

ABSTRACT
LapA of Pseudomonas fluorescens Pf0-1 belongs to a diverse family of cell surface-associated bacterial adhesins that are secreted via the type I secretion system (T1SS). We previously reported that the periplasmic protease LapG cleaves the N terminus of LapA at a canonical dialanine motif to release the adhesin from the cell surface under conditions unfavorable to biofilm formation, thus decreasing biofilm formation. Here, we characterize LapA as the first type I secreted substrate that does not follow the “one-step” rule of T1SS. Rather, a novel N-terminal element, called the retention module (RM), localizes LapA at the cell surface as a secretion intermediate. Our genetic, biochemical, and molecular modeling analyses support a model wherein LapA is tethered to the cell surface through its T1SS outer membrane TolC-like pore, LapE, until LapG cleaves LapA in the periplasm. We further demonstrate that this unusual retention strategy is likely conserved among LapA-like proteins, and it reveals a new subclass of T1SS ABC transporters involved in transporting this group of surface-associated LapA-like adhesins. These studies demonstrate a novel cell surface retention strategy used throughout the Proteobacteria and highlight a previously unappreciated flexibility of function for T1SS.
IMPORTANCE Bacteria have evolved multiple secretion strategies to interact with their environment. For many bacteria, the secretion of cell surface-associated adhesins is key for initiating contact with a preferred substratum to facilitate biofilm formation. Our work demonstrates that P. fluorescens uses a previously unrecognized secretion strategy to retain the giant adhesin LapA at its cell surface. Further, we identify likely LapA-like adhesins in various pathogenic and commensal proteobacteria and provide phylogenetic evidence that these adhesins are secreted by a new subclass of T1SS ABC transporters.
KEYWORDS: biofilm, Pseudomonas fluorescens, type I secretion system, adhesin, c-di-GMP
INTRODUCTION
The biofilm lifestyle is profoundly consequential to human health and industry, for better and for worse (1, 2). Although bacteria initiate surface contact and biofilm formation through a variety of strategies, many microbes need cell surface-associated protein adhesins to bind a surface. A family of giant type I secreted repeats-in-toxin (RTX)-containing adhesins have been shown to be critical for biofilm formation or surface binding by a variety of organisms, including Pseudomonas, Salmonella, Bordetella, Legionella, Vibrio, Shewanella, Desulfovibrio, and Marinomonas species (3–10). Cell surface display of these adhesins, which promotes biofilm formation, typically coincides with high intracellular cyclic di-GMP (c-di-GMP) levels.
Mechanistic studies from our lab on the giant RTX adhesin LapA of Pseudomonas fluorescens Pf0-1 identified a novel regulatory node, LapG/LapD, that links cell surface display of LapA to intracellular c-di-GMP levels. The ability of the periplasmic protease LapG to cleave LapA is inhibited when the effector, LapD, a transmembrane protein, is bound to cellular c-di-GMP. LapD sequesters LapG away from LapA when LapD is in its c-di-GMP-bound state. Conversely, when c-di-GMP levels decrease in the cell, LapD releases LapG. Free LapG in turn cleaves LapA from the cell surface, releasing the adhesin into the supernatant where it is unable to promote biofilm formation (11, 12).
In contrast, cell surface association and release of the RTX adhesin SiiE of Salmonella enterica, which does not encode lapG or lapD homologs, are controlled by the proton motive force (PMF). Here, the MotA- and MotB-like accessory proteins SiiA and SiiB likely utilize PMF to enhance SiiE retention (13). Thus, while SiiE and LapA belong to the same family of RTX adhesins, their cell surface regulation suggests they may localize to the cell surface via distinct mechanisms.
Although homologs of lapG and lapD are found throughout the Proteobacteria, the biofilm-promoting adhesins targeted by this regulatory node are largely unknown due to the low sequence identities between RTX adhesins. Two hallmarks of most LapG adhesin substrates characterized thus far are the presence of a N-terminal dialanine cleavage motif located roughly between residues 100 and 130 and a varied number of C-terminal RTX motifs (3, 14, 15).
Like other type I secretion system (T1SS) substrates, such as HlyA of Escherichia coli and CyaA of Bordetella bronchiseptica, LapA's C-terminal secretion signal and cognate T1SS machinery (LapEBC) are required for LapA cell surface localization and, thus, biofilm formation (6, 14). The T1SS is considered a one-step secretion strategy that lacks any periplasmic intermediate (16); thus, it is currently unclear how LapA localizes to the cell surface with an N-terminal dialanine cleavage motif that is accessible to the periplasmic protease LapG. Here, we demonstrate that LapA is not secreted in a canonical one-step T1SS fashion but rather tethers to the cell surface through its T1SS apparatus as a stalled secretion intermediate. We provide evidence that a cleavable retention module at the N terminus of LapA prohibits classical one-step secretion of the adhesin by forming a plug that tethers LapA to the outer membrane through its T1SS TolC-like pore, LapE. We also developed an algorithm to identify likely LapG substrates (i.e., LapA-like adhesins) in lapG- or lapD-containing genomes and provide evidence that this previously unappreciated retention strategy is broadly conserved in proteobacteria. Finally, phylogenetic analysis reveals that the ABC transporters of LapA-like substrates likely represent a distinct subclass of type I secretion systems.
RESULTS
Cell surface-associated LapA engages its T1SS.
To gain mechanistic insight into LapA cell surface localization, we first wanted to investigate the translocation status of cell surface-associated LapA. Surface-associated LapA is found in the outer membrane fraction; however, LapA lacks elements previously shown to be involved in the formation of outer membrane pores and translocation structures in target cell membranes (17–19). Thus, we considered the possibility that LapA is anchored to the cell surface by remaining in the T1SS apparatus as a secretion intermediate. This strategy could place LapA's N-terminal cleavage motif in the periplasm while displaying its C-terminal adhesive repeats toward the extracellular environment, and it is consistent with the C- to N-terminal T1SS secretion directionality (20). Although this retention mechanism is unheard of for T1SS substrates, the LapG substrate from Pseudomonas aeruginosa, CdrA, a two-partner secreted protein (TPS or T5bSS), likely uses a “cysteine hook” formed by an intramolecular disulfide bond to anchor itself to the cell surface through its outer membrane pore CdrB (21, 22). In support of our hypothesis, recent structural modeling suggests that the N terminus of the large RTX adhesin MpIBP of Marinomonas primoryensis could form a plug blocking secretion of the giant adhesin (23). To date, however, there are no experimental data to support this model.
Therefore, to determine if LapA is secreted via the classical one-step T1SS model or retained within its translocation machinery, we conducted a secretion competition experiment to compare the secretion of the C-terminal secretion domain of LapA tagged with a three-hemagglutinin (HA) epitope tag for Western blotting (HA-C235; Fig. 1A, far right). This tool allows us to discern if cell surface-associated LapA impacts the availability of LapEBC to secrete the HA-C235 peptide. That is, if the secretion pore (LapE) is occupied by LapA, we would expect that very little HA-C235 would be secreted. In contrast, if LapE is not occupied by LapA (due to deleting the adhesin, enhancing LapG activity to cleave LapA from the cell surface, or removing any potential N-terminal retention domains), we would expect increased secretion of the HA-C235 secretion substrate. Thus, we examined secretion of the HA-C235 protein in strains where LapA is locked at the cell surface (lapG mutant), or alternatively, in strains where LapA is continuously secreted into the supernatant (lapD mutant). In the lapD mutant, constitutive LapG activity removes LapA from the cell surface.
FIG 1.

LapA cell surface localization impacts LapEBC activity. (A) Scaled representation of LapA and HA-C235. (B and C) Biofilm formation (top) and Western blot analysis of HA-C235 in the supernatant (S) and whole-cell (WC) fractions (bottom) from the indicated strains. (D) Top, biofilm analysis of lapD mutants grown and washed in medium to activate (−EGTA) or inhibit (+EGTA) LapG activity. Bottom, Western blot analysis of the supernatant fraction for HA-C235 under the indicated growth and wash conditions. (E) Biofilm (top) and Western blot analysis (bottom) of the indicated strains. For panels B to E, the model substrate was detected with anti-HA antibody. (F) A model for how LapA and CdrA are localized to the cell surface. LapA is predicted to be anchored in the outer membrane via the outer membrane protein LapE, with an N-terminal “retention module” blocking the complete secretion of LapA. The previously described CdrA protein is anchored in the CdrB pore, and CdrA is retained by a cysteine hook that restricts its secretion through the CdrB pore. Left, when c-di-GMP levels are high, these proteins are anchored in their secretion pore. Right, upon cleavage of the N-terminal domains under low-c-di-GMP conditions, the adhesins are released from the cell surface. Also shown are the c-di-GMP receptor LapD and the LapG protease, which target the N terminus of LapA and the C terminus of CdrA.
Western blot analysis of the supernatant fraction indicates that like LapA, HA-C235 peptide secretion is LapEBC dependent. Deletion of the lapE gene, which codes for the outer membrane pore component of this T1SS, results in a loss of biofilm formation, as reported previously (6), and eliminates HA-C235 secretion. HA-C235 secretion is restored when HA-C235 and lapE are introduced into the lapE mutant and expressed as a transcriptional fusion from the pMQ72 expression vector (Fig. 1B).
We next assessed the level of secreted HA-C235 substrate in various mutant backgrounds to test our hypothesis that the occupancy of LapE by LapA would impact the secretion of our small model substrate. HA-C235 supernatant levels in the lapA lapG and lapD mutants are identical (Fig. 1C) and much higher than in the WT background (Fig. 1B), indicating that a lack of LapA and constitutive LapA secretion, respectively, does not impinge on HA-C235 secretion (Fig. 1B). However, when LapA is locked at the cell surface in the lapG mutant strain, HA-C235 secretion is nearly abolished (Fig. 1C). Together, these data indicate that cell surface localization, but not secretion, of LapA limits LapEBC availability for secreting other peptides.
Chemical inhibition of LapG locks LapA at the cell surface.
LapG is a calcium-dependent cysteine protease that can be chemically inhibited in vivo and in vitro with micromolar levels of EGTA (24). To determine if chemical inhibition of LapG in the lapD mutant background can block HA-C235 secretion by enhancing surface-associated LapA, we compared biofilm formation and HA-C235 secretion in the lapD mutant grown in the absence or presence of 500 μM EGTA (−EGTA or +EGTA, respectively). The addition of EGTA to the lapD mutant, which usually exhibits constitutive LapG activity, restores LapA cell surface localization and biofilm formation by inhibiting LapG activity (24). Here, each strain was subcultured for 5 h in K10T-1 medium with or without 500 μM EGTA (Fig. 1D, growth) and then gently pelleted and resuspended for 20 min in the indicated medium (Fig. 1D, wash). The supernatants were then collected and protein levels normalized to probe the HA-C235 protein via Western blot analysis. Biofilm formation was utilized to demonstrate LapG inhibition under +EGTA growth conditions. Biofilm assays were performed as described above for the Western blots, but for the biofilm assay, cells were grown statically for 5 h in K10T-1 in 96-well plates with or without 500 μM EGTA.
For cells grown and assayed in the absence of EGTA (−EGTA), which have abundant LapG activity and no cell surface LapA (24), there were no biofilm formation and robust secretion of the HA-C235 secretion substrate (Fig. 1D, left). In contrast, biofilm formation is restored in the lapD mutant grown in the presence of 500 μM EGTA (+EDTA), indicating that LapG activity is inhibited under these conditions (Fig. 1D, top). Consistent with our genetic evidence, chemical inhibition of LapG also blocked HA-C235 secretion, phenocopying the lapG mutant (Fig. 1C, lapG mutant, and Fig. 1D, center). Reactivation of LapG proteolysis by resuspending EGTA-grown cells (+EGTA) in K10T-1 medium lacking EGTA (−EGTA) partially restored HA-C235 secretion after a short 20-min incubation (Fig. 1D, right). Together, these data support the idea that LapA occupancy of the LapEBC secretion machinery can the block secretion of our model substrate, and they are consistent with the model of LapA localizing to the cell surface via anchoring in its T1SS machinery.
LapE overexpression rescues HA-C235 secretion in a lapG mutant background.
The LapEBC T1SS proteins form a tripartite complex (LapE-LapBC) to secrete LapA. Given that the secretion of HA-C235 is blocked when LapA is locked at the cell surface (i.e., a lapG mutant or addition of EGTA), we reasoned that a T1SS component(s) participating in cell surface localization of LapA is likely limiting. Therefore, overexpression of the limiting secretion component should allow additional secretion and thus should rescue HA-C235 secretion in a lapG mutant. Previous work from our lab comparing lapA, lapE, lapB, and lapC gene expression in the presence and absence of the biofilm-promoting nutrient phosphate indicates that lapE is the only gene downregulated under phosphate-limiting conditions that inhibits biofilm formation (25). Thus, we suspected that LapE might be the limiting component of the secretion machinery.
To determine if lapE overexpression can rescue HA-C235 secretion in a lapG mutant, we assayed for HA-C235 in the supernatant of the lapG mutant carrying a pMQ72-based HA-C235-lapE transcriptional fusion construct. This construct allows simultaneous expression of HA-C235 and LapE protein from the same plasmid, with the open reading frames coding for these proteins expressed from the same promoter. Western analysis of the supernatant fraction indicates that the coexpression of HA-C235 and LapE, but not HA-C235 alone, can stimulate HA-C235 secretion in a lapG mutant background (Fig. 1E). HA-C235 secretion in the lapA lapG mutant was used as a positive control for secretion of the model substrate. Together, these data are consistent with the model that LapA is retained on the cell surface by LapE, LapE levels are limiting, and retained LapA occupies the secretion pathway, thus blocking the secretion of any other substrate (Fig. 1F).
P. fluorescens LapA homologs contain a conserved N-terminal domain.
Previous work from our lab suggested that the N terminus of LapA may play a role in retaining the giant adhesin at the cell surface (14, 24). To gain insight into the retention mechanism of LapA, we used MUSCLE to align putative LapA-like proteins from closely related P. fluorescens strains and color-filled residues according to the default Clustal X color parameters in Jalview to highlight highly similar regions (see Fig. S1 and Table S1 in the supplemental material). We hypothesized that regions of high similarity may play functional roles in secretion and retention, while the low-identity regions may represent adhesive repeats that have evolved to bind specific substrata of importance to each microbe. Our analysis revealed these LapA homologs contain two highly similar regions: a C-terminal region that corresponds to LapA's type I secretion signal required for secretion via LapEBC (Fig. S1, blue box) (14) and an N-terminal region that extends ∼20 residues beyond the LapG dialanine cleavage motif (Fig. S1, red box, and Fig. 2A). Given the C- to N-terminal secretion directionality of T1S substrates (20), we speculated that the N terminus might be involved in retaining these LapA homologs at the cell surface.
FIG 2.

LapA's N terminus serves as a retention module. (A) The first ∼175 aa of the P. fluorescens LapA-like N termini (Fig. S1, red box) with LapG cleavage motif (arrowhead) and putative polyglycine linker (Poly-G). (B) Western blot analysis of supernatant (S) and whole-cell (WC) fractions for LapA. (C and D) Dot blot analysis of cell surface-associated LapA (CS) (C), and biofilm analysis of LapA N-terminal mutants (D). (D) n = 8. (Error bars indicate the standard error of the mean [SEM].) ***, P < 0.0001 (unpaired two-tailed t test). OD550, optical density at 550 nm.
The N terminus of LapA is required for surface retention.
Closer examination of the N terminus of P. fluorescens LapA homologs indicates that their sequence similarity breaks down shortly after a polyglycine region (Fig. 2A, Poly-G). Because polyglycine regions often serve as unstructured domain linkers, we hypothesized that the N-terminal region of LapA encompassing up to this linker may function as a retention module (1M–125S).
To test this idea, we engineered targeted deletions in this putative N-terminal domain. Analysis of the primary sequences and predicted secondary structures suggests that the N termini of Pseudomonas fluorescens LapA homologs may adopt similar secondary structures (Fig. S2A and C). We noticed regularly spaced glycine residues conserved in the N-terminal 125 amino acids (aa) of LapA and homologs (Fig. S2A and C), with a predicted β-sheet secondary structure between each glycine. Given that glycine residues are known to punctuate secondary structures, we used a “Gly-to-Gly” targeted truncation strategy to disrupt secondary structures within the N terminus of LapA, using the alignment from Fig. 2A as a guide.
LapA is typically undetectable in the supernatant and enriched at the cell surface of the lapG mutant (Fig. 2B and C, WT versus lapG). Therefore, candidate RM mutants were engineered into the hyper-biofilm-forming lapG gene deletion strain using unmarked allelic replacement, allowing us to decouple retention defects from LapG-mediated proteolysis. Biofilm formation and LapA localization were assayed by comparing each retention mutant to the parental lapG and Western blotting for LapA in the whole-cell (WC) and supernatant (S) fraction, as well as in cell surface (CS) fractions.
Consistent with our hypothesis, a LapA mutant lacking residues 31D–95A (Δ31D–95A) is produced at levels similar to that observed for wild-type LapA, but this mutant is released into the supernatant (Fig. 2B, blot S). Dot blot analysis of cell surface-associated LapA indicates this mutant variant does not localize to the cell surface (Fig. 2C), leading to the inability of this strain to form a biofilm (Fig. 2D). In support of our hypothesis that cell surface-associated LapA occupies its outer membrane pore LapE, this LapA retention mutant strain secrets HA-C235 at levels comparable to those in the lapA lapG and lapD mutant strains (Fig. 1C, far right). A retention defect phenotype was also observed in the Δ23V–95A RM mutant (Fig. S2B), a region that overlaps with the Δ31D–95A mutant, and Western analysis confirmed that this mutant is expressed at wild-type levels (data not shown). Interestingly, we were unable to detect the Δ02S–108A RM mutant via Western analysis (Fig. 2B and C). This mutant phenocopied a lapA mutant (Fig. 2B to D), suggesting the mutation destabilizes LapA. Together, these data support the idea that the N terminus of LapA functions as a retention module, contributing to the localization of LapA to the cell surface, and it also suggests that this region plays a critical, but unclear, role in LapA stability.
Biochemical characterization of the N-terminal module of LapA supports the proposed retention module model.
Thus far, our data indicate that the retention module is responsible for anchoring LapA at the cell surface, likely involving the LapE outer membrane channel as a conduit (Fig. 1F and 3A). Consequently, the N-terminal part of LapA comprising the first 160+ residues is predicted to contain a folded domain for retention and a narrow unfolded segment enabling the protein to traverse the LapE channel, a model similar to the one recently proposed for MpIBP (23). The following experiments were designed to test this model. In particular, to determine if the N terminus of LapA exhibits well-folded (retention module) and unfolded (LapE-traversing) properties, we probed the structure of the purified N-terminal fragment used in our in vitro cleavage experiments (1M–235V) in several ways.
FIG 3.

Biochemical characteristics of the LapA retention module. (A) Model for LapA retention in the LapE outer membrane conduit. Left, cartoon summary of LapA's domain architecture. Right, zoom in for modeling LapA surface retention module. Phyre prediction of the LapA retention module (template PDB: 5ix9) threaded through LapE, represented as TolC (PDB: 1ek9), with an LapG-targeting dialanine motif (TAAG) and regions critical for LapG binding (helix 1 and DPXXXLXX) exposed in the periplasm. Approximately 40 extended residues necessary to traverse the TolC channel are flanked by glycine-rich motifs (yellow). (B) Thermal melting. Purified LapA (residues 1 to 235) was subjected to thermal unfolding in the presence of SYPRO Orange, yielding a melting curve with a single unfolding phase at 85 ± 3.7°C. The plot shows the mean (continuous dark line) and standard deviation (gray lines) of 5 technical replicates. (C) SEC-MALS-SAXS data. LapA was subjected to size-exclusion chromatography (SEC) coupled to multiangle light scattering (MALS) and small-angle X-ray scattering (SAXS) analysis. The two plots show the elution peak from the MALS (top) and SAXS (bottom) sample cells. The SEC-MALS plot shows the signal of the light-scattering (LS) detector (gray line, left y axis) and molecular mass determination across the elution peak (black circles, right y axis). The two horizontal dashed lines indicate the monomer and dimer molecular weights of LapA based on the primary sequence. The SEC-SAXS plot shows the scattering intensity (gray line, left y axis) and radius of gyration (Rg; black squares, right y axis). (D) SAXS analysis. SAXS profiles collected across a size-exclusion chromatography peak were averaged. Background (buffer) scattering was subtracted using scattering data flanking the elution peak (top). The Guinier analysis estimates the protein Rg with 50.0 Å (qRg range, 0.57 to 1.30, where q is scattering vector). Inspection of the distance distribution function (middle) for LapA (gray solid curve) yields an Rg of 51.6 Å and a Dmax (maximum dimension) of 161 Å (Porod volume, 100,000 Å3; quality estimate, 78). For reference, the predicted distance distribution function for the retention domain homology model shown in panel A is shown (black dashed line). The corresponding Kratky plot for LapA (bottom) reveals features characteristic of partially unfolded proteins. (E) Limited proteolysis. Calcium-dependent LapA cleavage by LapG generates two distinct bands, a smaller fragment corresponding to an extended N-terminal retention domain (residues 1 to 108) and a larger C-terminal fragment (residues 109 to 235). Subtilisin and proteinase K converge over time to a single relatively protease-resistant fragment (*) with an apparent molecular mass similar to that predicted for the minimal retention domain (A). A representative gel of the results from at least three independent experiments is shown. Mol., molecular; term., terminal.
First, we assessed the stability of the purified protein fragment using thermal unfolding in the presence of an environmentally sensitive dye that interacts with hydrophobic regions as the protein unfolds. A folded protein is expected to show one (or more) unfolding transition(s), unlike fully disordered peptides that lack folded domains and typically lack significant cooperative changes in the exposure of hydrophobic residue upon melting. From the melting curves, it is apparent that LapA undergoes thermal unfolding at 85 ± 3.7°C (Fig. 3B), indicating this N-terminal fragment (or at least a part of it) adopts a highly thermostable fold.
Second, we assessed the conformation of the purified LapA fragment by size-exclusion chromatography (SEC)-coupled small-angle X-ray scattering (SAXS), an approach that reports on the folded state and overall dimensions of proteins in solution. X-ray solution scattering by LapA is recorded as the protein elutes from a gel filtration column. Coupling SAXS with SEC reduces radiation damage by avoiding repeated X-ray exposure of the same volume, while also benefiting from the size fractionation of the sample during SEC (Fig. 3C). In addition, the experimental setup included in-line multiangle light-scattering (MALS) detectors for the calculation of molecular weights of the eluting scattering particles, in this case, the N-terminal domain of LapA. Similar to the SEC profile during purification (not shown), the LapA fragment elutes in a single peak from a gel filtration column with a narrow distribution of radius of gyration (Rg) values across the peak, indicating a monodisperse species (Fig. 3C, bottom). The molecular mass values calculated based on light-scattering analysis fall between a monomer and dimer of LapA, with higher values at the front edge of the peak (Fig. 3C, top; molecular mass average ± standard deviation [SD], 40.8 ± 3.8 kDa). Such a molecular weight distribution suggests that purified LapA has a tendency to self-associate, existing as a transient monomer-dimer species under the conditions used here. The averaged X-ray scattering data collected across the elution peak (Fig. 3D, top) are used to extract structural information, including the overall dimensions of the protein in solution (26). Guinier analysis at very small scattering angles estimates the protein's radius of gyration (Rg) to be 50.0 ± 0.7 Å. A description of the data as a distance distribution or P(r) function yields a similar Rg as the Guinier analysis (51.6 Å) and a maximum diameter (Dmax) of the scattering units of 161 Å (Fig. 3D, middle). This analysis suggests that this LapA N-terminal fragment (or a portion of this fragment) has characteristics of intrinsically disordered proteins, an assessment that is based on the large Rg in proportion to the predicted molecular weight of the protein (26). Inspection of the Kratky plot generated from the scattering data is another common approach to estimate the folded state of a protein. In this analysis, a distinct peak is indicative of a compact folded protein, whereas unfolded or partially folded proteins feature a rise or plateau at the tail of the curve. The shape of the Kratky plot obtained for the LapA fragment does not reveal a distinct peak (Fig. 3D, bottom), supporting the notion that the N-terminal domain of LapA has characteristics of intrinsically disordered proteins. Thus, the combined experimental approaches described above indicate that the N terminus of LapA has both folded and unfolded features, which supports our overall model (Fig. 3A).
Last, we subjected the purified protein to limited proteolysis by subtilisin or proteinase K (Fig. 3E). In this assay, flexible and unfolded regions of a protein are more susceptible to proteolysis, whereas compactly folded regions resist degradation. The proteolysis patterns for LapA confirm the dual nature of LapA's solution state observed in the biophysical studies mentioned above: the proteolysis studies reveal a protease-stable domain fragment (asterisk in Fig. 3E) similar in molecular weight to the retention domain obtained after LapG cleavage, as well as a protease-sensitive part that is likely the region that is threaded through the TolC-like channel of LapE in cells displaying LapA at their surface (Fig. 3A).
ABC transporters of LapA-like adhesins form a distinct T1SS subgroup.
Previous bioinformatic studies have noted that LapG homologs are commonly encoded near T1SS ABC transporter genes (27). Because LapA retention does not fit the classical model of type I secretion, we were curious if the secretion machinery of LapA-like adhesins may contain features distinguishable from those of traditional T1SS. ABC transporters are found throughout all three domains of life but can often be functionally grouped based on common residues that are critical for secreting their substrate(s) (28–30). Studies of the HlyB component of the T1SS ABC transporter E. coli, which transports the RTX toxin HlyA, demonstrated that the HlyB ATPase contains an N-terminal domain that is critical for binding the C-terminal RTX motifs of HlyA (30). The N-terminal domain of HlyB resembles the C39 peptidase domain (C39) typically found to be N-terminally fused to bacteriocin ABC transporters, but HlyB lacks the catalytic cysteine residue required to cleave and activate immature bacteriocins during secretion (Fig. 4, bacteriocin transporters, cysteine highlighted in red) (31). Instead, the C39-like domain (CLD) of HlyB contains a tyrosine involved in binding HlyA that is conserved among many ABC transporters involved in secreting RTX toxins, including the ABC transporter secreting CyaB of B. bronchiseptica (Fig. 4, HlyB-like transporters, tyrosine highlighted in red). Thus, the C39 and CLD of bacteriocin and RTX transporters can be distinguished by their amino acid sequences (30).
FIG 4.

Phylogenetic analysis of ABC transporter subfamilies. Phylogenetic analysis of C39 peptidase domain or C39-like peptidase domains (CLD) from indicated ABC transporters (Table S2). Bootstrap values are indicated in red at noted branches. WebLogo results were generated by MUSCLE aligning members from each branch. WebLogo results were truncated, and characterized functional residues are indicated in red (Y for HlyB-like and C for bacteriocin transporters).
Interestingly, the ATPase component of the T1SS transporter of LapA, called LapB, contains an N-terminal CLD that lacks the residues critical for bacteriocin processing (C39) and RTX binding (Y20). To test if the CLD from lapB homologs can be used to distinguish transporters of adhesins that use an RM to localize these large proteins at the cell surface, we assessed the phylogenetic relationship between C39 and CLD sequences from bacteriocin and RTX toxins and the CLD from ABC transporters involved in adhesin retention. The N-terminal domains from several characterized RTX toxin and bacteriocin transporters were identified using InterPro (http://www.ebi.ac.uk/interpro/) and compared with putative LapB-like ABC transporters using the online phylogenetic analysis program phylogeny.fr, with default settings (http://www.phylogeny.fr). Three characterized LapB transporters (LapB, BB1189, and SO4318) and two ABC transporters encoded near lapGD homologs (Mar181_1244 and D782_4135) were chosen as potential LapB representatives for our analysis. Additionally, BLASTP was utilized to identify ABC transporters with N-terminal domains similar to LapB-CLD that are encoded in the Legionella pneumophila (LssB) and Pseudomonas aeruginosa (PA1876) genomes to determine if the LapB-CLD can predict transporters of LapA-like proteins. LssB transports RtxA, which is involved in L. pneumophila virulence, while PA1876 likely transports the cooperonic adhesin PA1874 and is involved in biofilm-specific antibiotic tolerance (32–34). Table S2 lists the species, UniProt identification, and CLD sequence information used for this analysis.
Our phylogenic analysis suggests that the ABC transporters of LapA-like adhesins form a distinct group of transporters, with good support, that lack the functional residues critical for RTX motif binding and bacteriocin processing (Fig. 4, LapB-like transporters WebLogo). These differences at the amino acid level likely reflect functional differences rather than the phylogenetic diversity of the organisms analyzed, because characterized RTX toxin and LapA-like adhesin ABC transporters encoded within the same genome cluster with their predicted substrate type. For example, the RTX toxin transporter CyaB (Fig. 4, orange circle) and LapA-like adhesin transporter BB1189 (red circle) encoded by B. bronchiseptica map to different clusters in this analysis. These data support the idea that adhesins with an N-terminal RM are secreted by a distinct group of T1SS ABC transporters.
Bioinformatic identification of LapG substrates.
Our genetic, biochemical, and bioinformatic data suggest that LapA belongs to a novel subfamily of RTX adhesins that are translocated via a distinct group of T1SS ABC transporters, retained at the cell surface through a periplasmic RM, and targeted by a periplasmic protease, LapG. Despite the importance of LapA-like proteins as key biofilm adhesins, it is difficult to identify these LapG substrates due to their relatively low sequence similarities. Additionally, open reading frame (ORF) analysis programs often overlook or misannotate these large and complex adhesins (5, 35, 36). To overcome the first limitation, we took advantage of the observation that the two proteins that control LapA localization, the c-di-GMP-receptor LapD and the LapD-regulated protease LapG, show high sequence similarity and functional conservation among microbes (3, 22, 37) and can be identified by their respective domain architectures (LapG, pfam06035; LapD, pfam16448). We utilized the NCBI Conserved Domains Database (CDD) and genome database to identify bacterial species containing lapDG homologs; ∼1,300 such lapGD-containing species spanning 120 genera were identified. Each annotated genome was investigated for proteins containing hallmarks of LapA: an N-terminal LapG cleavage site and C-terminal RTX motifs. To accomplish this task, we developed an algorithm to recognize large proteins (>1,000 aa) containing RTX motifs (DX[L/I]X4GXDX[L/I]XGGX3D) and a canonical N-terminal dialanine LapG cleavage motif ([T/A/P]AA[G/V]) between residues 80 and 150.
Although our approach is constrained to properly annotated LapA-like ORFs, we still identified approximately 500 putative LapG substrates in ∼50 genera throughout the Proteobacteria, including Legionella, Vibrio, and Marinomonas species (Table S3). Importantly, characterized LapG substrates LapA of P. fluorescens and BrtA of B. bronchiseptica were identified (3, 24). In support of our ABC transporter analysis (Fig. 4), our algorithm predicted RtxA, which is transported by the LapB-like ABC transporter LssB, to be a LapG substrate (Fig. 4, red circle). Interestingly, some species likely encode multiple LapG substrates, including P. fluorescens Pf0-1 (LapA and Pfl01_1463) and V. cholerae O395 (FrhA and VC0395_0388).
LapG substrates predicted in silico are processed in vitro.
LapG homologs can cleave a variety of LapA-like N termini from unrelated species in vitro and in vivo with various efficiencies (3, 37). To help validate the utility of our algorithm and confirm predicted LapG substrates, we cloned and expressed the N-terminal elements spanning the first ∼250 to 350 residues of the putative LapG-proteolyzed adhesins Pfl01_1463 from P. fluorescens and VC0395_0388 from Vibrio cholerae (Fig. 5A). Cell lysates of E. coli expressing C-terminally 6His-tagged, N-terminal fragments of Pfl01_1463 (residues 1M–240S, 117TAAG120 cleavage motif), or VC0395_0388 (1M–363G, 127AAAG130 cleavage motif) were mixed in equal concentrations with a lysate made from E. coli expressing P. fluorescens Pf0-1 LapG or empty vector (pMQ72). The LapG-dependent cleavage product was tracked via Western blotting, with the N terminus of LapA (1M–235V, 107TAAG110 cleavage motif) and an uncleavable variant (LapATRRG, AA108–109RR) serving as positive and negative controls, respectively (11).
FIG 5.
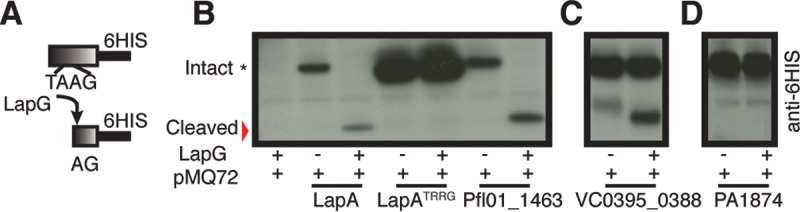
P. fluorescens Pf0-1 LapG in vitro cleavage analysis of LapA-like adhesins. (A) Overview of in vitro N-terminal LapG cleavage assay. (B to D) Intact and cleaved N termini from LapA(1M–272)-6His, Pfl01_01463(1M–240S)-6His, VC0395_0388(1M–363G)-6His, and PA1874(1M–251T)-6His, were visualized by Western blotting following 30 min of incubation with LapG-containing or empty vector control cell lysates.
Western blot analysis indicates that P. fluorescens Pf0-1 LapG can cleave both predicted substrates, although it is less efficient at cleaving VC0395_0388 (Fig. 5B and C). Unlike FrhA (4), a role for VC0395_0388 in infection and/or biofilm formation is currently unknown; however, these data implicate VC0395_0388 as a cell-surface-associated c-di-GMP-regulated biofilm-promoting adhesin.
Conversely, P. fluorescens Pf0-1 LapG is unable to cleave the N terminus of PA1874 (1M–251T, 137AAAIG141) (Fig. 5D), a large 238-kDa putative outer membrane adhesin encoded by Pseudomonas aeruginosa PAO1. Although the N terminus of PA1874 resembles LapA's RM (Fig. 6A), it was not detected by our algorithm because it contains a degenerate LapG cleavage motif (137AAAIG141). Instead, we manually identified PA1874 by its proximity to the LapB-like transporter PA1876 (Fig. 4) (34). Together, our in vitro cleavage analysis and ABC transporter bioinformatic analysis support the predictive power of our algorithm for detecting LapA-like LapG substrates, but they also may suggest that LapB-like transporters can transport adhesins that localize to the cell surface in a manner similar to that of LapA but are not cleaved by LapG.
FIG 6.

Low-identity retention modules are functionally conserved. (A) MUSCLE alignment of putative RMs from P. fluorescens, P. aeruginosa, and V. cholerae adhesins with residues colored according to the default Clustal X coloring scheme in Jalview. The putative polyglycine linker is boxed in red, while the canonical and degenerate LapG cleavage sites are boxed in green and yellow, respectively. (B) Dot blot analysis of surface-associated wild-type LapA and LapA chimeras (VC-Swap, PA-SWAP) in the wild-type (WT) and lapG mutant backgrounds. Pixel density is normalized to WT LapA in a WT background (y axis). (C) Biofilm formation of the strains from panel B. (D) Western blot analysis of LapA RM chimeras in the supernatant (S) and whole-cell (WC) fractions. For biofilm quantification, n = 8; error bars indicate the SEM. ***, P < 0.0001 (unpaired two-tailed t test).
LapG substrates predicted in silico have high-identity N- and C-terminal elements.
LapA homologs from P. fluorescens strains have high identity N- and C-terminal domains that correspond to their RM and type I secretion signals, respectively (Fig. S1 and S2). We next analyzed the predicted LapG substrates in L. pneumophila and B. bronchiseptica genomes identified by our algorithm to determine if these proteins have similar N- and C-terminal architectures. To accomplish this task, we used MUSCLE to align the predicted LapG substrates from the L. pneumophila and B. bronchiseptica genomes. In support of our retention model, the RtxA and BrtA homologs detected in L. pneumophila and B. bronchiseptica species show similar N- and C-terminal domains (Fig. S3 and S4, respectively). Although the degree of sequence similarity between these regions varies, LapA, RtxA, and BrtA homologs contain a conserved N-terminal region with spaced conserved glycine residues and a short linker sequence following the LapG cleavage site, which may encompass their respective retention modules. Additionally, PHYRE analysis (38) predicts that the putative RM of these adhesins may adopt secondary structures similar to that of LapA, despite their low sequence identity, suggesting these adhesins could tether through the outer membrane LapE pore, like LapA.
Evidence of a conserved retention strategy among LapB-like T1SS substrates.
Our ABC transporter bioinformatic analysis, RM comparisons, and in vitro cleavage results suggest that LapB-like T1SS may secrete two classes of adhesins that localize to the cell surface via a LapA-like mechanism: (i) those that are cleaved by LapG and removed from the cell surface in response to decreasing levels of c-di-GMP (LapA) and (ii) those that are retained at the cell surface similarly to LapA but are not targeted by LapG (PA1874). To further investigate this possibility, we engineered LapA chimera proteins where we replaced the RM of LapA (1M to 125S) with a representative RM from each of the above-mentioned classes. We chose VC0395_0388 and PA1874 for the first and second classes, respectively, because we have characterized these N termini in vitro (Fig. 5C and D). These chimeras are called VC-Swap and PA-Swap, respectively.
Using allelic exchange, we replaced the DNA encoding the N terminus of LapA of P. fluorescens with DNA corresponding to the amino acids detailed in the alignment in Fig. 6A in both the wild-type P. fluorescens and the lapG mutant background. Figure 6A also shows the LapG cleavage site in LapA and VC0395_0338 (boxed in green), the likely degenerate LapG cleavage site in PA1874 (boxed in yellow), and the putative polyglycine linker common to all three N termini (boxed in red).
Biofilm formation and chimera localization were compared to those of the parental wild-type and lapG mutant strains expressing the chimeras to determine if these chimera proteins were retained on the surface, could support biofilm formation in P. fluorescens, and/or were subject to LapG proteolysis to release the chimeric adhesins from the cell surface. The cell surface levels and biofilm phenotypes of the VC-Swap and PA-Swap chimeras are consistent with their differential susceptibilities to LapG cleavage in vitro (Fig. 6B and C). Western blot analysis of the VC-Swap chimera on the cell surface (Fig. 6B, CS) indicates that the deletion of the lapG gene enhances levels of the chimera at the cell surface, and more importantly, enhances biofilm formation (Fig. 6C). While we still detect some VC-Swap in the supernatant in a lapG mutant background (Fig. 6D), overall, the VC-Swap chimera mimics wild-type LapA in the parental wild-type and lapG backgrounds (Fig. 6B and C, WT). These data indicate that the RM from VC0395_0338 (Fig. 6A) can complement LapA localization and LapG-dependent release from the cell surface, suggesting that LapA and VC0395_0338 localize to the cell surface by a similar mechanism.
Conversely, PA-Swap chimera localization and corresponding biofilm formation were not impacted by LapG activity. Consistent with our in vitro LapG cleavage analysis (Fig. 5D), these data indicate that the PA1874 RM can complement LapA's RM for cell surface retention and biofilm formation but not for cleavage by LapG due the presence of a degenerate cleavage motif (Fig. 6, PA-Swap). Importantly, despite their low sequence identity with the RM of LapA, both chimeras are stable (Fig. 6D, WC), suggesting that these RMs also encode the information necessary for LapA stability (compare to Fig. 6B to D, Δ02S–108A).
DISCUSSION
Understanding the variety of mechanisms that govern secretion and surface display of adhesins is critical for combating or exploiting biofilm formation. In this study, we describe a novel cell surface localization strategy used by P. fluorescens Pf0-1 to anchor the biofilm-promoting giant adhesin LapA at its cell surface. To our knowledge, LapA is the first type I secreted substrate that does not follow the one-step rule of type I secretion. Our secretion competition analysis indicates that LapA is not secreted directly from the cell but rather occupies its T1SS TolC-like pore to associate with the cell surface. This surface attachment strategy is quite different from those of other type I secreted proteins, such as surface layer (S-layer) proteins, which bind cell surface epitopes to anchor at the cell surface (39). Bioinformatic analysis of LapA homologs from P. fluorescens strains suggests that these adhesins contain an N-terminal domain of approximately 120 to 150 amino acids that is punctuated by an unstructured polyglycine region. Mutational analysis of this LapA N-terminal domain indicates that this region is required for retention of the adhesion; thus, it is called the retention module (RM). Our biochemical analysis indicates the N terminus of LapA (residues 1M to 235V) is composed of two functionally distinct regions: (i) a well-structured N-terminal region that corresponds to the retention module and (ii) a poorly structured downstream region past the RM that may be threaded through the LapE outer membrane pore. Importantly, these N-terminal features are absent in adhesins, such as SiiE of S. enterica, which does not encode LapDG homologs.
Our bioinformatic and genetic data indicate that multiple adhesins throughout the Proteobacteria likely associate to the cell surface via this mechanism, suggesting that LapA represents a large subset of type I secreted substrates that do not follow the one-step secretion rule. To identify these likely LapG substrates, we developed an algorithm to identify large RTX adhesins with an N-terminal dialanine cleavage motif in lapGD-containing genomes. In addition to identifying known LapG substrates, this approach also revealed that some bacterial species encode multiple adhesins that are likely processed by LapG. To our surprise, P. fluorescens Pf0-1 encodes LapA, the focus of this study, as well as Pfl01_1463, a previously unknown LapG substrate. These LapA and Pfl01_1463 proteins have distinct domain architectures and differ in size (∼520 kDa versus ∼290 kDa, respectively); however, in vitro cleavage analysis indicates that Pfl01_1463 is indeed cleaved by LapG. These data suggest that P. fluorescens Pf0-1 and other organisms may utilize different adhesins to suit their current environment or the particular stage of biofilm formation.
How does the RM contribute to the cell surface localization of LapA? We propose that the RM stalls the final steps of LapA translocation, leaving LapA threaded through the TolC-like outer membrane pore, LapE, with its RM localized in the periplasm, accessible to LapG, and C-terminal adhesive repeats displayed at the cell surface (Fig. 1F and 3A). Our biochemical analysis of the proposed RM is consistent with the conclusion that a portion of this domain is well folded, thermostable, and sufficiently large to not be able to pass through the TolC-like LapE pore if the RM is folded. Presumably, the poorly folded domain is threaded through the LapE pore (Fig. 3A). Furthermore, while the models in Fig. 1F and 3A oppose the classical one-step paradigm detailed for over 25 years of T1SS literature, this model is consistent with our artificial HA-C235 substrate competition experiments, EGTA-mediated inhibition of LapG activity, and expression studies with the TolC-like LapE outer membrane protein. Given the abundance of LapA-like adhesins in pathogens and environmental microbes and the conserved role of c-di-GMP in regulating their cell surface localization, this means of secreting and anchoring an adhesin appears to be a general strategy used for cell-substratum and perhaps cell-cell adherence. Our bioinformatic analysis of LapB-like ABC transporters further supports this idea.
Bioinformatic analysis of ABC transporters involved in transporting characterized LapA-like RTX adhesins indicates that these transporters form a distinct subgroup of ABC transporters. Importantly, by combining our ABC transporter and LapG substrate prediction analyses, we can identify candidate T1SS ABC transporters and their likely cognate adhesin substrate. These predictions could be quite useful for identifying the mechanism of biofilm formation by poorly characterized microbes.
While the CLD of HlyB binds the C-terminal RTX motifs of HlyA to facilitate HlyA transport, LapB lacks the residues shown to be critical for HlyB-CLD binding. In toxins, such as HlyA, the RTX motifs have been shown to be critical for secretion and are thought to discourage premature folding of the toxin in the cytoplasm. This strategy seems well suited for small toxins; however, it is unclear if or how C-terminal RTX motifs could discourage cytoplasmic folding of giant adhesins. In the case of LapA, the N terminus and C-terminal RTX motifs are separated by >5,000 amino acid residues. Additionally, the deletion of RTX motifs of LapA does not profoundly inhibit biofilm formation (14), suggesting an alternative role for the RTX in LapA. Given that both the C39 and degenerate CLD have been shown to bind glycine-rich regions within their substrates, it is possible that the LapB-CLD binds LapA's glycine-rich linker to discourage folding, which could act as an antinucleation event.
Based on the recently reported Marinomonas MpIBP structural modeling (23) and the data presented here and reported previously (14), we built a model of the LapA retention module (Fig. 1F and 3A). Our genetic and structural analyses of the N terminus of LapA are in good agreement with the proposed N-terminal domain of MpIBP being localized to the periplasm. Interestingly, the proposed model for the MpIBP adhesin of Marinomonas (22) indicates that the LapG proteolysis site may be partially obscured by the TolC pore. Our mutational analysis also suggests that regions not included in the MpIBP nuclear magnetic resonance (NMR) structure are important for retention and LapG processing (14). In contrast, we propose that the LapG processing site is accessible in the periplasm, a conclusion consistent with our modeling (Fig. 3A), as well as our previous biochemical and genetic studies (11, 14, 22). For example, regions C-terminal to the RM may be important for stalling or aligning LapA within the LapE pore (Fig. 3A, far right between 117GGxxGGG123 and 159GG160). Further studies will be required to obtain a detailed structural picture of the adhesion anchored in the secretion pore.
MATERIALS AND METHODS
Strains and media.
The P. fluorescens and E. coli strains listed in Table S4 in the supplemental material were grown on lysogeny broth at 30°C and 37°C, respectively. Gentamicin was used when appropriate (10 μg/ml for E. coli and 30 μg/ml for P. fluorescens). For biofilm and LapA localization analyses, P. fluorescens strains were subcultured in K10T-1 for 6 h statically and with rotation, respectively (25).
In silico prediction of LapG substrates.
LapG and LapD homologs were defined as ORF-coding proteins with the pfam06035 and pfam16448 domains, respectively. The NCBI Conserved Domains Database (CDD) was utilized to generate a list of lapG- and lapD-containing bacteria, and the programming language R was used to determine the intersecting lapDG-containing bacteria. The protein annotations of these genomes were downloaded from the NCBI genome database, and each annotated locus was interrogated for the presence of a LapG cleavage site within amino acids 80 to 150 ([T/A/P]AA[G/V]) and at least one RTX motif (DX[L/I]X4GXDX[L/I]XGGX3D).
In vitro LapG cleavage analysis.
Crude extracts of E. coli S17 carrying the indicated pMQ72-based (40) plasmids were prepared by sonication. Protein levels were normalized after protein quantification with a bicinchoninic acid assay (Pierce). Cell lysates were mixed in equal amounts and incubated for 30 min at 30°C. The assay was terminated by adding an equal volume of 2× SDS-PAGE dye. Western blot analysis against the 6His epitope was used to detect the N-terminal fragment. All strains were grown with 0.4% arabinose to induce gene transcription.
Constructs for deletion mutants.
Briefly, ∼500 to 1,000 bp immediately upstream and downstream of the region targeted for deletion or exchange was cloned into pMQ30 using yeast cloning and transformed into E. coli S17 for conjugation into the indicated P. fluorescens Pf0-1 strain. Allelic replacement was performed as described previously (14, 40). All primers used in this study are listed in Table S5.
Static biofilm assay.
The static biofilm assay was performed and quantified as described previously (14). P. fluorescens strains were grown as indicated in “Strains and media,” above, in 96-well plates. Cell growth was discarded, and wells were stained with 0.1% crystal violet and destained with a 30% methanol and 10% acetic mixture. Biofilm intensity was quantified by measuring the absorbance at 550 nm.
LapA localization.
Whole-cell, supernatant, and cell surface localization analyses were performed as previously using an HA-tagged variant of LapA (14). Pf0-1 strains were subcultured as detailed in “Strains and media,” above. Cells were normalized and washed once in K10T-1. For cell surface analysis, 5-μl aliquots were spotted onto nitrocellulose. The remaining cells were resuspended in 1× SDS buffer as a whole-cell fraction. Filter-sterilized supernatants were concentrated in 30-molecular-weight cutoff (MWCO) centrifugal filters (catalog no. UFC803096; Millipore) and the protein quantified using the bicinchoninic acid assay (Pierce). Western blot analysis against the HA epitope was used to detect LapA.
HA-C235 localization.
Strains were subcultured for 5.5 h, washed, and then resuspended for 20 min in indicated medium under noninducing conditions. For chemical inhibition assays, K10T-1 medium was supplemented with 500 μM EGTA when noted. Whole-cell and supernatant fractions were prepped as for LapA localization. Western blot analysis against the HA epitope was used to detect HA-C235.
Thermal melting assay.
The N-terminal module of LapA (residues 1 to 235) was purified as described previously (24). A fluorescence-based thermal melting assay using SYPRO Orange (Invitrogen, Carlsbad, CA) was used to generate protein melting curves (41). The reaction mixture contains 200 μM protein, 25 mM Tris-HCl (pH 7.5), 100 mM NaCl, 2 mM dithiothreitol (DTT), and 5× SYPRO Orange. A 20-μl reaction mixture was melted in a MicroAmp 384-well plate (Applied Biosystems, Foster City, CA) using a ViiA 7 real-time PCR system (Applied Biosystems). The protocol was initiated with a 5-min incubation at 10°C, followed by increasing the temperature to 99.9°C at the rate of 0.03°C/s; the excitation and emission wavelengths were 485 ± 15 and 625 ± 10 nm, respectively. Data from 5 technical replicates were normalized, averaged, and plotted using GraphPad Prism (GraphPad, La Jolla, CA).
SEC-MALS-SAXS analysis.
Purified LapA (residues 1 to 235; 10 mg/ml) was injected onto a Superdex 200 10/300 column (GE Life Sciences) equilibrated in SAXS buffer (25 mM Tris-HCl [pH 7.5], 300 mM NaCl). The gel filtration column was in-line with the sample cells of light-scattering detector (Dawn Heleos II; Wyatt, Santa Barbara, CA), refractive index detector (Optilab T-rEX; Wyatt, Santa Barbara, CA), and SAXS. Each frame of scattering data was collected for 2 s, and frames corresponding to a single protein peak were averaged and background corrected by using the program BioXTAS RAW (42). Averaged and buffer-subtracted data were analyzed using Primus (43) and GNOM (44). Molecular weights were determined based on in-line SEC-MALS, as reported previously (11).
Limited proteolysis.
LapG was purified as described previously (37). Subtilisin and proteinase K were purchased from Sigma (Carlsbad, CA). Purified LapA (40 μM) was incubated with LapG (1.6 μM) in protease buffer (25 mM Tris-HCl [pH 7.5], 500 mM NaCl) in the absence or presence of 20 mM EGTA for 90 min at room temperature. In parallel reactions, LapA (40 μM) was incubated with either subtilisin (1 μM) or proteinase K (1 μM) over a time course of 30 min on ice. Aliquots were taken at the indicated time points. Reactions were stopped by the addition of protease inhibitors (Complete; Sigma, Carlsbad, CA) and SDS-PAGE sample buffer, followed by incubation at 99°C for 5 min. Proteolysis patterns were resolved on 15% SDS-PAGE gels stained with Coomassie blue.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Richard Gillilan and Jesse Hopkins for their support of SAXS data collection at Cornell's High Energy Synchrotron Source (CHESS). CHESS is supported by the National Science Foundation (NSF) and NIH/NIGMS via NSF award DMR-1332208, and the MacCHESS resource is supported by NIH/NIGMS award GM-103485. Our work was supported by the NIH via grant R01GM123609 (to H.S. and G.A.O.) and a Cornell Sloan Fellowship (to M.E.F.).
We thank C. Boyd for contributing one of the strains used in this study.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00734-17.
REFERENCES
- 1.Hall CW, Mah T-F. 2017. Molecular mechanisms of biofilm-based antibiotic resistance and tolerance in pathogenic bacteria. FEMS Microbiol Rev 41:276–301. doi: 10.1093/femsre/fux010. [DOI] [PubMed] [Google Scholar]
- 2.Liu T, Yu YY, Deng XP, Ng CK, Cao B, Wang JY, Rice SA, Kjelleberg S, Song H. 2015. Enhanced Shewanella biofilm promotes bioelectricity generation. Biotechnol Bioeng 112:2051–2059. doi: 10.1002/bit.25624. [DOI] [PubMed] [Google Scholar]
- 3.Ambrosis N, Boyd CD, O'Toole GA, Fernández J, Sisti F. 2016. Homologs of the LapD-LapG c-di-GMP effector system control biofilm formation by Bordetella bronchiseptica. PLoS One 11:1–16. doi: 10.1371/journal.pone.0158752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Syed KA, Beyhan S, Correa N, Queen J, Liu J, Peng F, Satchell KJF, Yildiz F, Klose KE. 2009. The Vibrio cholerae flagellar regulatory hierarchy controls expression of virulence factors. J Bacteriol 191:6555–6570. doi: 10.1128/JB.00949-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo S, Garnham CP, Whitney JC, Graham LA, Davies PL. 2012. Re-evaluation of a bacterial antifreeze protein as an adhesin with ice-binding activity. PLoS One 7:e0048805. doi: 10.1371/journal.pone.0048805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hinsa SM, Espinosa-Urgel M, Ramos JL, O'Toole GA. 2003. Transition from reversible to irreversible attachment during biofilm formation by Pseudomonas fluorescens WCS365 requires an ABC transporter and a large secreted protein. Mol Microbiol 49:905–918. doi: 10.1046/j.1365-2958.2003.03615.x. [DOI] [PubMed] [Google Scholar]
- 7.Cirillo SLG, Bermudez LE, El-Etr SH, Duhamel GE, Cirillo JD. 2001. Legionella pneumophila entry gene rtxA is involved in virulence. Infect Immun 69:508–517. doi: 10.1128/IAI.69.1.508-517.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu C, Cheng Y-Y, Yin H, Song X-N, Li W-W, Zhou X-X, Zhao L-P, Tian L-J, Han J-C, Yu H-Q. 2013. Oxygen promotes biofilm formation of Shewanella putrefaciens CN32 through a diguanylate cyclase and an adhesin. Sci Rep 3:1945. doi: 10.1038/srep01945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barlag B, Hensel M. 2015. The giant adhesin SiiE of Salmonella enterica. Molecules 20:1134–1150. doi: 10.3390/molecules20011134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De León KB, Zane GM, Trotter VV, Krantz GP, Arkin AP, Butland GP, Walian PJ, Fields MW, Wall JD. 2017. Unintended laboratory-driven evolution reveals genetic requirements for biofilm formation by Desulfovibrio vulgaris Hildenborough. mBio 8:e01696-. doi: 10.1128/mBio.01696-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newell PD, Boyd CD, Sondermann H, O'Toole GA. 2011. A c-di-GMP effector system controls cell adhesion by inside-out signaling and surface protein cleavage. PLoS Biol 9:e1000587. doi: 10.1371/journal.pbio.1000587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newell PD, Monds RD, O'Toole GA. 2009. LapD is a bis-(3′,5′)-cyclic dimeric GMP-binding protein that regulates surface attachment by Pseudomonas fluorescens Pf0-1. Proc Natl Acad Sci U S A 106:3461–3466. doi: 10.1073/pnas.0808933106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wille T, Wagner C, Mittelstädt W, Blank K, Sommer E, Malengo G, Döhler D, Lange A, Sourjik V, Hensel M, Gerlach RG. 2014. SiiA and Siib are novel type I secretion system subunits controlling SPI4-mediated adhesion of Salmonella enterica. Cell Microbiol 16:161–178. doi: 10.1111/cmi.12222. [DOI] [PubMed] [Google Scholar]
- 14.Boyd CD, Smith TJ, El-Kirat-Chatel S, Newell PD, Dufrêne YF, O'Toole GA. 2014. Structural features of the Pseudomonas fluorescens biofilm adhesin LapA required for LapG-dependent cleavage, biofilm formation, and cell surface localization. J Bacteriol 196:2775–2788. doi: 10.1128/JB.01629-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou G, Yuan J, Gao H. 2015. Regulation of biofilm formation by BpfA, BpfD, and BpfG in Shewanella oneidensis. Front Microbiol 6:1–11. doi: 10.3389/fmicb.2015.00790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koronakis V, Hughes C, Koronakis E. 1991. Energetically distinct early and late stages of HlyB/HlyD-dependent secretion across both Escherichia coli membranes. EMBO J 10:3263–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gadeberg OV, Orskov I. 1984. In vitro cytotoxic effect of alpha-hemolytic Escherichia coli on human blood granulocytes. Infect Immun 45:255–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fullner Satchell KJ. 2007. MARTX, multifunctional autoprocessing repeats-in-toxin toxins. Infect Immun 75:5079–5084. doi: 10.1128/IAI.00525-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Satchell KJF. 2011. Structure and function of MARTX toxins and other large repetitive RTX proteins. Annu Rev Microbiol 65:71–90. doi: 10.1146/annurev-micro-090110-102943. [DOI] [PubMed] [Google Scholar]
- 20.Lenders MHH, Weidtkamp-Peters S, Kleinschrodt D, Jaeger K-E, Smits SHJ, Schmitt L. 2015. Directionality of substrate translocation of the hemolysin A type I secretion system. Sci Rep 5:12470. doi: 10.1038/srep12470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borlee BR, Goldman AD, Murakami K, Samudrala R, Wozniak DJ, Parsek MR. 2010. Pseudomonas aeruginosa uses a cyclic-di-GMP-regulated adhesin to reinforce the biofilm extracellular matrix. Mol Microbiol 75:827–842. doi: 10.1111/j.1365-2958.2009.06991.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cooley RB, Smith TJ, Leung W, Tierney V, Borlee BR, O'Toole GA, Sondermann H. 2016. Cyclic di-GMP-regulated periplasmic proteolysis of a Pseudomonas aeruginosa type Vb secretion system substrate. J Bacteriol 198:66–76. doi: 10.1128/JB.00369-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo S, Stevens CA, Vance TDR, Olijve LLC, Graham LA, Campbell RL, Yazdi SR, Escobedo C, Bar-dolev M, Yashunsky V, Braslavsky I, Langelaan DN, Smith SP, Allingham JS, Voets IK, Davies PL. 2017. Structure of a 1.5-MDa adhesin that binds its Antarctic bacterium to diatoms and ice. Sci Adv 3:e1701440. doi: 10.1126/sciadv.1701440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boyd CD, Chatterjee D, Sondermann H, O'Toole GA. 2012. LapG, required for modulating biofilm formation by Pseudomonas fluorescens Pf0-1, is a calcium-dependent protease. J Bacteriol 194:4406–4414. doi: 10.1128/JB.00642-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Monds RD, Newell PD, Gross RH, O'Toole GA. 2007. Phosphate-dependent modulation of c-di-GMP levels regulates Pseudomonas fluorescens Pf0-1 biofilm formation by controlling secretion of the adhesin LapA. Mol Microbiol 63:656–679. doi: 10.1111/j.1365-2958.2006.05539.x. [DOI] [PubMed] [Google Scholar]
- 26.Kikhney AG, Svergun DI. 2015. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett 589:2570–2577. doi: 10.1016/j.febslet.2015.08.027. [DOI] [PubMed] [Google Scholar]
- 27.Ginalski K, Kinch L, Rychlewski L, Grishin N. 2004. BTLCP proteins: a novel family of bacterial transglutaminase-like cysteine proteinases. Trends Biochem Sci 29:389–392. doi: 10.1016/j.tibs.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 28.Kanonenberg K, Schwarz CKW, Schmitt L. 2013. Type I secretion systems–a story of appendices. Res Microbiol 164:596–604. doi: 10.1016/j.resmic.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 29.ter Beek J, Guskov A, Slotboom DJ. 2014. Structural diversity of ABC transporters. J Gen Physiol 143:419–435. doi: 10.1085/jgp.201411164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lecher J, Schwarz CKW, Stoldt M, Smits SHJ, Willbold D, Schmitt L. 2012. An RTX transporter tethers its unfolded substrate during secretion via a unique N-terminal domain. Structure 20:1778–1787. doi: 10.1016/j.str.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 31.Håvarstein LS, Diep DB, Nes IF. 1995. A family of bacteriocin ABC transporters carry out proteolytic processing of their substrates concomitant with export. Mol Microbiol 16:229–240. doi: 10.1111/j.1365-2958.1995.tb02295.x. [DOI] [PubMed] [Google Scholar]
- 32.Fuche F, Vianney A, Andrea C, Doublet P, Gilbert C. 2015. Functional type 1 secretion system involved in Legionella pneumophila virulence. J Bacteriol 197:563–571. doi: 10.1128/JB.02164-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cirillo SLG, Yan L, Samrakandi MM, Cirillo JD. 2002. Regulation of the Legionella pneumophila rtxA gene in amoebae. Microbiology 148:1667–1677. doi: 10.1099/00221287-148-6-1667. [DOI] [PubMed] [Google Scholar]
- 34.Zhang L, Mah TF. 2008. Involvement of a novel efflux system in biofilm-specific resistance to antibiotics. J Bacteriol 190:4447–4452. doi: 10.1128/JB.01655-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chatterjee R, Nag S, Chaudhuri K. 2008. Identification of a new RTX-like gene cluster in Vibrio cholerae. FEMS Microbiol Lett 284:165–171. doi: 10.1111/j.1574-6968.2008.01199.x. [DOI] [PubMed] [Google Scholar]
- 36.Cirillo SLG, Lum J, Cirillo JD. 2000. Identification of novel loci involved in entry by Legionella pneumophila. Microbiology 146:1345–1359. [DOI] [PubMed] [Google Scholar]
- 37.Chatterjee D, Boyd CD, O'Toole GA, Sondermann H. 2012. Structural characterization of a conserved, calcium-dependent periplasmic protease from Legionella pneumophila. J Bacteriol 194:4415–4425. doi: 10.1128/JB.00640-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kelly LA, Mezulis S, Yates C, Wass M, Sternberg M. 2015. The Phyre2 web portal for protein modelling, prediction, and analysis. Nat Protoc 10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ford MJ, Nomellini JF, Smit J. 2007. S-layer anchoring and localization of an S-layer-associated protease in Caulobacter crescentus. J Bacteriol 189:2226–2237. doi: 10.1128/JB.01690-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shanks RMQ, Caiazza NC, Hinsa SM, Toutain CM, O'Toole GA. 2006. Saccharomyces cerevisiae-based molecular tool kit for manipulation of genes from Gram-negative bacteria. Appl Environ Microbiol 72:5027–5036. doi: 10.1128/AEM.00682-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lo MC, Aulabaugh A, Jin G, Cowling R, Bard J, Malamas M, Ellestad G. 2004. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem 332:153–159. doi: 10.1016/j.ab.2004.04.031. [DOI] [PubMed] [Google Scholar]
- 42.Hopkins JB, Gillilan RE, Skou S. 2017. BioXTAS RAW: improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J Appl Crystallogr 50:1545–1553. doi: 10.1107/S1600576717011438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. 2003. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J Appl Crystallogr 36:1277–1282. doi: 10.1107/S0021889803012779. [DOI] [Google Scholar]
- 44.Svergun DI. 1992. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr 25:495–503. doi: 10.1107/S0021889892001663. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.