was exploited [48]. A particularly impressive extension of these ideas is the work of the Cravatt group [52] who designed a fragment library where each fragment contained a photoactivatable diazirine and an alkyne group. The fragments were incubated with cells and exposed to light so that the fragments were covalently linked to their binding partner protein. Click chemistry was then used to tag the labelled protein with a fluorescent moiety and subsequent MS identified the targets labelled.
Fragments as enzyme activators
There are well-established targets such as nuclear receptors or G-protein coupled receptors, where the aim in lead discovery is to identify agonists – i.e. compounds that modulate the conformation of the protein to generate biological function. However, there are very few examples where small molecules activate an enzyme activity. Recent work has demonstrated that non-covalent binding of a fragment can increase enzyme activity [53] and covalent attachment of the fragment is being explored to provide improved industrial enzymes (unpublished). There are a variety of physiological processes where activation of activity could be beneficial – and this could be attractive for therapeutic intervention, as it may be possible to achieve some benefit with just a small increase of activity.
Computational approaches
There have been massive increases in the speed of computation over recent years. This has helped advance the use of computational chemistry methods in FBLD but arguably, there have not been any major advances in the underlying methods or computational models that are used. If a structure is available for the protein target, and the ligand binds without major conformational changes, then computational docking of ligands will generate a number of poses (positions and orientations) for the ligand that includes the actual pose, but it remains a challenge to correctly calculate the binding energy and thus recognize that pose. The situation is even more challenging for fragments, as the number of interactions made (and the binding affinity) is much smaller. One interesting new idea is that it is the activation energy for dissociation (off-rate) that dominates the energetics of binding. This has been explored in a dynamic undocking approach called DUCK [54] where the work required to break a key interaction is estimated as a surrogate for the binding affinity.
One area that has received considerable attention in recent years is using molecular simulation methods to map the protein surface for which functional groups will bind, such as the programs FTMAP [55] and MDmix [56]. These can be seen as an extension of the earlier GRID [14] and MCSS [15] methods, but incorporating flexibility into the protein target and exploiting the massive increase in available computational power. It is arguable how predictive these methods can be, but they do provide tools to explore flexibility and generate ideas about which functional groups could be useful to incorporate as a fragment is optimized.
Final remarks
In many ways, a fragment is no different to any other small molecule that binds to a protein – the main advantage is that the chance of a fragment binding is much greater than a larger molecule. The main challenges have been in developing the methods and experience in both identifying binding of such weak compounds and the methods for optimizing the fragments to lead compounds. The main advantage for the medicinal chemist is that because the chemistry is starting small and there are usually many fragments to consider, there are more opportunities to make better quality decisions in generating a lead compound with optimal properties. This has provided novel, selective lead compounds for conventional targets beyond those identified through more conventional, HTS approaches. More striking are the opportunities offered for challenging targets. Here, the biophysical methods, in particular NMR, are able to identify and validate binding of fragments to the target – often providing starting points for lead discovery when other techniques, such as HTS, have failed.
For drug discovery, a major impact of FBLD methods has been driving development of tools (particularly biophysical methods) and experience in characterizing binding of ligands to proteins. This has developed a mindset that enables problem solving in early lead discovery – in particular characterizing whether and how a ligand is interacting with a protein. This can be particularly important when working with new classes of target where establishing the relationship between ligand binding and functional activity can be a challenge.
For the academic researcher, the methods are attractive since binders can be identified using a small library, not requiring the massive outlay in compounds and automation that is required for HTS; most of the biophysical techniques required for fragment screening are also available in most institutions. As stressed earlier, it is relatively easy to identify fragments that bind to most binding sites on most targets; what is difficult is knowing what to do with them. What recent research has shown, however, is that imaginative use of fragment-based methods can deliver new approaches and results in chemical biology – i.e. chemical tools to probe, understand and modulate biological systems and function.
Summary
There is great experience in design of fragment libraries and methods for finding fragments that bind.
Fragments can be found for most targets including those that fail in HTS.
Fragments explore the chemical space of what can bind to a target, providing new chemical entities even for well-characterized targets such as protein kinases.
Fragments that bind with millimolar affinity can be evolved into potent lead compounds.
There are now many compounds in clinical trials that were discovered from fragment methods – and two products on the market treating patients.
There are emerging ideas of using fragments to probe biological systems – as an additional tool in chemical biology.
Abbreviations
- CLL
chronic lymphocytic leukaemia
- CPMG
Carr–Purcell–Meiboom–Gill sequence
- FBLD
fragment-based lead discovery
- HSQC
heteronuclear single quantum correlation spectroscopy
- HTS
high-throughput screening
- LOGSY
ligand-observed gradient spectroscopy
- MCSS
multiple copy simultaneous search
- MS
mass spectrometry
- MSCS
multiple solvent crystal structure
- MST
microscale thermophoresis
- MW
molecular weight
- NMR
nuclear magnetic resonance
- SAR
structure–activity relationship
- SPR
surface plasmon resonance
- STD
saturation transfer difference
Funding
B.L. is supported by the European Union’s Horizon2020 MSCA Programme under grant agreement 675899 (FRAGNET); research in the R.E.H. group is additionally supported by research grants from the BBSRC and institutional infrastructure support provided by the Wellcome Trust and EPSRC.
Author Contribution
R.E.H. conceived the general structure of the essay, B.L. constructed the figures, both authors contributed to the writing of the essay.
Competing Interests
The authors declare that there are no competing interests associated with the manuscript.
References
- 1.Nasiri H.R., Bell N.M., McLuckie K.I.E., Husby J., Abell C., Neidle S. et al. (2014) Targeting a c-MYC G-quadruplex DNA with a fragment library. Chem. Commun. 50, 1704–1707 [DOI] [PubMed] [Google Scholar]
- 2.Shuker S.B., Hajduk P.J., Meadows R.P. and Fesik S.W. (1996) Discovering high-affinity ligands for proteins: SAR by NMR. Science 274, 1531–1534 [DOI] [PubMed] [Google Scholar]
- 3.Congreve M., Carr R., Murray C. and Jhoti H. (2003) A ‘rule of three’ for fragment-based lead discovery? Drug Discov. Today 8, 876–877 [DOI] [PubMed] [Google Scholar]
- 4.Erlanson D.A., Ballinger M.D. and Wells J.A. (2006) Tethering. Fragment-Based Approaches in Drug Discovery, pp. 285–310, Wiley-VCH Verlag GmbH & Co. KGaA [Google Scholar]
- 5.Hubbard R.E., Davis B., Chen I. and Drysdale M.J. (2007) The SeeDs approach: integrating fragments into drug discovery. Curr. Top. Med. Chem. 7, 1568–1581 [DOI] [PubMed] [Google Scholar]
- 6.Souers A.J., Leverson J.D., Boghaert E.R., Ackler S.L., Catron N.D., Chen J. et al. (2013) ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 19, 202–208 [DOI] [PubMed] [Google Scholar]
- 7.Bollag G., Tsai J., Zhang J., Zhang C., Ibrahim P., Nolop K. et al. (2012) Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 11, 873–886 [DOI] [PubMed] [Google Scholar]
- 8.Erlanson D.A., Fesik S.W., Hubbard R.E., Jahnke W. and Jhoti H. (2016) Twenty years on: the impact of fragments on drug discovery. Nat. Rev. Drug Discov. 15, 605–619 [DOI] [PubMed] [Google Scholar]
- 9.Renaud J.P., Chung C.W., Danielson U.H., Egner U., Hennig M., Hubbard R.E. et al. (2016) Biophysics in drug discovery: impact, challenges and opportunities. Nat. Rev. Drug Discov. 15, 679–695 [DOI] [PubMed] [Google Scholar]
- 10.Johnson C.N., Erlanson D.A., Murray C.W. and Rees D.C. (2017) Fragment-to-lead medicinal chemistry publications in 2015. J. Med. Chem. 60, 89–99 [DOI] [PubMed] [Google Scholar]
- 11.Erlanson D.A. and Jahnke W. (2016) Fragment-Based Drug Discovery: Lessons and Outlook, Wiley [Google Scholar]
- 12.Hubbard R.E. (2015) Fragment based lead discovery. In The Handbook of Medicinal Chemistry: Principles and Practice: The Royal Society of Chemistry, 10.1039/9781782621836-00122 [DOI] [Google Scholar]
- 13.Jencks W.P. (1981) On the attribution and additivity of binding energies. Proc. Natl. Acad. Sci. U.S.A. 78, 4046–4050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goodford P.J. (1985) A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 28, 849–857 [DOI] [PubMed] [Google Scholar]
- 15.Miranker A. and Karplus M. (1991) Functionality maps of binding sites: a multiple copy simultaneous search method. Proteins 11, 29–34 [DOI] [PubMed] [Google Scholar]
- 16.Ringe D. and Mattos C. (2006) Location of Binding Sites on Proteins by the Multiple Solvent Crystal Structure Method. Fragment-based Approaches in Drug Discovery, pp. 67–88, Wiley-VCH Verlag GmbH & Co. KGaA [Google Scholar]
- 17.English A.C., Done S.H., Caves L.S., Groom C.R. and Hubbard R.E. (1999) Locating interaction sites on proteins: the crystal structure of thermolysin soaked in 2% to 100% isopropanol. Proteins 37, 628–640 [PubMed] [Google Scholar]
- 18.Hajduk P.J. and Greer J. (2007) A decade of fragment-based drug design: strategic advances and lessons learned. Nat. Rev. Drug Discov. 6, 211–219 [DOI] [PubMed] [Google Scholar]
- 19.Nienaber V.L., Richardson P.L., Klighofer V., Bouska J.J., Giranda V.L. and Greer J. (2000) Discovering novel ligands for macromolecules using X-ray crystallographic screening. Nat. Biotechnol. 18, 1105–1108 [DOI] [PubMed] [Google Scholar]
- 20.Boehm H.J., Boehringer M., Bur D., Gmuender H., Huber W., Klaus W. et al. (2000) Novel inhibitors of DNA gyrase: 3D structure based biased needle screening, hit validation by biophysical methods, and 3D guided optimization. A promising alternative to random screening. J. Med. Chem. 43, 2664–2674 [DOI] [PubMed] [Google Scholar]
- 21.Hann M.M., Leach A.R. and Harper G. (2001) Molecular complexity and its impact on the probability of finding leads for drug discovery. J. Chem. Inf. Comput. Sci. 41, 856–864 [DOI] [PubMed] [Google Scholar]
- 22.Hopkins A.L., Groom C.R. and Alex A. (2004) Ligand efficiency: a useful metric for lead selection. Drug Discov. Today 9, 430–431 [DOI] [PubMed] [Google Scholar]
- 23.Baurin N., Aboul-Ela F., Barril X., Davis B., Drysdale M., Dymock B. et al. (2004) Design and characterization of libraries of molecular fragments for use in NMR screening against protein targets. J. Chem. Inf. Comput. Sci. 44, 2157–2166 [DOI] [PubMed] [Google Scholar]
- 24.Keseru G.M., Erlanson D.A., Ferenczy G.G., Hann M.M., Murray C.W. and Pickett S.D. (2016) Design principles for fragment libraries: maximizing the value of learnings from pharma fragment-based drug discovery (fbdd) programs for use in academia. J. Med. Chem. 59, 8189–8206 [DOI] [PubMed] [Google Scholar]
- 25.Lau W.F., Withka J.M., Hepworth D., Magee T.V., Du Y.J., Bakken G.A. et al. (2011) Design of a multi-purpose fragment screening library using molecular complexity and orthogonal diversity metrics. J. Comput.-Aided Mol. Des. 25, 621–636 [DOI] [PubMed] [Google Scholar]
- 26.Hubbard R.E. and Murray J.B. (2011) Experiences in fragment-based lead discovery. Methods Enzymol. 493, 509–531 [DOI] [PubMed] [Google Scholar]
- 27.Meiby E., Simmonite H., le Strat L., Davis B., Matassova N., Moore J.D. et al. (2013) Fragment screening by weak affinity chromatography: comparison with established techniques for screening against HSP90. Anal. Chem. 85, 6756–6766 [DOI] [PubMed] [Google Scholar]
- 28.Schiebel J., Krimmer S.G., Rower K., Knorlein A., Wang X., Park A.Y. et al. (2016) High-throughput crystallography: reliable and efficient identification of fragment hits. Structure 24, 1398–1409 [DOI] [PubMed] [Google Scholar]
- 29.Pearce N.M., Krojer T., Bradley A.R., Collins P., Nowak R.P., Talon R. et al. (2017) A multi-crystal method for extracting obscured crystallographic states from conventionally uninterpretable electron density. Nat. Comm. 8, 15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fischer M., Leech A.P. and Hubbard R.E. (2011) Comparative assessment of different histidine-tags for immobilization of protein onto surface plasmon resonance sensorchips. Anal. Chem. 83, 1800–1807 [DOI] [PubMed] [Google Scholar]
- 31.Linke P., Amaning K., Maschberger M., Vallee F., Steier V., Baaske P. et al. (2016) An Automated Microscale Thermophoresis Screening Approach for Fragment-Based Lead Discovery. J. Biomol. Screen 21, 414–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schulz M.N., Landstrom J. and Hubbard R.E. (2013) MTSA-a Matlab program to fit thermal shift data. Anal. Biochem. 433, 43–47 [DOI] [PubMed] [Google Scholar]
- 33.Vivat V., Atmanene C., Denis Z., Van Dorsselaer A. and Sanglier-Cianférani S. (2010) Native MS: An ESI way to support structure- and fragment-based drug discovery. J. Biomol. Screen 21, 414–421 [DOI] [PubMed] [Google Scholar]
- 34.Chen X., Qin S., Chen S., Li J., Li L., Wang Z. et al. (2015) A Ligand-observed mass spectrometry approach integrated into the fragment based lead discovery pipeline. Sci. Rep. 5, article:8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gossert A.D. and Jahnke W. (2016) NMR in drug discovery: a practical guide to identification and validation of ligands interacting with biological macromolecules. Prog. Nucl. Magn. Reson. Spectrosc. 97, 82–125 [DOI] [PubMed] [Google Scholar]
- 36.Giannetti A.M. (2011) From experimental design to validated hits a comprehensive walk-through of fragment lead identification using surface plasmon resonance. Methods Enzymol. 493, 169–218 [DOI] [PubMed] [Google Scholar]
- 37.Davies D.R., Begley D.W., Hartley R.C., Staker B.L. and Stewart L.J. (2011) Predicting the success of fragment screening by x-ray crystallography. Methods Enzymol. 493, 91–114 [DOI] [PubMed] [Google Scholar]
- 38.Schiebel J., Radeva N., Krimmer S.G., Wang X., Stieler M., Ehrmann F.R. et al. (2016) Six biophysical screening methods miss a large proportion of crystallographically discovered fragment hits: a case study. ACS Chem. Biol. 11, 1693–1701 [DOI] [PubMed] [Google Scholar]
- 39.Chen I.J. and Hubbard R.E. (2009) Lessons for fragment library design: analysis of output from multiple screening campaigns. J. Comput.-Aided Mol. Des. 23, 603–620 [DOI] [PubMed] [Google Scholar]
- 40.Hajduk P.J., Huth J.R. and Fesik S.W. (2005) Druggability indices for protein targets derived from NMR-based screening data. J. Med. Chem. 48, 2518–2525 [DOI] [PubMed] [Google Scholar]
- 41.Oltersdorf T., Elmore S.W., Shoemaker A.R., Armstrong R.C., Augeri D.J., Belli B.A. et al. (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681 [DOI] [PubMed] [Google Scholar]
- 42.Hubbard R.E. (2008) Fragment approaches in structure-based drug discovery. J. Synchrotron Radiat. 15, 227–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hajduk P.J. (2006) SAR by NMR: putting the pieces together. Mol. Interv. 6, 266–272 [DOI] [PubMed] [Google Scholar]
- 44.Howard S., Berdini V., Boulstridge J.A., Carr M.G., Cross D.M., Curry J. et al. (2009) Fragment-based discovery of the pyrazol-4-yl urea (AT9283), a multitargeted kinase inhibitor with potent aurora kinase activity. J. Med. Chem. 52, 379–388 [DOI] [PubMed] [Google Scholar]
- 45.Roughley S., Wright L., Brough P., Massey A. and Hubbard R.E. (2011) Hsp90 inhibitors and drugs from fragment and virtual screening. Top. Curr. Chem. 317, 61–82 [DOI] [PubMed] [Google Scholar]
- 46.Bertrand S.M., Ancellin N., Beaufils B., Bingham R.P., Borthwick J.A., Boullay A.B. et al. (2015) The discovery of in vivo active mitochondrial branched-chain aminotransferase (BCATm) inhibitors by hybridizing fragment and HTS hits. J. Med. Chem. 58, 7140–7163 [DOI] [PubMed] [Google Scholar]
- 47.Ludlow R.F., Verdonk M.L., Saini H.K., Tickle I.J. and Jhoti H. (2015) Detection of secondary binding sites in proteins using fragment screening. Proc. Natl. Acad. Sci. U.S.A. 112, 15910–15915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ostrem J.M., Peters U., Sos M.L., Wells J.A. and Shokat K.M. (2013) K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maurer T., Garrenton L.S., Oh A., Pitts K., Anderson D.J., Skelton N.J. et al. (2012) Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl. Acad. Sci. U.S.A. 109, 5299–5304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun Q., Burke J.P., Phan J., Burns M.C., Olejniczak E.T., Waterson A.G. et al. (2012) Discovery of Small Molecules that Bind to K-Ras and Inhibit Sos-Mediated Activation. Angew. Chem., Int. Ed. 51, 6140–6143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller R.M., Paavilainen V.O., Krishnan S., Serafimova I.M. and Taunton J. (2013) Electrophilic fragment-based design of reversible covalent kinase inhibitors. J. Am. Chem. Soc. 135, 5298–5301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parker C.G., Galmozzi A., Wang Y., Correia B.E., Sasaki K., Joslyn C.M. et al. (2017) Ligand and target discovery by fragment-based screening in human cells. Cell 168, 527–541, e29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Darby J.F., Landstrom J., Roth C., He Y., Davies G.J. and Hubbard R.E. (2014) Discovery of selective small-molecule activators of a bacterial glycoside hydrolase. Angew. Chem., Int. Ed. Engl. 53, 13419–13423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ruiz-Carmona S., Schmidtke P., Luque F.J., Baker L., Matassova N., Davis B. et al. (2017) Dynamic undocking and the quasi-bound state as tools for drug discovery. Nat. Chem. 9, 201–206 [DOI] [PubMed] [Google Scholar]
- 55.Kozakov D., Grove L.E., Hall D.R., Bohnuud T., Mottarella S.E., Luo L. et al. (2015) The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 10, 733–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alvarez-Garcia D. and Barril X. (2014) Molecular simulations with solvent competition quantify water displaceability and provide accurate interaction maps of protein binding sites. J. Med. Chem. 57, 8530–8539 [DOI] [PubMed] [Google Scholar]