

Abstract
In an in vitro cytopathic effect inhibition assay with the H3N2 influenza virus A/Hong Kong/68 (HK/68), the bark extract of Burkea africana was found to be a promising antiviral lead with an IC50 value of 5.5 μg/mL without noteworthy cytotoxicity in Madin Darby canine kidney cells. After several chromatographic steps, triterpene saponins of the lupane and oleanane types were identified as the bioactive principles. In total, eight new triterpene saponins (1–8) with four so far undescribed aglycone structures were isolated and characterized via HRESIMS, GC-MS, and 1D and 2D NMR spectroscopy. Their anti-influenza virus activity on HK/68 and the 2009 pandemic H1N1 strain A/Jena/8178/09 revealed the most potent effects by compounds 7 and 8, with IC50 values between 0.05 and 0.27 μM. This is the first time triterpene saponins have been reported as constituents of the investigated plant material.
Burkea africana Hook. is known under various names such as “wild syringa”, “seringa tree”, and “Rhodesian ash” in English or “mpulu” (Tsonga), “monato” (Tswana), “mufhulu” (Venda), and “wildesering” in Afrikaans. It is a flat-topped tree up to 20 m high, belonging to the family Leguminosae and widely distributed in the tropical and subtropical African regions. This species is difficult to grow and not yet planted on a commercial scale.1
Traditionally, the roots of B. africana are used to treat toothache and stomach pain. The bark, rich in plant polyphenols, is used for leather tanning or pounded as a fish stupefiant and an arrow poison antidote. Bark decoctions are used for the treatment of colds, coughs, and stomach obstruction, while infusions are applied in the treatment of gonorrhea and syphilis.2 The stem bark is documented as possessing antidiarrheal, antioxidant, and antibacterial activity against Salmonella typhi(3−5) and to inhibit the hyaluronidase of the black-necked spitting cobra, Naja nigricollis.6
In the literature, little can be found on the phytochemical composition of B. africana.7 Described compound classes, identified solely by using simple color reactions or by TLC comparison, comprise alkaloids, cardiac glycosides, flavonoids, phenolic acids, terpenoids and tannins, and both hydrolyzable polyphenols and proanthocyanidins.1,4,7 To date, no triterpene saponins have been reported for this species.
Triterpene saponins are distributed widely as plant secondary metabolites with great structural diversity, which is also reflected in the variety of reported biological and pharmacological properties, such as antidiabetic, antifungal, cytotoxic, hepatoprotective, anticancer, chemopreventive, antiallergic, immunomodulatory, immunostimulatory, molluscicidal, hemolytic, and anti-inflammatory activities.8−11 Moreover, antiviral activities against, for example, human respiratory syncytial virus (HRSV), human immunodeficiency virus (HIV), herpes simplex type 1 and 2 viruses (HSV), human rhinovirus (RV), tobacco mosaic virus (TMV), or hepatitis B virus (HBV) have been described.12−17 So far, there are only a few reports on the anti-influenza virus activity of triterpene saponins.18 Recently, Song et al. described the anti-influenza virus (H1N1) activity of uralsaponins isolated from Glycyrrhiza uralensis,17 and Chen et al. demonstrated the anti-influenza A activity of saikosaponin A in vitro and in vivo.19
In this study, eight triterpene saponins (1–8) were isolated from the ethanolic stem bark extract of B. africana and identified by means of HRESIMS, GC-MS, and 1D and 2D NMR spectroscopic methods. Herein, the isolation, structural elucidation, and the anti-influenza properties of these compounds are described.
Results and Discussion
Screening for Anti-Influenza Natural Materials
To identify anti-influenza natural leads, some 160 lead-like enhanced extracts of various plant and fungal species were prepared as described recently.20,21 Briefly, defatted materials were extracted successively with dichloromethane and methanol. The two resulting extracts were combined in order to cover a wide and drug-like range of polarity within one extract. Finally, tannin depletion via polyamide gel was carried out in order to remove common assay interfering compounds.
In a cytopathic effect (CPE) inhibitory assay with the H3N2 influenza virus A/Hong Kong/68 (HK/68) in Madin Darby canine kidney (MDCK) cells, the bark extract of B. africana (BA-E) was identified as a strong inhibitor of the CPE with an IC50 of 5.5 μg/mL. Moreover, this extract was not cytotoxic to MDCK cells (Table S1, Supporting Information).
Assessment of Anti-Influenza Virus Activity of B. africana Bark
For the phytochemical workup, an ethanolic extract of the bark material of B. africana was generated in a larger scale and chromatographically separated via polyamide to obtain 10 fractions, whereby fractions A3 to A10 contained increasing amounts of tannins as monitored via TLC. The fractions were well tolerated by MDCK cells (Table S1 and Figure S1, Supporting Information). In the CPE assay, the tannin-rich fractions A3 to A10 showed activity rising parallel to their polarity. This might have been related to the increasing presence of polyphenols, which have been previously described as anti-influenza agents.22−24 Since the focus of this study was to identify novel anti-influenza scaffolds, polyphenol-enriched fractions were not considered in the subsequent phytochemical workup. Hence, the tannin-depleted fractions (A1 and A2), which showed distinct antiviral effects against IAV HK/68 with IC50 values of 4.0 and 3.0 μg/mL, respectively, were pooled for further phytochemical workup.
Microfractionation of the Tannin-Depleted Fraction: Activity Profiling and Dereplication
With a small aliquot of a tannin-depleted fraction, a time-based microfractionation via flash chromatography was performed to give 13 fractions (B1 to B13). These were then tested in the CPE inhibition assay as well as in the cytotoxicity assay (Table S2 and Figure S1, Supporting Information). Fractions B7 to B9 displayed potent anti-influenza virus activity with IC50 values of 0.17, 0.13, and 0.17 μg/mL, respectively. Moreover, the calculated selectivity indices (SI, CC50/IC50) for B7 to B9 were between 6 and 14, suggesting a virus-specific activity of the contained compounds.
Dereplication of the bioactive microfractions using HRESIMS suggested triterpenoid saponins as the major constituents based on characteristic neutral losses of carbohydrate residues and fragment ions typical for C30 aglycones.
Isolation and Identification of Triterpene Saponins from B. africana
For the targeted isolation of triterpene saponins, 28 g of the tannin-depleted extract were dissolved in water and partitioned sequentially between petroleum ether, ethyl acetate (EtOAc), and n-butanol. By using HRESIMS analysis, the EtOAc fraction was found to be enriched with the triterpene saponins of interest. Hence, this fraction was subjected to several chromatographic steps including size exclusion, supercritical fluid, and flash CC, resulting in the isolation of eight pure triterpene saponins (1–8). Their structures were identified by means of HRESIMS as well as 1D and 2D NMR methods.

The full assignments of the 13C and 1H NMR spectroscopic data of isolated compounds 1–8 are listed in Tables 1 to 4. The respective NMR and HRESIMS spectra are provided in the Supporting Information.
Table 1. 13C NMR Spectroscopic Data of 1–4 (125 MHz, CD3OD, δ in ppm, Multiplicities Determined by APT Experiments).
| position | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| 1 | 47.7, CH2 | 47.7, CH2 | 47.8, CH2 | 47.9, CH2 |
| 2 | 68.2, CH | 68.2, CH | 68.3, CH | 68.3, CH |
| 3 | 96.5, CH | 96.7, CH | 96.5, CH | 96.9, CH |
| 4 | 41.7, C | 41.8, C | 41.8, C | 41.8, C |
| 5 | 56.9, CH | 56.9, CH | 57.0, CH | 56.9, CH |
| 6 | 19.3, CH2 | 19.3, CH2 | 19.2, CH2 | 19.3, CH2 |
| 7 | 35.4, CH2 | 35.4, CH2 | 34.1, CH2 | 34.2, CH2 |
| 8 | 41.9, C | 41.9, C | 42.7, C | 42.6, C |
| 9 | 51.9, CH | 51.9, CH | 53.2, CH | 53.1, CH |
| 10 | 38.9, C | 39.0, C | 39.2, C | 39.1, C |
| 11 | 22.2, CH2 | 22.2, CH2 | 22.4, CH2 | 22.5, CH2 |
| 12 | 26.8, CH2 | 26.8, CH2 | 26.3, CH2 | 26.3, CH2 |
| 13 | 39.6, CH | 39.5, CH | 40.2, CH | 40.2, CH |
| 14 | 43.6, C | 43.6, C | 47.4, C | 47.4, C |
| 15 | 30.8, CH2 | 30.8, CH2 | 36.5, CH2 | 36.5, CH2 |
| 16 | 33.3, CH2 | 33.5, CH2 | 24.3, CH2 | 24.3, CH2 |
| 17 | 57.4, C | 57.6, C | 57.3, C | 57.4, C |
| 18 | 50.3, CH | 50.4, CH | 50.7, CH | 50.7, CH |
| 19 | 48.4, CH | 48.4, CH | 48.4, CH | 48.4, CH |
| 20 | 151.9, C | 152.1, C | 152.0, C | 152.1, C |
| 21 | 31.6, CH2 | 31.7, CH2 | 31.6, CH2 | 31.7, CH2 |
| 22 | 38.1, CH2 | 38.3, CH2 | 38.0, CH2 | 38.1, CH2 |
| 23 | 28.4, CH3 | 28.3, CH3 | 28.4, CH3 | 28.6, CH3 |
| 24 | 17.3, CH3 | 17.2, CH3 | 17.3, CH3 | 17.8, CH3 |
| 25 | 17.9, CH3 | 17.9, CH3 | 18.4, CH3 | 18.3, CH3 |
| 26 | 16.6, CH3 | 16.6, CH3 | 17.0, CH3 | 17.0, CH3 |
| 27 | 15.0, CH3 | 15.1, CH3 | 61.0, CH2 | 60.9, CH2 |
| 28 | 180.1, C | 180.6, C | 180.2, C | 180.6, C |
| 29 | 19.5, CH3 | 19.5, CH3 | 19.6, CH3 | 19.6, CH3 |
| 30 | 110.2, CH2 | 110.1, CH2 | 110.2, CH2 | 110.2, CH2 |
| 1′ | 104.8, CH | 104.8, CH | 104.8, CH | 104.8, CH |
| 2′ | 82.1, CH | 81.9, CH | 82.1, CH | 80.3, CH |
| 3′ | 78.0, CH | 77.2, CH | 78.0, CH | 78.3, CH |
| 4′ | 71.2, CH | 78.3, CH | 71.2, CH | 71.1, CH |
| 5′ | 78.5, CH | 76.9, CH | 78.5, CH | 78.6, CH |
| 6′ | 62.2, CH2 | 61.4, CH2 | 62.2, CH2 | 63.2, CH2 |
| 1″ | 105.8, CH | 105.7, CH | 105.8, CH | 104.2, CH |
| 2″ | 76.0, CH | 76.0, CH | 76.0, CH | 76.1, CH |
| 3″ | 78.0, CH | 78.0, CH | 78.0, CH | 78.1, CH |
| 4″ | 70.9, CH | 71.2, CH | 71.0, CH | 72.0, CH |
| 5″ | 67.1, CH2 | 67.0, CH2 | 67.1, CH2 | 77.9, CH |
| 6″ | 62.3, CH2 | |||
| 1‴ | 102.6, CH | |||
| 2‴ | 72.4, CH | |||
| 3‴ | 72.2, CH | |||
| 4‴ | 73.7, CH | |||
| 5‴ | 70.5, CH | |||
| 6‴ | 17.8, CH3 |
Table 4. 1H NMR Spectroscopic Data of 5–8 (500 MHz, CD3OD, δ in ppm).
| position | 5 δH (J in Hz) | 6 δH (J in Hz) | 7 δH (J in Hz) | 8 δH (J in Hz) |
|---|---|---|---|---|
| 1 | 1.70, m | 0.89, m | 0.97, m | 0.97, m |
| 2.01, m | 2.01, m | 1.61, m | 1.61, m | |
| 2 | 3.73, m | 3.73, d (9.2) | 1.69, m | 1.69, m |
| 1.80, m | 1.78, m | |||
| 3 | 2.96, d (9.4) | 2.97, m | 3.11, dd (11.6, 4.4) | 3.11, dd (11.5, 4.6) |
| 5 | 0.84, m | 0.87, m | 0.78, m | 0.77, m |
| 6 | 1.43, m | 1.43, m | 1.41, m | 1.40, m |
| 1.57, m | 1.58, m | 1.58, m | 1.57, m | |
| 7 | 1.20, m | 1.34, m | 1.32, m | 1.32, m |
| 1.38, m | 1.53, m | 1.51, m | 1.52, m | |
| 9 | 1.62, m | 1.65, m | 1.60, m | 1.58, m |
| 11 | 1.59, m | 1.81, m | 1.81, m | 1.81, m |
| 2.11, m | 2.10, m | 2.10, m | ||
| 12 | 5.25, t (3.4) | 5.32, t (3.3) | 5.31, t (3.3) | 5.31, t (3.5) |
| 15 | 1.06, m | 1.12, m | 1.12, m | 1.12, m |
| 1.80, m | 1.77, m | 1.76, m | 1.77, m | |
| 16 | 1.94, m | 1.64, m | 1.91, m | 1.91, m |
| 1.96, m | ||||
| 18 | 2.87, dd (13.6, 4.1) | 2.98, m | 2.98, dd (13.8, 4.0) | 2.98, dd (13.9, 3.7) |
| 19 | 0.87, m | 1.33, m | 1.32, m | 1.31, m |
| 1.12, m | 1.95, m | 1.95, m | 1.95, m | |
| 21 | 1.33, m | 4.93, m | 4.93, dd (11.7, 5.0) | 4.93, dd (11.7, 5.1) |
| 1.53, m | ||||
| 22 | 1.74, m | 1.76, m | 1.76, m | 1.80, m |
| 1.82, m | ||||
| 23 | 1.11, s | 1.12, s | 1.05, s | 1.05, s |
| 24 | 0.90, s | 0.91, s | 0.84, s | 0.84, s |
| 25 | 1.02, s | 1.02, s | 0.96, s | 0.96, s |
| 26 | 0.83, s | 0.83, s | 0.83, s | 0.83, s |
| 27 | 1.15, s | 1.19, s | 1.19, s | 1.18, s |
| 29 | 0.90, s | 0.94, s | 0.94, s | 0.94, s |
| 30 | 0.95, s | 1.10, s | 1.10, s | 1.10, s |
| 32 | 6.52, d (16.0) | 6.52, d (16.0) | 6.52, d (16.0) | |
| 33 | 7.67, d (16.0) | 7.67, d (16.0) | 7.67, d (16.0) | |
| 35 | 7.62, m | 7.62, m | 7.62, m | |
| 36 | 7.40, m | 7.40, m | 7.40, m | |
| 37 | 7.40, m | 7.40, m | 7.40, m | |
| 38 | 7.40, m | 7.40, m | 7.40, m | |
| 39 | 7.62, m | 7.62, m | 7.62, m | |
| 1′ | 4.44, d (7.8) | 4.45, d (7.8) | 4.38, d (7.1) | 4.40, d (7.4) |
| 2′ | 3.59, dd (9.0, 7.8) | 3.59, dd (9.0, 7.8) | 3.42, m | 3.46, m |
| 3′ | 3.72, t (9.0) | 3.72, t (9.0) | 3.49, m | 3.61, m |
| 4′ | 3.63, dd (10.8, 9.0) | 3.63, dd (11.3, 9.0) | 3.49, m | 3.62, m |
| 5′ | 3.42, m | 3.42, m | 3.18, m | 3.21, m |
| 3.83, m | 3.97, m | |||
| 6′ | 3.67, dd (12.1, 3.9) | 3.67, dd (12.4, 3.9) | ||
| 3.81, m | 3.80, m | |||
| 1″ | 4.65, d (7.6) | 4.65, d (7.7) | 4.51, d (7.7) | 4.54, d (7.7) |
| 2″ | 3.22, dd (9.1, 7.6) | 3.22, dd (8.9, 7.7) | 3.46, m | 3.46, m |
| 3″ | 3.31, m | 3.31, m | 3.31, m | 3.31, m |
| 4″ | 3.44, m | 3.43, m | 3.49, m | 3.47, m |
| 5″ | 3.14, t (10.9) | 3.15, t (10.9) | 3.16, m | 3.16, m |
| 3.80, m | 3.80, m | 3.81, m | 3.83, m | |
| 1‴ | 4.86, m | 4.86, m | 5.14, d (1.5) | 5.15, d (1.4) |
| 2‴ | 3.82, m | 3.82, m | 3.93, dd (3.5, 1.5) | 3.93, dd (3.6, 1.6) |
| 3‴ | 3.61, t (8.7) | 3.61, t (8.8) | 3.70, dd (9.5, 3.5) | 3.70, dd (9.5, 3.5) |
| 4‴ | 3.40, m | 3.40, m | 3.39, t (9.5) | 3.38, t (9.4) |
| 5‴ | 3.98, dq (9.3, 6.2, 6.2, 6.2) | 3.99, dq (9.5, 6.2, 6.2, 6.2) | 4.01, dq (9.5, 6.2, 6.2, 6.2) | 3.91, dq (9.4, 6.1, 6.1, 6.1) |
| 6‴ | 1.26, d (6.2) | 1.26, d (6.2) | 1.24, d (6.2) | 1.25, d (6.1) |
| 1′′′′ | 4.79, d (1.4) | |||
| 2′′′′ | 3.75, dd (3.5, 1.4) | |||
| 3′′′′ | 3.67, dd (9.4, 3.5) | |||
| 4′′′′ | 3.39, t (9.4) | |||
| 5′′′′ | 4.01, dq (9.4, 6.1, 6.1, 6.1) | |||
| 6′′′′ | 1.26, d (6.1) |
The absolute configuration of the sugar residues of the saponins was determined by hydrolysis and subsequent conversion of the sugars to chiral diastereomers using (R)-(−)-2-butanol. After silylation with hexamethyldisilazane (HMDS) and trimethylchlorosilane (TMCS), these monosaccharide units were analyzed via GC-MS. The extracted ion chromatogram of the specific trimethylsilylated (R)-(−)-2-butyl glycoside fragments (m/z 175) allowed for the selective detection of the chiral diastereomers. The chromatograms obtained were compared to chromatograms of authentic reference sugars with known absolute configuration: d-glucose (d-Glc), d-xylose (d-Xyl), and l-rhamnose (l-Rha) (Figures S122–S125, Supporting Information).
Compound 1 was isolated as a white, amorphous powder with the molecular formula C41H66O13, obtained by HRESIMS from the [M – H]− ion at m/z 765.4437 (calcd for C41H65O13–, 765.4431, Δ = −0.8 ppm). The HRESIMS/MS spectrum of the [M + HCOO]− ion showed the loss of a terminal pentose with 132 Da and of a hexose (162 Da) to yield a Y0– ion at m/z 471.3485, corresponding to a C30H48O4 aglycone. These findings were confirmed by the MS/MS of the [M + Na]+ ion. In addition, C2 and B2 ions (the intact carbohydrate chain) of the [M + Na]+ ion indicated that the hexose and pentose units are arranged as a disaccharide, suggesting the presence of a monodesmosidic triterpene saponin. This was demonstrated in the 13C and 1H NMR spectra: among the 41 identified carbons, 11 could be assigned to the two sugar residues, with characteristic anomeric resonances [δC 104.8 (C-1′), δH 4.41 (H-1′); δC 105.8 (C-1″), δH 4.60 (H-1″)]. The remaining 30 carbons corresponded to the sapogenin, for which the 1H NMR spectrum exhibited resonances of six methyl singlets at δH 0.87, 0.92, 0.96, 1.00, 1.09, and 1.69. Additionally, it displayed two vinyl proton signals at δH 4.59 and δH 4.71 (H-30a/b). The 13C NMR spectrum showed resonances for one carboxylic acid carbon (δC 180.1, C-28), two oxygenated methines (δC 68.2, C-2; δC 96.5, C-3), two sp2 carbons (δC 151.9, C-20; δC 110.2, C-30), and six methyl moieties (δC 15.0, 16.6, 17.3, 17.9, 19.5, 28.4). The HSQC spectrum was used to classify the remaining carbons as five methines, nine methylenes, and five quaternary carbons. By comparison with the literature, the aglycone was identified as alphitolic acid.25 An increased δ value of C-3 at 96.5 ppm and the HMBC correlation of the anomeric hexose H-1′ with C-3 of the aglycone suggested the presence of a disaccharide unit at C-3. The linkage of the pentose unit to the hexose was established from the HMBC correlation between the resonances of H-1″ (δH 4.60) and C-2′ (δC 82.1). Both sugars were determined to be in the pyranose form, and according to the large coupling constants of the anomeric protons (J = 5.7 Hz, H-1′; J = 5.5 Hz, H-1″), their orientation was deduced to be β. By NMR data comparison of the sugar residues of 1 and 2, they were identified as Glc and Xyl units. Hydrolysis of 1 and conversion of the sugars to chiral diastereomers followed by GC-MS analysis confirmed their absolute configuration as d-glucose and d-xylose. Based on these data, compound 1 was identified as 3-O-β-d-xylopyranosyl-(1→2)-β-d-glucopyranosylalphitolic acid.
Constituent 2, having a molecular formula of C47H76O17 (m/z 911.5004 [M – H]−; calcd for C47H75O17–, 911.5010, Δ = 0.6 ppm), was also obtained as a white, amorphous powder. The MS/MS and NMR spectra were closely comparable to those of 1, indicating the same sapogenin and sugar residues with the difference of an additional deoxyhexose unit (loss of 146 Da). Detection of a C2 fragment ion at m/z 481.1520 (intact carbohydrate chain), a Y1α (loss of a deoxyhexose), a Y1β (loss of a pentose), and a Y1α/1β (combined loss of a deoxyhexose and a pentose) fragment ion upon CID of the [M + Na]+ ion demonstrated the presence of a branched trisaccharide unit with the pentose and deoxyhexose moieties both being terminal. The findings from the HRESIMS analysis were confirmed by the NMR data, which displayed six additional carbons to the otherwise analogous resonances for the sapogenin alphitolic acid with the Xyl and Glc unit analogous to 1. The δ value of the additional anomeric carbon [δC 102.6 (C-1‴), δH 4.86 (H-1‴)] and an extra methyl moiety [δC 17.8 (C-1‴), δH 1.24 (H-1‴)] indicated the presence of a deoxyhexose substituent. GC-MS analysis of the hydrolyzed and derivatized sugars confirmed the identity of d-glucose, d-xylose, and l-rhamnose. Based on the HMBC correlation between the resonances of H-1′′′ and C-4′, the sugar linkage between Rha and Glc was established as 1→4. The anomeric proton of the Rha unit showed a broad singlet in the 1H NMR spectrum and strong HMBC correlation with the carbons at positions 3‴ (δC 72.2) and 5‴ (δC 70.5), which indicated an α-orientation. Accordingly, compound 2 was determined as 3-O-β-d-xylopyranosyl-(1→2)-[α-rhamnopyranosyl-(1→4)]-β-d-glucopyranosylalphitolic acid.
The triterpene saponin 3, again a white, amorphous powder, was found to have one oxygen more than compound 1 from the HRESIMS, which indicated a molecular formula of C41H66O14 (m/z 781.4381 [M – H]−; calcd for C41H65O14–, 781.4380, Δ = −0.2 ppm). The fragmentation pattern clearly located the additional oxygen on the aglycone. The NMR data of 3 were generally in accordance with those of 1, but in contrast, the sapogenin of 3 showed only five methyl groups in the NMR spectra. Instead, C-27 was characterized by an oxygenated methylene group (δC 61.0, δH 3.76/4.15). This was confirmed by a significant increase of the δ values of positions C-14 (δC 47.4) and C-15 (δC 36.5, δH 1.46/1.72) and a slight increase in positions C-13 (δC 40.2, δH 2.40) and C-8 (δC 42.7). So far, this sapogenin has not been described in the literature and was assigned as 27-hydroxyalphitolic acid. GC-MS analysis and the NMR spectra indicated that 3 contains the same sugar moiety as compound 1, and consequently, it was assigned as 3-O-β-d-xylopyranosyl-(1→2)-β-d-glucopyranosyl-27-hydroxyalphitolic acid.
Compound 4 was isolated as a whitish, amorphous powder with the molecular formula C42H68O15 (m/z 811.4485 [M – H]−; calcd for C42H67O15–, 811.4485, Δ = 0.1 ppm). The resonances derived from the NMR spectra revealed the same aglycone as in compound 3. The only difference between compounds 3 and 4 was found in the second sugar moiety of the glycoside, where an additional carbon signal in the 13C NMR spectrum and slightly different chemical shifts of the neighboring carbons indicated the replacement of the pentose unit by a hexose moiety. This was confirmed by the MS/MS data, which showed a disaccharide consisting of two hexoses. HMBC correlations again supported a 1→2 linkage between the sugar units. After hydrolysis, conversion of the sugars to chiral diastereomers followed by GC-MS analysis indicated the presence of d-glucose. Therefore, both sugar units were assigned as d-glucose, and accordingly, 4 was identified as 3-O-β-d-glucopyranosyl-(1→2)-β-d-glucopyranosyl-27-hydroxyalphitolic acid.
Saponin 5 was obtained as white powder showing an identical elemental formula to compound 2 ([M – H]−m/z 911.5000, calcd for C47H75O17–, 911.5010, Δ = 1.1 ppm), with the same fragmentation pattern by HRESIMS/MS analysis, but differences in the relative abundance of the fragment ions. When compared to the spectroscopic data of 2, compound 5 revealed matching resonances for the trisaccharide unit bound to C-3 and for rings A and B of the sapogenin (C-1 to C-10). Hydrolysis of 5 and conversion of the sugars to chiral diastereomers followed by GC-MS analysis indicated the presence of d-glucose, d-xylose, and l-rhamnose. For the aglycone, the 13C NMR spectrum exhibited resonances of a carboxylic acid carbon (182.6, C-28), two oxygenated carbons at positions C-2 (δC 68.0) and C-3 (δC 96.7), seven sp3 carbons at δ 17.1, 17.5, 17.8, 24.0, 26.4, 28.5, and 33.7, and two sp2 carbons at δ 123.2 (C-12) and 145.6 (C-13). By comparison with data from the literature,26 the sapogenin of 5 was identified as maslinic acid. Hence, 5 was determined as 3-O-β-d-xylopyranosyl-(1→2)-[α-l-rhamnopyranosyl-(1→4)]-β-d-glucopyranosylmaslinic acid.
For compound 6, which was obtained as yellowish crystals, a molecular formula of C56H82O19 was determined by HRESIMS (m/z 1057.5382 [M – H]−; calcd for C56H81O19–, 1057.5378, Δ = −0.4 ppm). Fragmentation of the [M – H]− ion showed the presence of the same sugar moiety as in 5 but an aglycone with the elemental formula C39H54O6. The loss of 148.0527 Da, corresponding to C9H8O2, from this aglycone indicated the presence of a cinnamoyl ester. This could be confirmed by interpretation of the NMR spectroscopic data, where six characteristic resonances for a cinnamoyloxy moiety could be assigned. One ester carbon (δC 168.2), two olefinic carbons (δC 118.9, δH 6.52; δC 146.3, δH 7.68), and six aromatic carbons with one of them being quaternary (δC 135.7; 2 × δC 129.3, δH 7.62; 2 × δC 130.1, δH 7.42; δC 131.6, δH 7.40) were assigned. The downfield shift of the C-21 carbon (δC 76.9, δH 4.93) and its neighboring atoms [δC 36.3 (C-20); δC 37.9, δH 1.76/1.82 (C-22)] pointed toward an attachment of the cinnamoyl residue at position C-21. Since the remaining resonances and GC-MS data of chiral butyl diastereomers (Figure S125, Supporting Information) were in accordance with those of 5, compound 6 was assigned as 3-O-β-d-xylopyranosyl-(1→2)-[α-l-rhamnopyranosyl-(1→4)]-β-d-glucopyranosyl-21-cinnamoyloxymaslinic acid. This is the first time this aglycone has been described.
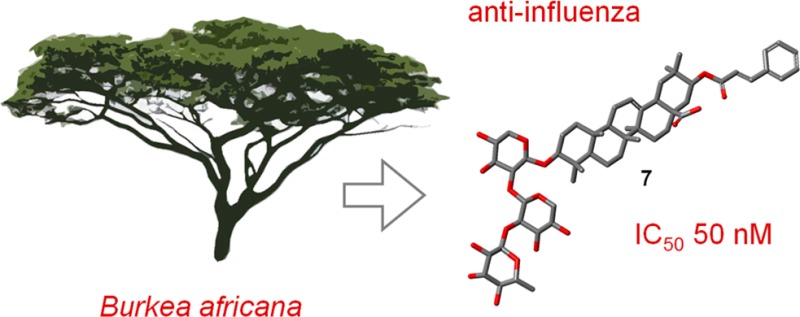
Compound 7 was isolated as white, amorphous powder with the elemental formula C55H80O17 (HRESIMS m/z 1011.5329 [M – H]−; calcd for C55H79O17–, 1011.5323, Δ = −0.6 ppm). When compared to the spectroscopic data of 6, the sapogenin of 7 differed only by the lack of a hydroxy group at position 2. This was corroborated by the upfield shift of C-2 (δC 27.2), its additional proton signal (δH 1.69/1.80), and the upfield shifts of the signals of the neighboring atoms [δC 39.8 (C-1); δC 90.7 (C-3); δC 40.3 (C-4); δC 37.9 (C-10)]. For the saccharide unit, the HRESIMS analysis was consistent with the presence of a linear trisaccharide, with a pentose bound directly to the aglycone, followed by an intermediate pentose and a terminal deoxyhexose moiety. Sixteen carbon resonances of the 13C NMR spectrum could be assigned to two pentose units and one deoxyhexose, with δ values of 106.0, 106.2, and 102.5 for the anomeric carbons of pentose′, pentose″, and of the deoxyhexose, respectively. HMBC correlations between C-1′ of the first pentose unit with C-3 of the sapogenin supported the attachment of the trisaccharide at this position by analogy with compounds 1–6. The interglycosidic linkages were determined as 1→2 between the two pentoses as well as between the deoxyhexose and the pentose, according to the correlations of the HMBC spectrum. Hydrolysis of 7 and conversion of the sugars to chiral diastereomers followed by GC-MS analysis indicated the presence of d-xylose and l-rhamnose. Compound 7 was identified as 3-O-α-l-rhamnopyranosyl-(1→2)-β-d-xylopyranosyl-(1→2)-β-d-xylopyranosyl-21-cinnamoyloxyoleanolic acid.
Compound 8, a white, amorphous powder, was assigned the molecular formula C61H90O21 (HRESIMS m/z 1157.5902 [M – H]−; calcd for C61H89O21, 1157.5902, Δ = 0.0 ppm). The isolate 8 revealed the same MS/MS and NMR profiles as compound 7 in terms of the aglycone. Concerning the glycosidic residues, a linear trisaccharide unit (pentose, pentose, deoxyhexose) analogous to that of compound 7 was indicated from the comparable NMR and MS/MS data. Additionally, a second terminal deoxyhexose (additional fragment loss of 146 Da in the negative-ion mode) was detected by HRESIMS/MS analysis. The fragmentation pattern in the MS/MS spectrum of the [M + Na]+ ion, where in addition to the “loss” of a terminal deoxyhexose further cleavage of both a pentose and a second terminal deoxyhexose unit could be observed, was supportive of a linkage of the second deoxyhexose to the innermost pentose. The HMBC correlation between the anomeric proton of the additional deoxyhexose (δH 4.78) and position C-4 of the first pentose (δC 75.3) was consistent with these findings. After hydrolysis and conversion to chiral diastereomers, GC-MS analysis indicated the presence of d-xylose and l-rhamnose. Accordingly, 8 was assigned as 3-O-α-l-rhamnopyranosyl-(1→2)-β-d-xylopyranosyl-(1→2)-[α-l-rhamnopyranosyl-(1→4)]-β-d-xylopyranosyl-21-cinnamoyloxyoleanolic acid.
Anti-Influenza Virus Activity of Triterpene Saponins from B. africana and their Aglycones
Besides the extract and fractions, all isolated compounds (1–8) including their aglycones were tested for their anti-influenza virus potential in the CPE inhibition assay using HK/68. Additionally, an isolate of the H1N1 influenza pandemic 2009 (A(H1N1)pdm09), the influenza virus A/Jena/8178/09 (Jena/8178), was included in these studies to further prove the antiviral activity of the isolated compounds against a currently circulating H1N1 strain (Table 5).
Table 5. Cytotoxicity and Anti-Influenza A Virus Activity of Compounds 1 to 8 and Their Aglyconesa.
| IC50 [μM] of CPE |
selectivity
index |
||||
|---|---|---|---|---|---|
| code | CC50 [μM] in MDCK cells | IAVb HK/68 | IAVb Jena/8178 | IAVb HK/68 [CC50/IC50] | IAVb Jena/8178 [CC50/IC50] |
| 1 | >100 | n.a.c | 38.6 ± 15.7 | ||
| 2 | 10.4 ± 0.52 | 1.1 ± 0.44 | 1.9 ± 0.08 | 11 | 6 |
| 3 | >100 | n.a.c | n.a.c | ||
| 4 | >100 | n.a.c | 43.0 ± 11.8 | >2 | |
| 5 | 15.5 ± 1.69 | 1.7 ± 0.07 | n.a.c | 9 | |
| 6 | 5.8 ± 0.69 | 1.8 ± 0.23 | 1.8 ± 0.03 | 3 | 3 |
| 7 | 1.5 ± 0.80 | 0.05 ± 0.02 | 0.27 ± 0.13 | 31 | 6 |
| 8 | 1.2 ± 0.10 | 0.17 ± 0.18 | 0.16 ± 0.07 | 7 | 7 |
| alphitolic acid | 49.9 ± 6.68 | n.a.c | n.a.c | ||
| 27-hydroxyalphitolic acid | >100 | n.a.c | 26.5 ± 4.71 | >4 | |
| maslinic acid | 23.3 ± 2.93 | n.a.c | n.a.c | ||
| 21-cinnamoyloxy-maslinic acid | >100 | 11.3 ± 7.35 | 6.9 ± 2.97 | >9 | >14 |
| 21-cinnamoyloxy-oleanolic acid | >100 | 8.9 ± 3.95 | 6.8 ± 4.12 | >11 | >15 |
| oseltamivir | 0.003 ± 0.001 | 0.064 ± 0.013 | |||
Their 50% mean cytotoxic concentration (CC50) in MDCK cells and their 50% inhibition concentration (IC50) determined against HK/68 and Jena/8178 in the CPE inhibition assay in MDCK cells are presented (n = 3).
Influenza A virus (IAV).
No activity (n.a.).
Concerning the tested lupane-type triterpene saponins 1 to 4, those comprising a disaccharide chain, i.e., 1, 3, and 4, showed only weak or no anti-influenza virus activity, whereas compound 2, comprising a branched trisaccharide moiety, is active with IC50 values of 1.1 and 1.9 μM against HK/68 or Jena/8178, respectively.
The oleanane-type triterpene saponins 5 and 6, both comprising a branched trisaccharide moiety, were identified as potent anti-influenza virus agents with IC50 values against HK/68 and/or Jena/8178 in the low micromolar range. The most pronounced anti-influenza virus effect was observed for the oleanane-type triterpene saponins 7 (linear trisaccharide residue) and 8 (branched tetrasaccharide moiety), which reduced the CPE of HK/68 or Jena/8178 by 50% at nanomolar concentrations of 0.05 and 0.27 μM or 0.17 and 0.16 μM, respectively.
Since after oral intake the sugar moieties of triterpene saponins are most likely removed in the gastrointestinal tract,27 we were further interested in the anti-influenza virus activity of the aglycones of 1 to 8 obtained by hydrolysis. As a general trend, they showed better compatibility and lower or no antiviral activity compared to their glycosidic counterparts. Intriguingly, the sapogenins of the most potent compounds 6, 7, and 8, namely, the so far unknown 21-p-methoxycinnamoyloxymaslinic acid and 21-p-methoxycinnamoyloxyoleanolic acid, lost their cytotoxicity in MDCK cells, while still demonstrating an anti-influenza virus activity against both tested strains in the range of 7–11 μM.
An anti-influenza mechanism via the inhibition of the viral surface protein neuraminidase can be excluded (data not shown). Recent studies performed with a series of similar synthetic 3-O-β-chacotriosyl oleanane- and ursane-type triterpenoids identified this compound class as entry inhibitors targeting viral hemagglutinin.28−31 The chacotriose residue, a branched trisaccharide moiety comprising two rhamnose units and one glucose unit, was reported to be essential for the observed bioactivity, which is in agreement with our results of at least three sugars as a mandatory feature. Whether linear or branched sugar chains are more preferable for anti-influenza virus activity of the investigated triterpene saponins could not be concluded in this study. Thus, an interaction with the viral envelope protein hemagglutinin being a key factor for viral entry into the host cell could be involved in the anti-influenza activity of the so far undiscovered triterpene saponins (1 to 8) from B. africana bark. However, this is only a hypothesis, which warrants further investigation through another study.
Experimental Section
General Experimental Procedures
Optical rotations were measured at 20 °C in MeOH on a PerkinElmer 341 polarimeter (PerkinElmer Waltham, MA, USA). FTIR spectra were acquired on a Bruker Tensor 27 spectrometer (Bruker, Billerica, MA, USA). 1D and 2D NMR experiments were performed by using a Bruker Avance 500 NMR spectrometer (UltraShield) (Bruker, MA, USA) with a 5 mm switchable probe (TCI Prodigy CryoProbe, 5 mm, triple resonance inverse detection probe head) with z-axis gradients and automatic tuning and matching accessory (Bruker BioSpin). The samples (∼5 mg) were measured at 298 K in fully deuterated methanol referenced to the residual nondeuterated solvent signals. The resonance frequency for 1H NMR was 500.13 MHz and for 13C NMR 125.75 MHz. Standard 1D and gradient-enhanced 2D experiments, such as double quantum filtered (DQF) COSY, NOESY, HSQC, and HMBC, were used as supplied by the manufacturer. High-resolution mass spectrometric (HRMS) analyses were performed on a maXis HD ESI-Qq-TOF mass spectrometer (Bruker Daltonics, Bremen, Germany) equipped with electrospray ionization (ESI) in the positive and negative modes: capillary voltage, 2.0 to 4.5 kV (individually optimized); nebulizer, 0.4 bar (N2); dry gas flow, 4 L/min (N2); dry temperature, 200 °C. Samples were directly infused into the ESI source at a flow rate of 3 μL/min. Fragment ion spectra of the [M + H]+, the [M + Na]+, and either the [M – H]− or the [M + HCOO]− ion were recorded, whereby the collision energy was manually optimized. The sum formulas of the ions were determined using Bruker Compass DataAnalysis 4.2 based on the mass accuracy (Δ m/z ≤ 2 ppm for MS1 and ≤ 3 ppm for MS/MS) and isotopic pattern matching (SmartFormula algorithm). GC-MS analysis was performed on a QP2010 GC-MS (Shimadzu, Kyoto, Japan) equipped with a Phenomenex ZB-5 capillary column (65 m × 0.25 mm i.d. × 0.25 μm) under the following conditions: carrier gas, He 5.0 bar; flow rate, 3.0 mL/min; splitless injection mode; temperature gradient, 100–270 °C at a rate of 3 °C/min; injector and interface temperature, 270 °C; MS, ion source temperature 250 °C; electron-impact ionization at 70 eV; scan range, m/z 40–500. Flash chromatography was performed on an Interchim puriFlash 4250 system (Montluçon, France), equipped with an evaporative light scattering detector (ELSD), a photodiode array (PDA), and a fraction collector, controlled by Interchim Software. PuriFlash C18 HQ columns (15 μm, 55 g, and 35 g) served as stationary phase. Semipreparative supercritical fluid chromatography was performed on a Waters Prep-15 System (Waters, Milford, MA, USA) equipped with an ELSD, a PDA, and a fraction collector. Waters Viridis Prep Silica 2-ethylpyridine columns (5 μm; 10 × 250 mm) served as the stationary phase, and data were analyzed using MassLynx. The mobile phase consisted of a supercritical CO2/organic modifier (MeOH–H2O, 95:5) gradient (temperature, 30 °C; flow rate, 15 mL/min). The fractions obtained from all chromatographic steps were analyzed by TLC (mobile phase: toluene–EtOAc–MeOH–EtOH–HCOOH–H2O, 5:4:0.5:0.5:1:0.25) for polyamide fractions or EtOAc–MeOH–H2O (8:1.3:0.7) for all other fractions; stationary phase: Merck silica gel 60 PF254, detected after derivatization with vanillin/H2SO4 (5% in MeOH) under both visible light and UV254 and UV366. HPLC analysis was performed on a Shimadzu Nexera Prominence system (Kyoto, Japan), equipped with a PDA detector, and LabSolutions Lite software on a ThermoQuest Hypersil BDS C18 column (5 μm, 4.6 × 250 mm) with a MeOH/H2O gradient as mobile phase (0 min 20%/80%, 15 min 50%/50%, 20 min 75%/25%, 40 min 82%/18%, 42 min 98%/2%, 50 min 98%/2%). Conditions: temperature, 40 °C; flow rate, 1 mL/min; injection volume, 10 μL; detection wavelengths, 210, 254, 280, 325 nm. UPLC analysis was performed on a Waters Acquity UPLC system (H-class) equipped with a binary solvent manager, a sample manager, a column manager, a PDA detector, and an ELSD. The stationary phase was a Waters Acquity UPLC BEH Phenyl column (1.7 μm, 2.1 × 100 mm), and data were analyzed with Waters Empower 3. The mobile phase consisted of a MeCN/H2O gradient (0 min 40%/60%, 5 min 65%/35%, 5.1 min 98%/2%, 7.9 min 98%/2%, 8 min 40%/60%). Conditions: temperature, 40 °C; flow rate, 0.4 mL/min; injection volume, 1 μL; detection wavelengths, 254, 280, 325 nm; and full range scan, 190–400 nm. (Ultrahigh-)gradient grade solvents and deuterated solvents from Merck (Darmstadt, Germany) were used. Reference sugars were obtained from Merck (d-glucose, d-xylose, l-rhamnose) with a purity of ∼98%. HMDS and TMCS were obtained from Sigma-Aldrich with a purity of 99.9%.
Plant Material
The bark of B. africana was collected in March 2015, northwest of Zeerust, South Africa. It was identified by P. C. Zietsman (Botany Department, National Museum, Bloemfontein, South Africa). The GPS coordinates of the collection location were as follows: latitude 25°11′38.5″; longitude 25°57′63.2′′. A voucher specimen (no. 5858) was deposited at the Department of Chemistry, University of the Free State, Bloemfontein, South Africa. A material transfer agreement is in place.
Extraction and Fractionation of Extract BA-E via Polyamide
An ethanol extract of the bark material of B. africana (∼160 g, BA-E) was prepared. For tannin depletion, BA-E was subjected to column chromatography (CC) over polyamide (Carl Roth GmbH & Co. KG, Karlsruhe, Germany) (∼600 g; 10 × 160 cm). Before sample application, the polyamide was washed with 5.5 L of H2O followed by 5.0 L of MeOH. The sample was dissolved in MeOH and was subjected to the column and eluted with 15 L of MeOH, followed by 9 L of MeOH–acetone–H2O (80:16:4) and 18 L of acetone–H2O (7:3), to yield 10 fractions (A1 to A10).
Microfractionation of Fractions A1 + A2
For the generation of microfractions, 250 mg of the combined fractions A1+2 were dissolved in MeOH and subjected to CC to yield 13 subfractions (B1 to B13), by applying a MeOH–H2O gradient (0 min 5%/95%, 15 min 30%/70%, 20 min 45%/55%, 60 min 80%/20%, 70 min 100%/0%).
Isolation of Triterpene Saponins
The remaining amount of the pooled fraction A1+2 (28.0 g) was suspended in water and successively partitioned between petroleum ether, EtOAc, and BuOH, respectively. The EtOAc fraction (3.08 g) was passaged over Sephadex LH-20 and eluted with MeOH to yield 22 fractions (C1 to C22). Fraction C4 (419.5 mg) was subjected to semipreparative supercritical fluid chromatography and further divided in 11 fractions (D1 to D11). D5 was identified as compound 5 (12.9 mg), and fraction D6 as compound 2 (23.9 mg). Fraction C5 (1073.2 mg) was separated into 30 subfractions (E1 to E30) by flash CC. Fractions E11, E3, E2, E14, E20, and E18 were identified as compounds 1 (29.0 mg), 3 (58.6 mg), 4 (98.8 mg), 6 (12.4 mg), 7 (17.7 mg), and 8 (23.9 mg), respectively. The purity of the isolated compounds was determined by HPLC-PDA-MS to be >98%.
3-O-β-d-Xylopyranosyl-(1→2)-β-d-glucopyranosylalphitolic acid (1):
yellowish, amorphous powder; [α]20D +3.3 (c 0.10, MeOH); 1H and 13C NMR, see Tables 1 and 2; HRESIMS m/z 765.4437 [M – H]− (calcd for C41H65O13–, 765.4431, Δ = −0.8 ppm).
Table 2. 1H NMR Spectroscopic Data of 1–4 (500 MHz, CD3OD, δ in ppm).
| position | 1 δH (J in Hz) | 2 δH (J in Hz) | 3 δH (J in Hz) | 4 δH (J in Hz) |
|---|---|---|---|---|
| 1 | 0.82, m | 0.83, m | 0.85, m | 0.86, m |
| 2.06, dd (12.7, 4.7) | 2.05, dd (12.9, 4.5) | 2.08, dd (12.7, 4.8) | 2.09, dd (12.6, 4.8) | |
| 2 | 3.71, ddd (10.7, 9.4, 4.5) | 3.69, ddd (12.4, 9.4, 4.5) | 3.73, ddd (11.2, 9.4, 4.8) | 3.74, m |
| 3 | 2.96, d (9.4) | 2.94, d (9.4) | 2.96, d (9.4) | 3.01, d (9.4) |
| 5 | 0.80, m | 0.78, m | 0.89, m | 0.90, m |
| 6 | 1.42, m | 1.42, m | 1.40, m | 1.40, m |
| 1.54, m | 1.54, m | 1.54, d (13.1) | 1.54, d (13.2) | |
| 7 | 1.41, m | 1.40, m | 1.41, m | 1.41, m |
| 2.23, dt (12.9, 3.2, 3.2) | 2.23, dt (12.8, 3.6, 3.6) | |||
| 9 | 1.37, m | 1.36, m | 1.43, m | 1.44, m |
| 11 | 1.30, m | 1.30, m | 1.24, m | 1.24, m |
| 1.47, m | 1.45, m | 1.47, m | 1.47, m | |
| 12 | 1.06, m | 1.04, m | 0.83, m | 0.83, m |
| 1.74, d (12.2) | 1.73, m | 1.70, m | 1.70, m | |
| 13 | 2.33, dt (12.2, 12.2, 3.5) | 2.36, dt (12.1, 12.1, 3.6) | 2.42, dt (12.4, 12.4, 3.0) | 2.43, dt (12.5, 12.5, 3.2) |
| 15 | 1.16, m | 1.15, m | 1.46, m | 1.46, m |
| 1.53, m | 1.53, m | 1.72, m | 1.72, m | |
| 16 | 1.41, m | 1.38, m | 1.27, m | 1.28, m |
| 2.24, dt (12.8, 3.0, 3.0) | 2.24 dt (12.9, 2.7, 2.7) | 1.84, dt (13.6, 3.0, 3.0) | 1.83, dt (13.6, 2.4, 2.4) | |
| 18 | 1.62, t (11.3) | 1.61, t (11.3) | 1.74, t (11.4) | 1.74, t (11.3) |
| 19 | 3.03, dt (10.7,10.7, 4.7) | 3.05, dt (10.8,10.8, 4.7) | 3.04, dt (10.6,10.6, 4.5) | 3.04, dt (11.0, 11.0, 4.5) |
| 21 | 1.37, m | 1.36, m | 1.39, m | 1.38, m |
| 1.93, m | 1.92, m | 1.94, m | 1.93, m | |
| 22 | 1.45, m | 1.41, m | 1.46, m | 1.44, m |
| 1.89, m | 1.89, m | 1.93, m | 1.92, m | |
| 23 | 1.09, s | 1.08, s | 1.09, s | 1.12, s |
| 24 | 0.87, s | 0.87, s | 0.88, s | 0.90, s |
| 25 | 0.93, s | 0.92, s | 0.95, s | 0.95, s |
| 26 | 0.96, s | 0.97, s | 0.97, s | 0.97, s |
| 27 | 1.00, s | 1.00, s | 3.77, d (12.5) | 3.77, d (12.5) |
| 4.15, d (12.5) | 4.15, d (12.5) | |||
| 29 | 1.70, s | 1.70, s | 1.70, s | 1.70, s |
| 30 | 4.60, d (1.8) | 4.59, d (1.7) | 4.60, d (1.4) | 4.60, d (1.6) |
| 4.71, d (1.8) | 4.71, d (1.7) | 4.71, d (1.4) | 4.71, d (1.6) | |
| 1′ | 4.42, d (7.6) | 4.43, d (7.8) | 4.42, d (7.5) | 4.45, d (7.8) |
| 2′ | 3.53, dd (9.1, 7.6) | 3.58, dd (9.2, 7.8) | 3.54, dd (9.2, 7.5) | 3.69, m |
| 3′ | 3.34, m | 3.71, t (9.2) | 3.32, m | 3.25, m |
| 4′ | 3.58, t (8.8) | 3.63, t (9.2) | 3.59, t (8.7) | 3.60, m |
| 5′ | 3.43, m | 3.40, t (9.2) | 3.44, m | 3.35, m |
| 6′ | 3.67, dd (11.9, 4.9) | 3.66, m | 3.67, dd (11.8, 5.1) | 3.59, m |
| 3.85, dd (11.9, 1.7) | 3.80, m | 3.86, dd (11.8, 1.5) | 3.83, dd (11.8, 2.2) | |
| 1″ | 4.60, d (7.6) | 4.65, d (7.7) | 4.61, d (7.7) | 4.75, d (7.8) |
| 2″ | 3.22, dd (9.1, 7.6) | 3.21, dd (9.1, 7.6) | 3.22, dd (9.1, 7.7) | 3.23, dd (9.7, 7.8) |
| 3″ | 3.30, m | 3.30, m | 3.30, m | 3.35, m |
| 4″ | 3.35, m | 3.43, m | 3.37, m | 3.18, t (9.3) |
| 5″ | 3.13, dd (11.4, 10.4) | 3.14, t (11.0) | 3.13, dd (11.5, 10.4) | 3.35, m |
| 3.80, dd (11.4, 5.4) | 3.79, m | 3.80, dd (11.5, 5.4) | ||
| 6″ | 3.66, dd (11.8, 5.2) | |||
| 3.87, dd (11.8, 1.2) | ||||
| 1‴ | 4.86, d (1.6) | |||
| 2‴ | 3.82, m | |||
| 3‴ | 3.61, m | |||
| 4‴ | 3.40, t (9.4) | |||
| 5‴ | 3.98, dq (9.4, 6.2, 6.2, 6.2) | |||
| 6‴ | 1.24, d (6.2) |
3-O-β-d-Xylopyranosyl-(1→2)-[α-l-rhamnopyranosyl-(1→4)]-β-d-glucopyranosylalphitolic acid (2):
white, amorphous powder; [α]20D +2.9 (c 0.10, MeOH); 1H and 13C NMR, see Tables 1 and 2; HRESIMS m/z 911.5004 [M – H]− (calcd for C47H75O17–, 911.5010, Δ = 0.6 ppm).
3-O-β-d-Xylopyranosyl-(1→2)-β-d-glucopyranosyl-27-hydroxyalphitolic acid (3):
white, amorphous powder; [α]20D +3.7 (c 0.10, MeOH); 1H and 13C NMR, see Tables 1 and 2; HRESIMS m/z 781.4381 [M – H]− (calcd for C41H65O14–, 781.4380, Δ = −0.2 ppm).
3-O-β-d-Glucopyranosyl-(1→2)-β-d-glucopyranosyl-27-hydroxyalphitolic acid (4):
yellowish, amorphous powder; [α]20D −1.4 (c 0.10, MeOH); 1H and 13C NMR, see Tables 1 and 2; HRESIMS m/z 811.4485 [M – H]− (calcd for C42H67O15–, 811.4485, Δ = 0.1 ppm).
3-O-β-d-Xylopyranosyl-(1→2)-[α-l-rhamnopyranosyl-(1→4)]-β-d-glucopyranosylmaslinic acid (5):
white, amorphous powder; [α]20D +8.8 (c 0.10, MeOH); 1H and 13C NMR, see Tables 3 and 4; HRESIMS m/z 911.5000 [M – H]− (calcd for C47H75O17–, 911.5010, Δ = 1.1 ppm).
Table 3. 13C NMR Spectroscopic Data of 5–8 (125 MHz, CD3OD, δ in ppm, Multiplicities Determined by APT Experiments).
| position | 5 | 6 | 7 | 8 |
|---|---|---|---|---|
| 1 | 47.4, CH2 | 47.4, CH2 | 39.8, CH2 | 39.8, CH2 |
| 2 | 68.0, CH | 67.9, CH | 27.2, CH2 | 27.2, CH2 |
| 3 | 96.7, CH | 96.7, CH | 90.7, CH | 90.6, CH |
| 4 | 41.8, C | 41.7, C | 40.3, C | 40.4, C |
| 5 | 56.8, CH | 56.7, CH | 57.0, CH | 57.1, CH |
| 6 | 19.4, CH2 | 19.3, CH2 | 19.3, CH2 | 19.3, CH2 |
| 7 | 35.0, CH2 | 33.9, CH2 | 34.0, CH2 | 34.0, CH2 |
| 8 | 40.5, C | 40.5, C | 40.0, C | 40.5, C |
| 9 | 49.0, CH | 49.1, CH | 49.1, CH | 49.0, CH |
| 10 | 38.8, C | 38.7, C | 37.9, C | 37.9, C |
| 11 | 24.1, CH2 | 25.2, CH2 | 25.3, CH2 | 25.3, CH2 |
| 12 | 123.2, CH | 124.1, CH | 124.3, CH | 124.4, CH |
| 13 | 145.6, C | 144.0, C | 143.9, C | 143.9, C |
| 14 | 42.9, C | 42.9, C | 42.9, C | 42.9, C |
| 15 | 28.9, CH2 | 28.7, CH2 | 28.8, CH2 | 28.8, CH2 |
| 16 | 24.6, CH2 | 24.6, CH2 | 24.5, CH2 | 24.5, CH2 |
| 17 | 47.8, C | 49.3, C | 49.3, C | 49.4, C |
| 18 | 42.8, CH | 42.1, CH | 42.2, CH | 42.2, CH |
| 19 | 47.5, CH2 | 47.7, CH2 | 47.8, CH2 | 47.8, CH2 |
| 20 | 31.7, C | 36.3, C | 36.3, C | 36.3, C |
| 21 | 33.9, CH2 | 76.9, CH | 76.9, CH | 76.9, CH |
| 22 | 33.9, CH2 | 37.9, CH2 | 37.9, CH2 | 37.9, CH2 |
| 23 | 28.5, CH3 | 28.5, CH3 | 28.2, CH3 | 28.2, CH3 |
| 24 | 17.5, CH3 | 17.5, CH3 | 16.6, CH3 | 16.6, CH3 |
| 25 | 17.1, CH3 | 17.1, CH3 | 16.0, CH3 | 16.0, CH3 |
| 26 | 17.8, CH3 | 17.7, CH3 | 17.7, CH3 | 17.7, CH3 |
| 27 | 26.4, CH3 | 26.2, CH3 | 26.2, CH3 | 26.2, CH3 |
| 28 | 182.6, C | 180.0, C | 180.2, C | 180.2, C |
| 29 | 33.7, CH3 | 29.2, CH3 | 29.2, CH3 | 29.2, CH3 |
| 30 | 24.0, CH3 | 18.8, CH3 | 18.8, CH3 | 18.8, CH3 |
| 31 | 168.2, C | 168.2, C | 168.2, C | |
| 32 | 118.9, CH | 119.0, CH | 119.0, CH | |
| 33 | 146.3, CH | 146.3, CH | 146.3, CH | |
| 34 | 135.7, C | 135.7, C | 135.7, C | |
| 35 | 129.3, CH | 129.3, CH | 129.3, CH | |
| 36 | 130.1, CH | 130.1, CH | 130.1, CH | |
| 37 | 131.6, CH | 131.6, CH | 131.5, CH | |
| 38 | 130.1, CH | 130.1, CH | 130.1, CH | |
| 39 | 129.3, CH | 129.3, CH | 129.3, CH | |
| 1′ | 104.8, CH | 104.8, CH | 106.0, CH | 106.0, CH |
| 2′ | 81.9, CH | 81.9, CH | 83.0, CH | 83.0, CH |
| 3′ | 77.2, CH | 77.2, CH | 77.8, CH | 76.1, CH |
| 4′ | 78.3, CH | 78.2, CH | 71.0, CH | 75.3, CH |
| 5′ | 76.9, CH | 76.9, CH | 66.4, CH2 | 63.5, CH2 |
| 6′ | 61.4, CH2 | 61.4, CH2 | ||
| 1″ | 105.7, CH | 105.7, CH | 106.2, CH | 106.2, CH |
| 2″ | 76.0, CH | 76.0, CH | 83.5, CH | 83.5, CH |
| 3″ | 78.0, CH | 78.0, CH | 76.8, CH | 76.8, CH |
| 4″ | 71.2, CH | 71.2, CH | 69.9, CH | 69.9, CH |
| 5″ | 67.0, CH2 | 67.0, CH2 | 67.2, CH2 | 67.2, CH2 |
| 1‴ | 102.7, CH | 102.6, CH | 102.5, CH | 102.5, CH |
| 2‴ | 72.4, CH | 72.4, CH | 72.3, CH | 72.3, CH |
| 3‴ | 72.2, CH | 72.1, CH | 72.2, CH | 72.2, CH |
| 4‴ | 73.3, CH | 73.7, CH | 74.0, CH | 74.0, CH |
| 5‴ | 70.5, CH | 70.5, CH | 70.0, CH | 70.0, CH |
| 6‴ | 17.8, CH3 | 17.8, CH3 | 17.9, CH3 | 17.8, CH3 |
| 1′′′′ | 99.7, CH | |||
| 2′′′′ | 72.4, CH | |||
| 3′′′′ | 72.0, CH | |||
| 4′′′′ | 73.9, CH | |||
| 5′′′′ | 69.9, CH | |||
| 6′′′′ | 17.9, CH3 |
3-O-β-d-Xylopyranosyl-(1→2)-[α-l-rhamnopyranosyl-(1→4)]-β-d-glucopyranosyl-21-cinnamoyloxymaslinic acid (6):
yellowish crystals; [α]20D +21.1 (c 0.10, MeOH); 1H and 13C NMR, see Tables 3 and 4; HRESIMS m/z 1057.5382 [M – H]− (calcd for C56H81O19–, 1057.5378, Δ = −0.4 ppm).
3-O-α-l-Rhamnopyranosyl-(1→2)-β-d-xylopyranosyl-(1→2)-β-d-xylopyranosyl-21-cinnamoyloxyoleanolic acid (7):
white, amorphous powder; [α]20D +27.7 (c 0.10, MeOH); 1H and 13C NMR, see Tables 3 and 4; HRESIMS m/z 1011.5329 [M – H]− (calcd for C55H79O17–, 1011.5323, Δ = −0.6 ppm).
3-O-α-l-Rhamnopyranosyl-(1 →2)-β-d-xylopyranosyl-(1 →2)-[α-l-rhamnopyranosyl-(1 →4)]-β-d-xylopyranosyl-21-cinnamoyloxyoleanolic acid (8):
white, amorphous powder; [α]20D −0.4 (c 0.10, MeOH); 1H and 13C NMR, see Tables 3 and 4; HRESIMS m/z 1157.5902 [M – H]− (calcd for C61H89O21–, 1157.5902, Δ = 0.0 ppm).
Determination of Absolute Configurations of Sugars
The determination of the absolute configuration of the sugar moieties was performed as described previously.32 Briefly, compounds 1–8 (2 mg) were hydrolyzed with 0.5 mL of Kiliani reagent (CH3COOH–HClconc–H2O, 1.75/0.5/2.75) at 100 °C for 2 h. After cooldown, each solution was extracted five times with 1 mL of ethyl acetate (5 mL of ethyl acetate in total). The aqueous phase, containing the hydrolyzed sugars, was evaporated to dryness. (R)-(−)-2-Butanol (0.45 mL) and concentrated HCl (0.05 mL) were added to the residue and heated to 100 °C for 15 h to give the corresponding diastereomeric 2-butylglycosides. The samples were evaporated to dryness and redissolved in 150 μL of pyridine. Silylated derivatives were prepared by adding 40 μL of HMDS and 50 μL of TMCS. The samples were heated for 10 min at 50 °C in a tightly sealed reaction vial to form volatile derivatives. Authentic reference sugars, i.e., d-glucose, d-xylose, and l-rhamnose, were treated by the same procedure. The (R)-(−)-2-butyl glycosides were separated by GC-MS analysis. For the selective detection of trimethylsilylated (−)-2-butyl glycosides the fragment m/z 175 was chosen, resulting in peaks at tR = 29.2, 31.0 (l-Rha), 32.0, 34.3 (d-Xyl), and 38.0, 40.3 (d-Glc) min, respectively. In comparison with reference sugars, for 1 and 3d-Xyl and d-Glc were proven. Compounds 2, 5, and 6 revealed the presence of l-Rha, d-Xyl, and d-Glc. Compounds 7 and 8 contained l-Rha and d-Xyl, and 4 contained d-Glc. As an example, the chromatograms of sugars derived from hydrolysis of 6 and respective reference sugars are provided as Supporting Information.
Cell Culture and Viruses
H1N1 influenza virus A/Jena/8178/09 (Jena/8178/09; isolated from nasal swabs of an influenza patient during the pandemic of 2009)33 and the H3N2 strain A/HongKong/68 (HK/68; strain collection of the Institute of Medical Microbiology, Jena, Germany) were used in antiviral studies. They were propagated in MDCK cells (Friedrich-Loeffler Institute, Riems, Germany) in serum-free Eagle’s minimum essential medium, 2 μg/mL trypsin, and 1.2 mM bicarbonate.34 Titers of virus stocks were determined according to Reed and Muench (1938) in MDCK cells.35
Determination of Cytotoxicity and Cytopathic Effect Inhibition
The 50% cytotoxic concentration (CC50) as well as 50% inhibitory concentration (IC50, inhibition of virus-induced CPE) was determined on 2-day-old confluent MDCK cell monolayers grown in 96-well plates as described previously (maximum tested concentration: 100 μg/mL for extracts/fractions and 100 μM for pure compounds).36 Cytotoxicity was analyzed 72 h after adding the samples. CPE inhibition was measured 48 h after infection. A multiplicity of infection of 0.0008 and 0.003 TCID50/cell of Jena/8178 (A(H1N1)pdm09) and HK/68 (H3N2), respectively, resulted in a complete CPE at this time point. Each concentration was tested twice. The mean CC50 and IC50 values are given from at least three experiments.
Acknowledgments
This article is dedicated to the memory of our deceased collaboration partner and friend J. H. van der Westhuizen (Department of Chemistry, University of the Free State, South Africa). The authors thank G. Taylor (Innovation and Business Development Team, University of the Free State, South Africa) for his support in the feasability of this study, P. C. Zietsman (Botany Department, National Museum, Bloemfontein) for the collection and identification of the plant material, G. Reznicek and S. Glasl-Tazreiter (Department of Pharmacognosy, University of Vienna) for their valuable support in GC-MS measurements, B. Jahn (Institute of Medical Microbiology, Jena University Hospital, Germany) for technical assistance, and A. Smolag (Department of Pharmacognosy, University of Vienna) for assistance in phytochemical processing. This work was supported by the Austrian Science Fund (FWF: P24587) and the European Social Fund (ESF & TMWAT Project 2011 FGR 0137).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnatprod.7b00774.
Cytotoxicity and anti-influenza A virus activity of extract and fractions; IR, HRESIMS/MS, 1H NMR, 13C APT NMR, HSQC, HMBC, and HH-COSY spectra of isolated compounds; GC-MS chromatograms of the sugars with corresponding reference sugars (PDF)
Author Contributions
# C. E. Mair and U. Grienke contributed equally to this work.
The authors declare no competing financial interest.
Dedication
Dedicated to Dr. Susan Band Horwitz, of Albert Einstein College of Medicine, Bronx, NY, for her pioneering work on bioactive natural products.
Supplementary Material
References
- Houérou L.http://www.fao.org/ag/agp/agpc/doc/GBASE/data/pf000374.htm, accessed on 10/07/17.
- van Wyk B. E.; Gericke N.. People’s Plants. A Guide to Useful Plants of Southern Africa; Briza Publications: Pretoria, 2007; pp 182, 286. [Google Scholar]
- Tanko Y.; Iliya B.; Mohammed A.; Mahdi M. A.; Musa K. Y. Arch. Appl. Sci. Res. 2011, 3, 122–130. [Google Scholar]
- Mathisen E.; Diallo D.; Andersen O. M.; Malterud K. E. Phytother. Res. 2002, 16, 148–153. 10.1002/ptr.936. [DOI] [PubMed] [Google Scholar]
- Mbatchou V. C.; Aggrey I.; Oyelude E. O. Can. J. Pure Appl. Sci. 2011, 5, 1623–1629. [Google Scholar]
- Molander M.; Nielsen L.; Soegaard S.; Staerk D.; Roensted N.; Diallo D.; Chifundera K. Z.; van Staden J.; Jager A. K. J. Ethnopharmacol. 2014, 157, 171–180. 10.1016/j.jep.2014.09.027. [DOI] [PubMed] [Google Scholar]
- Cordier W.; Gulumian M.; Cromarty A. D.; Steenkamp V. BMC Complementary Altern. Med. 2013, 13, 116. 10.1186/1472-6882-13-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacaille-Dubois M. A.; Wagner H. Phytomedicine 1996, 2, 363–386. 10.1016/S0944-7113(96)80081-X. [DOI] [PubMed] [Google Scholar]
- Dinda B.; Debnath S.; Mohanta B. C.; Harigaya Y. Chem. Biodiversity 2010, 7, 2327–2580. 10.1002/cbdv.200800070. [DOI] [PubMed] [Google Scholar]
- Netala V. R.; Ghosh S. B.; Bobbu P.; Anitha D.; Tartte V. Int. J. Pharm. Pharm. Sci. 2014, 7, 5. [Google Scholar]
- Garai S. Nat. Prod. Chem. Res. 2014, 2, 148. [Google Scholar]
- Ge Y.; Luo H.; Zheng Y.; Chen X.; Geng Z. Nongyaoxue Xuebao 2015, 17, 300–306. [Google Scholar]
- Zhou M.; Xu M.; Ma X.-X.; Zheng K.; Yang K.; Yang C.-R.; Wang Y.-F.; Zhang Y.-J. Planta Med. 2012, 78, 1702–1705. 10.1055/s-0032-1315209. [DOI] [PubMed] [Google Scholar]
- Lueckemeyer D. D.; Mueller V. D. M.; Moritz M. I. G.; Stoco P. H.; Schenkel E. P.; Barardi C. R. M.; Reginatto F. H.; Simoes C. M. O. Phytother. Res. 2012, 26, 535–540. 10.1002/ptr.3590. [DOI] [PubMed] [Google Scholar]
- Wang S.; Li J.; Huang H.; Gao W.; Zhuang C.; Li B.; Zhou P.; Kong D. Biol. Pharm. Bull. 2009, 32, 132–135. 10.1248/bpb.32.132. [DOI] [PubMed] [Google Scholar]
- He Z.; Qiao C.; Han Q.; Wang Y.; Ye W.; Xu H. Tetrahedron 2005, 61, 2211–2215. 10.1016/j.tet.2004.12.032. [DOI] [Google Scholar]
- Song W.; Si L.; Ji S.; Wang H.; Fang X.-M.; Yu L.-Y.; Li R.-Y.; Liang L.-N.; Zhou D.; Ye M. J. Nat. Prod. 2014, 77, 1632–1643. 10.1021/np500253m. [DOI] [PubMed] [Google Scholar]
- Rao G. S.; Sinsheimer J. E.; Cochran K. W. J. Pharm. Sci. 1974, 63, 471–473. 10.1002/jps.2600630341. [DOI] [PubMed] [Google Scholar]
- Chen J.; Zhao Y.; Ling F.; Qi W.; Zeng Z.; Liao M.; Liu Y.; Duan M.; Xiao K.; Li Q.; Li B.; Lu C.; Chen W.; Zhao Y.; Li B. Oncotarget 2015, 6, 42541–42556. 10.18632/oncotarget.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz J. M.; Mair C. E.; Oettl S. K.; Saxena P.; Scheel O.; Schuster D.; Hering S.; Rollinger J. M. Planta Med. 2016, 82, 1009–1015. 10.1055/s-0042-105572. [DOI] [PubMed] [Google Scholar]
- Camp D.; Davis R. A.; Campitelli M.; Ebdon J.; Quinn R. J. J. Nat. Prod. 2012, 75, 72–81. 10.1021/np200687v. [DOI] [PubMed] [Google Scholar]
- Yang Z. F.; Bai L. P.; Huang W. B.; Li X. Z.; Zhao S. S.; Zhong N. S.; Jiang Z. H. Fitoterapia 2014, 93, 47–53. 10.1016/j.fitote.2013.12.011. [DOI] [PubMed] [Google Scholar]
- Bahramsoltani R.; Sodagari H. R.; Farzaei M. H.; Abdolghaffari A. H.; Gooshe M.; Rezaei N. Expert Rev. Anti-Infect. Ther. 2016, 14, 57–80. 10.1586/14787210.2016.1120670. [DOI] [PubMed] [Google Scholar]
- Theisen L. L.; Erdelmeier C. A.; Spoden G. A.; Boukhallouk F.; Sausy A.; Florin L.; Muller C. P. PLoS One 2014, 9, e88062. 10.1371/journal.pone.0088062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre M. C.; Delporte C.; Backhouse N.; Erazo S.; Letelier M. E.; Cassels B. K.; Silva X.; Alegría S.; Negrete R. Bioorg. Med. Chem. 2006, 14, 5673–5677. 10.1016/j.bmc.2006.04.021. [DOI] [PubMed] [Google Scholar]
- Acebey-Castellon I. L.; Voutquenne-Nazabadioko L.; Huong D. T. M.; Roseau N.; Bouthagane N.; Muhammad D.; Le Magrex Debar E.; Gangloff S. C.; Litaudon M.; Sevenet T.; Nguyen V. H.; Lavaud C. J. Nat. Prod. 2011, 74, 163–168. 10.1021/np100502y. [DOI] [PubMed] [Google Scholar]
- Yu K.; Chen F.; Li C. Curr. Drug Metab. 2012, 13, 577–598. 10.2174/1389200211209050577. [DOI] [PubMed] [Google Scholar]
- Song G.; Yang S.; Zhang W.; Cao Y.; Wang P.; Ding N.; Zhang Z.; Guo Y.; Li Y. J. Med. Chem. 2009, 52, 7368–7371. 10.1021/jm900275m. [DOI] [PubMed] [Google Scholar]
- Song G.; Shen X.; Li S.; Li Y.; Liu Y.; Zheng Y.; Lin R.; Fan J.; Ye H.; Liu S. Eur. J. Med. Chem. 2015, 93, 431–442. 10.1016/j.ejmech.2015.02.029. [DOI] [PubMed] [Google Scholar]
- Song G.; Shen X.; Li S.; Li Y.; Si H.; Fan J.; Li J.; Gao E.; Liu S. Eur. J. Med. Chem. 2016, 119, 109–121. 10.1016/j.ejmech.2016.04.061. [DOI] [PubMed] [Google Scholar]
- Li S.; Jia X.; Shen X.; Wei Z.; Jiang Z.; Liao Y.; Guo Y.; Zheng X.; Zhong G.; Song G. Bioorg. Med. Chem. 2017, 25, 4384–4396. 10.1016/j.bmc.2017.06.025. [DOI] [PubMed] [Google Scholar]
- Reznicek G.; Susman O. Sci. Pharm. 1993, 61, 35–45. [Google Scholar]
- Walther E.; Xu Z.; Richter M.; Kirchmair J.; Grienke U.; Rollinger J. M.; Krumbholz A.; Saluz H. P.; Pfister W.; Sauerbrei A.; Schmidtke M. Front. Microbiol. 2016, 7, 357. 10.3389/fmicb.2016.00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer K.; Richter M.; Wutzler P.; Schmidtke M. Antiviral Res. 2009, 82, 34–41. 10.1016/j.antiviral.2009.01.006. [DOI] [PubMed] [Google Scholar]
- Reed L. J.; Muench H. Am. J. Epidemiol. 1938, 27, 493–497. 10.1093/oxfordjournals.aje.a118408. [DOI] [Google Scholar]
- Schmidtke M.; Schnittler U.; Jahn B.; Dahse H.; Stelzner A. J. Virol. Methods 2001, 95, 133–143. 10.1016/S0166-0934(01)00305-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.