

ABSTRACT
Clustered regularly interspaced short palindromic repeats (CRISPR) and the CRISPR-associated proteins (Cas) provide bacteria and archaea with adaptive immunity to specific DNA invaders. Mycobacterium tuberculosis encodes a type III CRISPR-Cas system that has not been experimentally explored. In this study, we found that the CRISPR-Cas systems of both M. tuberculosis and Mycobacterium bovis BCG were highly upregulated by deletion of Rv2837c (cnpB), which encodes a multifunctional protein that hydrolyzes cyclic di-AMP (c-di-AMP), cyclic di-GMP (c-di-GMP), and nanoRNAs (short oligonucleotides of 5 or fewer residues). By using genetic and biochemical approaches, we demonstrated that the CnpB-controlled transcriptional regulation of the CRISPR-Cas system is mediated by an Orn-like activity rather than by hydrolyzing the cyclic dinucleotides. Additionally, our results revealed that tuberculosis (TB) complex mycobacteria are functional in processing CRISPR RNAs (crRNAs), which are also more abundant in the ΔcnpB strain than in the parent strain. The elevated crRNA levels in the ΔcnpB strain could be partially reduced by expressing Escherichia coli orn. Our findings provide new insight into transcriptional regulation of bacterial CRISPR-Cas systems.
IMPORTANCE Clustered regularly interspaced short palindromic repeats (CRISPR) and the CRISPR-associated proteins (Cas) provide adaptive immunity to specific DNA invaders. M. tuberculosis encodes a type III CRISPR-Cas system that has not been experimentally explored. In this study, we first demonstrated that the CRISPR-Cas systems in tuberculosis (TB) complex mycobacteria are functional in processing CRISPR RNAs (crRNAs). We also showed that Rv2837c (CnpB) controls the expression of the CRISPR-Cas systems in TB complex mycobacteria through an oligoribonuclease (Orn)-like activity, which is very likely mediated by nanoRNA. Since little is known about regulation of CRISPR-Cas systems, our findings provide new insight into transcriptional regulation of bacterial CRISPR-Cas systems.
KEYWORDS: Mycobacterium tuberculosis, Rv2837c, Orn, CRISPR, nanoRNA, c-di-AMP
INTRODUCTION
Clustered regularly interspaced short palindromic repeats (CRISPR) and the CRISPR-associated (cas) genes have been discovered in approximately one-half of bacteria and most archaea that have been sequenced (1–3). The CRISPR-Cas systems provide these cells with prokaryotic adaptive immunity to invasion of mobile genetic elements, including plasmids and viruses (1–3). CRISPRs harbor arrays of conserved short repetitive DNA sequences (repeats), which are interspaced by unique DNA fragments (spacers) adapted from foreign invaders. The cas genes are typically clustered in an operon adjacent to the CRISPR arrays. The proteins encoded by these genes include nucleases, helicases, DNA- and RNA-binding proteins, and polymerases (4). Traditionally, CRISPR-Cas systems have been classified into three major types (5, 6). A new classification was expanded to encompass six types, among which types I, II, and III have been extensively studied (6–8).
The CRISPR-Cas system-mediated defense process can be divided into three phases: (i) spacer acquisition, (ii) CRISPR expression, and (iii) CRISPR interference (1–3). The components of bacterial CRISPR-Cas systems and their roles in gene editing have been extensively characterized in recent years. In contrast, transcriptional regulation of CRISPR-Cas systems is overall poorly understood. In several bacterial species, CRISPR-Cas systems are regulated by transcription factors, such as cyclic AMP (cAMP) receptor protein (CRP), histone-like nucleoid-structuring (H-NS) protein, leucine-responsive regulatory protein (LRP), and regulator of leucine biosynthesis operon (LeuO) (9–15).
Mycobacterium tuberculosis is the etiologic agent of tuberculosis (TB), which causes approximately 8 to 9 million new cases and around 1.5 million deaths annually according to the recent annual TB reports of the World Health Organization (WHO). Although M. tuberculosis has been recognized for over a century, the biology of the pathogen remains largely unknown. The CRISPR-Cas systems in mycobacteria have been bioinformatically analyzed (16, 17), whereas the biology of these systems has not been experimentally explored in TB complex mycobacteria. M. tuberculosis encodes a type III CRISPR-Cas system, which is composed of two CRISPR arrays that harbor 24 and 18 repeats, respectively. The 36-bp repeat sequences in both CRISPR arrays are highly conserved among TB complex mycobacteria. The arrangement of the CRISPR arrays of Mycobacterium bovis is similar to that of M. tuberculosis except that M. bovis consists of 25 and 17 repeats, respectively, within the two CRISPR arrays. Mycobacterium bovis BCG (BCG) was originally derived from M. bovis and is the currently available attenuated vaccine strain against M. tuberculosis. BCG harbors 30 and 19 repeats, respectively, within the two CRISPR arrays (16). Therefore, BCG possesses several distinct spacers compared to both M. tuberculosis and M. bovis.
We have previously reported that M. tuberculosis Rv3586 (disA) encodes a diadenylate cyclase that converts ATP into cyclic di-AMP (c-di-AMP) (18). M. tuberculosis Rv2837c (cnpB) encodes a phosphodiesterase that specifically cleaves c-di-AMP into AMP (19). We also demonstrated that DisA is the sole diadenylate cyclase in M. tuberculosis, as a ΔdisA ΔcnpB strain does not process detectable c-di-AMP (20). Deletion of cnpB resulted in significant virulence attenuation in a mouse pulmonary infection model (19, 21), which was very likely due to significantly elevated c-di-AMP levels, as overexpression of M. tuberculosis disA also led to a similar outcome (22). An earlier study demonstrated that CnpB functions similarly to an Escherichia coli oligoribonuclease (Orn) that hydrolyzes 2-mer to 5-mer nanoRNAs (short oligonucleotides of 5 or fewer residues), except that CnpB prefers 2-mer nanoRNA as a substrate (23). Additionally, a recent report showed that CnpB also degrades cyclic di-GMP (c-di-GMP) (24), although we showed that CnpB prefers c-di-AMP to c-di-GMP, according to an in vitro enzymatic kinetics analysis (19). In this study, we performed a transcriptome-sequencing (RNA-Seq) analysis using M. tuberculosis wild type (WT) and ΔcnpB to determine genes that were differentially expressed between the two strains. Surprisingly, we found that the RNA reads of the CRISPR-Cas system in the ΔcnpB strain were much higher than those in the WT. We further determined the molecular basis of the CnpB-mediated control of the CRISPR-Cas systems in TB complex mycobacteria. Our findings reveal that CnpB controls the CRISPR-Cas systems through an Orn-like activity, which is very likely mediated by nanoRNA rather than by hydrolyzing c-di-AMP or c-di-GMP.
RESULTS
RNA-Seq analysis revealed that CnpB controls expression of the CRISPR-Cas systems in M. tuberculosis and BCG in a c-di-AMP-independent manner.
We have previously shown that M. tuberculosis CnpB is functional as a c-di-AMP phosphodiesterase and that deletion of cnpB significantly elevates c-di-AMP levels within M. tuberculosis (19). In this study, we initially attempted to determine c-di-AMP-mediated gene regulation in M. tuberculosis by comparing the expression profiles of the WT and ΔcnpB strains using RNA-Seq. Overall, 26 genes were upregulated and 35 genes were downregulated significantly in the mutant compared to the WT, using ≥2 -fold change as a cutoff (Table 1). Interestingly, all the cas genes were highly upregulated in the ΔcnpB strain compared to the WT (Fig. 1A and B). It has been reported that the M. tuberculosis CRISPR-Cas system consists of two CRISPR arrays and nine cas genes in the locus (16, 17) (Fig. 1C). We used csm3 as a representative to validate the RNA-Seq results by reverse transcription (RT)-PCR. The results showed that csm3 expression was significantly elevated in the ΔcnpB strain, which is consistent with the RNA-Seq data. The expression of csm3 in the ΔcnpB strain was reduced to the WT level by complementation of the mutant with cnpB (Fig. 1D), indicating that the altered expression of the CRISPR-Cas system is CnpB specific. A similar result was also observed with the BCG strains (Fig. 1E). Surprisingly, the expression of csm3 was not altered by deletion of disA, which encodes the c-di-AMP synthase, in both the WT and ΔcnpB genetic backgrounds (Fig. 1D and E), indicating that the regulation of the CRISPR-Cas systems of M. tuberculosis and BCG by CnpB is independent of c-di-AMP.
TABLE 1.
Gene regulation by deletion of cnpB in M. tuberculosisa
| Gene | Function | Fold change (log2) | P value |
|---|---|---|---|
| Rv2913c | N-Acyl-d-glutamate amidohydrolase | −2.1 | 0.00005 |
| Rv0792c | FAD-dependent NAD(P)-disulfide oxidoreductase | −1.9 | 0.00005 |
| Rv2912c | Transcriptional regulator; TetR family | −1.8 | 0.00005 |
| Rv0791c | N5,N10-Methylene tetrahydromethanopterin reductase | −1.8 | 0.00005 |
| Rv2009 | Hypothetical protein | −1.6 | 0.00005 |
| Rv2466c | Hypothetical protein | −1.6 | 0.00005 |
| Rv0654 | Lignostilbene-α,β-dioxygenase | −1.5 | 0.00005 |
| Rv1807 | PPE family protein | −1.4 | 0.00005 |
| Rv3252c | Alkane-1 monooxygenase | −1.4 | 0.00005 |
| Rv3463 | N5,N10-methylene tetrahydromethanopterin reductase | −1.4 | 0.00005 |
| Rv1285 | Sulfate adenylyltransferase subunit 2 | −1.3 | 0.00005 |
| Rv1808 | PPE family protein | −1.3 | 0.00005 |
| Rv0768 | Aldehyde dehydrogenase | −1.3 | 0.00005 |
| Rv1168c | PPE family protein | −1.2 | 0.00005 |
| Rv3054c | NADPH-quinone oxidoreductase | −1.2 | 0.00005 |
| Rv0251c | Heat shock protein Hsp | −1.1 | 0.00005 |
| Rv0331 | Oxidoreductase (flavoprotein) | −1.1 | 0.00005 |
| Rv1169c | PE family of proteins | −1.1 | 0.00005 |
| Rv0105c | LSU ribosomal protein L28p | −1.1 | 0.00002 |
| Rv2010 | Toxin 1; PIN domain | −1.1 | 0.00005 |
| Rv3174 | Putative oxidoreductase | −1.1 | 0.00160 |
| Rv3615c | RD1-dependent secreted antigen | −1.1 | 0.00005 |
| Rv1130 | 2-Methylcitrate dehydratase | −1.1 | 0.00005 |
| Rv1554 | Fumarate reductase subunit C | −1.0 | 0.00165 |
| Rv0384c | ClpB protein | −1.0 | 0.00005 |
| Rv2025c | Cobalt-zinc-cadmium resistance protein | −1.0 | 0.00005 |
| Rv1286 | Sulfate adenylyltransferase subunit 1 | −1.0 | 0.00035 |
| Rv1805c | Hypothetical protein | −1.0 | 0.00005 |
| Rv3614c | ESX-1 secretion system protein | −1.0 | 0.00005 |
| Rv1552 | Succinate dehydrogenase flavoprotein subunit | −1.0 | 0.00005 |
| Rv3616c | ESX-1 secretion system protein | −1.0 | 0.00005 |
| Rv1809 | PPE family protein | −1.0 | 0.00005 |
| Rv1639c | Hypothetical protein | −1.0 | 0.00005 |
| Rv3824c | Polyketide synthase-associated protein PapA1 | −1.0 | 0.00005 |
| Rv3171c | Nonheme haloperoxidase Hpx | −1.0 | 0.00005 |
| Rv2006 | Uncharacterized glycosyl hydrolase | 1.0 | 0.00005 |
| Rv1757c | Mobile element protein | 1.0 | 0.00040 |
| Rv3000 | YbbM seven-transmembrane-helix protein | 1.0 | 0.00005 |
| Hypothetical protein | 1.1 | 0.00005 | |
| Rv1370c | Mobile element protein | 1.1 | 0.00055 |
| Rv2815c | Mobile element protein | 1.1 | 0.00010 |
| Rv0342 | Isoniazid-inducible protein IniA | 1.1 | 0.00005 |
| Hypothetical protein | 1.1 | 0.00005 | |
| Rv1955 | Hypothetical protein | 1.2 | 0.00005 |
| Rv2835c | Glycerol-3-phosphate ABC transporter UgpA | 1.3 | 0.00005 |
| Rv3862c | Transcriptional regulator WhiB-like WhiB6 | 1.3 | 0.00010 |
| Rv2450c | Putative saccharopine dehydrogenase | 1.3 | 0.00005 |
| Rv2816c | CRISPR-associated protein Cas2 | 1.5 | 0.00005 |
| Rv0341 | Isoniazid-inducible protein IniB | 1.8 | 0.00005 |
| Rv2836c | Putative DNA damage-inducible protein F | 2.4 | 0.00005 |
| Rv2817c | CRISPR-associated protein Cas1 | 3.2 | 0.00005 |
| Hypothetical protein | 3.7 | 0.00005 | |
| Hypothetical protein | 4.9 | 0.00005 | |
| Rv2819c | CRISPR-associated protein; Csm5 family | 5.0 | 0.00005 |
| Rv2818c | CRISPR-associated protein Csm6 | 5.3 | 0.00005 |
| Rv2822c | CRISPR-associated protein; Csm2 family | 5.5 | 0.00005 |
| Rv2823c | CRISPR-associated protein; Csm1 family | 5.5 | 0.00005 |
| Rv2820c | CRISPR-associated RAMP protein; Csm4 family | 5.5 | 0.00005 |
| Rv2824c | CRISPR-associated protein Cas6 | 5.6 | 0.00005 |
| Rv2821c | CRISPR-associated RAMP Csm3 | 5.7 | 0.00005 |
| Hypothetical protein | 5.8 | 0.00005 |
Genes listed without gene names were not annotated in the M. tuberculosis H37Rv genome reported by Cole et al. (25). The mean fold change and the P value of each gene were directly exported from the analysis with RNA-Rocket.
FIG 1.

Expression controlled by CnpB in M. tuberculosis. (A) RNA reads of all the genes in WT and ΔcnpB strains determined using RNA-Seq were plotted. The cas genes are indicated in red. The data plotted are the means of three biological repeats analyzed using RNA-Rocket. (B) RNA fold changes of the cas genes in M. tuberculosis ΔcnpB compared to those in the WT determined using RNA-Seq. The data shown are the means analyzed from three biological repeats (P = 5 × 10−5 for all the cas genes). (C) Genetic organization of the CRISPR-Cas system in M. tuberculosis. The size of each gene (in base pairs) is indicated above the gene. The size of each intergenic region (in base pairs) is also indicated between the related adjacent genes. Negative numbers indicate overlaps between the adjacent genes. (D) Results of RT-PCR of csm3 in M. tuberculosis WT, ΔdisA, and ΔcnpB strains and the complemented mutants (ΔdisA ΔdisC and ΔcnpB ΔcnpC). (E) Results of RT-PCR of csm3 in BCG WT, ΔcnpB, complemented ΔcnpB (ΔcnpB ΔcnpC), and ΔdisA ΔcnpB strains. (D and E) The gel images shown are representative of three repeat experiments. The PCRs of sigA served as controls for normalization of the cDNAs. The relative expression of csm3 versus that of sigA was quantitated using ImageJ. The bar graphs show the means of three independent experiments. The error bars indicate the standard errors of the means (SEM). Note that the M. tuberculosis ΔdisA ΔcnpB strain exhibited a result similar to that for the double mutant of BCG, and therefore, the result for the M. tuberculosis double mutant is not shown. *, P < 0.05; **, P < 0.01 compared to the WT.
Determination of the transcription start site of the cas operon.
In order to characterize the expression of the cas genes, we cloned a 197-bp DNA fragment upstream of the cas6 open reading frame (ORF) into a promoterless lacZ reporter plasmid based on the annotation of the M. tuberculosis genome (25) and transformed this cas6-lacZ reporter plasmid into BCG WT and ΔcnpB strains. Neither engineered strain exhibited β-galactosidase activity (data not shown), which is inconsistent with the RNA-Seq results. We noticed putative −35 (TTGACC), −10 (TATTCT), and Shine-Dalgarno (SD) (AGGAGA) sequences downstream of the annotated translation start codon (Fig. 2A). Therefore, we hypothesized that the translation start codon for cas6 was misannotated and the actual translation started 156 bp downstream from the site annotated (Fig. 2A). To verify our prediction, we first performed RT-PCRs using three different forward primers (P1 to P3) and one reverse primer (P4), as indicated in Fig. 2A. The results showed that only P3 and P4 amplified a fragment from the cDNA, whereas no amplicon was detected using either P1 or P2 as a forward primer (Fig. 2B). This observation indicates that the transcript of cas6 is much shorter at the 5′ end than annotated, which is consistent with our prediction. We further examined the transcription start site of cas6 by using 5′ rapid amplification of cDNA ends (RACE), which was detected 131 nucleotides (nt) downstream from the annotated translational start codon (Fig. 2A). Based on these analyses, we constructed another cas6-lacZ reporter fusion, which exhibited low expression in BCG WT and the ΔdisA strain but robust expression in the ΔcnpB and ΔdisA ΔcnpB strains compared to the gyrB-lacZ reporter control (Fig. 2C). This observation is consistent with the results of RT-PCR, the transcription start site mapping, and the RNA-Seq analysis. Therefore, CnpB controls the transcription of the CRISPR-Cas systems of TB complex mycobacteria.
FIG 2.

Transcriptional analysis of cas6. (A) DNA sequence upstream of cas6. ttg, annotated start codon of cas6; atg, start codon of cas6 that we predicted. Putative −10, −35, and SD sequences are marked. The transcription start site mapped using 5′-RACE is indicated by the asterisk. The primers (P1 to P4) used for the RT-PCRs in panel B are also indicated. (B) RT-PCR analysis of cas6 transcripts using the primers indicated in panel A. sigA was used as a positive control. serC represents RT-PCR with primers flanking an intergenic region between serC and Rv0885, which served as a negative control without transcript. The data shown are a representative of three repeat experiments. (C) β-Galactosidase assays of BCG WT, ΔdisA, ΔcnpB, and ΔdisA ΔcnpB harboring the vector control (lacZ) or promoter fusions for gyrB and cas6. The promoter fusion with cas6 was constructed based on our sequence analysis results. Bacteria were grown 7 days prior to the assays. lac, a promoterless control; gyrB, a constitutive expression control; cas6, lacZ fused with the cas6 promoter. The data shown are the means of three repeat experiments. The error bars indicate SEM.
The upregulation of the cas genes in the ΔcnpB strain is caused by the loss of an Orn-like activity of CnpB.
We previously reported that CnpB is capable of hydrolyzing c-di-AMP into AMP (19). However, our new findings clearly indicate that the regulation of the cas genes by CnpB is not mediated by c-di-AMP (Fig. 1). It has been reported that CnpB can also cleave c-di-GMP (24) and nanoRNA (23). Therefore, we determined whether the regulation of the cas genes by CnpB is mediated by c-di-GMP or nanoRNA.
It has been shown that Rv1357c encodes a c-di-GMP phosphodiesterase (26). We constructed a ΔRv1357c mutant in M. tuberculosis and determined the expression of csm3 in ΔRv1357c by RT-PCR. The results showed that csm3 expression was not altered by deletion of Rv1357c in M. tuberculosis (Fig. 3A), indicating that the regulation of the cas operon is not mediated by c-di-GMP.
FIG 3.
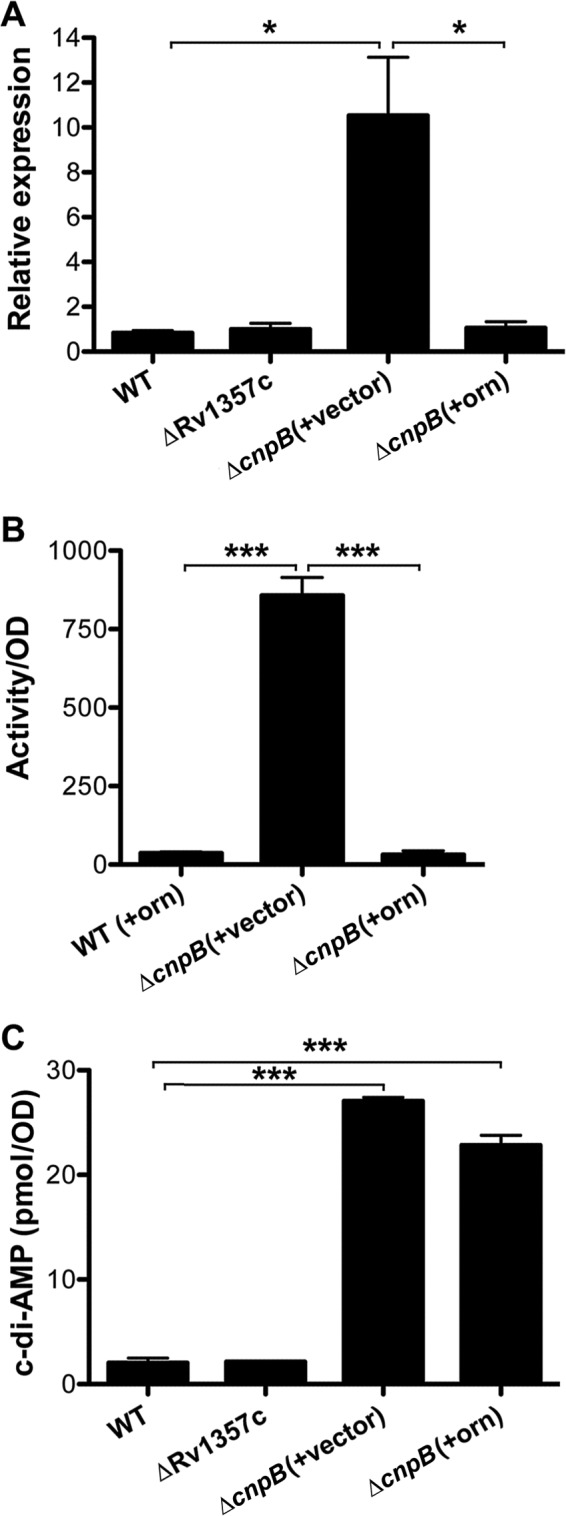
Regulation of the cas genes by CnpB. (A) Results of RT-PCR of csm3 in M. tuberculosis WT, ΔRv1357c, ΔcnpB expressing orn (+orn), and ΔcnpB harboring the control vector (+vector) strains. The PCR bands were quantitatively analyzed using ImageJ. The relative expression of csm3 was normalized by that of sigA. (B) β-Galactosidase assays of cas6 promoter-reporter fusion in orn-expressing (+orn) BCG WT and ΔcnpB strains. The BCG ΔcnpB strain bearing an expression vector (+vector) was used as a control. The cas6 promoter fusion was constructed based on our mapping results. Note that the expression of the promoter fusion in BCG WT harboring the vector control was indistinguishable from that in the WT expressing orn, and therefore, it is not shown. (C) Determination of intrabacterial c-di-AMP levels in M. tuberculosis WT, ΔRv1357c, ΔcnpB expressing orn (+orn), and ΔcnpB harboring the control vector (+vector) strains. Samples were prepared from 7-day cultures and analyzed using ELISA. The data shown are the means of three repeat experiments. The error bars indicate SEM. *, P < 0.05; ***, P < 0.001.
E. coli Orn is an oligoribonuclease that cleaves 2-mer to 5-mer nanoRNAs. CnpB is a homolog of Orn in terms of nanoRNA cleavage (23). We constructed a plasmid that overexpresses E. coli orn and transformed this recombinant plasmid and the control vector, respectively, into M. tuberculosis ΔcnpB. RT-PCR with these strains showed that the upregulation of csm3 in ΔcnpB was corrected by expressing orn but not by the transformation of the control vector (Fig. 3A). Furthermore, we transformed the orn-expressing and control plasmids individually into BCG ΔcnpB, which harbors the cas6-lacZ reporter fusion. As a result, the high activity of β-galactosidase in the ΔcnpB strain was dramatically reduced by the expression of orn but not by the transformation of the control vector (Fig. 3B). These results indicate that CnpB controls the expression of the cas genes by an activity similar to that of E. coli Orn.
To exclude the possibility that Orn also cleaves c-di-AMP, we determined bacterial c-di-AMP levels in an M. tuberculosis WT strain and ΔcnpB strains harboring the orn-expressing plasmid or the empty vector as a control. As expected, expression of orn did not significantly reduce the c-di-AMP levels of ΔcnpB compared to the levels in a ΔcnpB strain bearing the empty vector (Fig. 3C). Furthermore, we purified the Orn protein overexpressed in a recombinant E. coli strain and incubated the protein with c-di-AMP, c-di-GMP, or phosphoadenylyl adenosine (pApA) as a 2-mer nanoRNA. The catalytic products were then separated by high-performance liquid chromatography (HPLC). The results showed that Orn cleaved pApA into AMP similarly to CnpB but did not hydrolyze either c-di-AMP or c-di-GMP, which differs from CnpB (Fig. 4; see Fig. S1 in the supplemental material). Since the common function between CnpB and Orn is cleavage of nanoRNA, we conclude that CnpB controls the expression of the cas genes in TB complex mycobacteria through an Orn-like activity, which is very likely mediated by nanoRNA.
FIG 4.
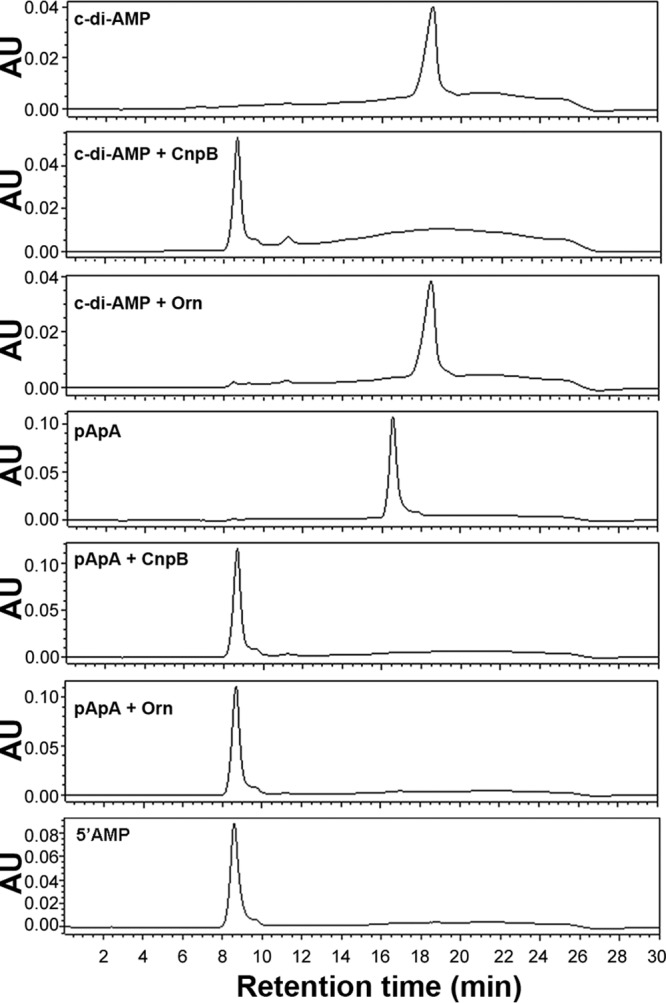
Enzymatic activities of CnpB and Orn determined using HPLC. Purified proteins were incubated with c-di-AMP or pApA. Purified c-di-AMP and pApA were individually analyzed as standards. AU, absorbance units.
CnpB controls the levels of crRNAs in TB complex mycobacteria.
It has been reported that the M. tuberculosis CRISPR-Cas system consists of two CRISPR arrays and nine cas genes in the locus (16, 27). However, whether mycobacteria are able to process CRISPR RNA (crRNA) and how the crRNA levels are controlled are unexplored. Our RNA-Seq data showed much higher reads for not only the cas genes but also the CRISPR arrays in the ΔcnpB strain than in the WT (Fig. 1A and 5). We explored whether precursor crRNA (pre-crRNA) or processed crRNA is upregulated by deletion of cnpB. Since M. tuberculosis and BCG possess similar CRISPR-Cas systems, except that the numbers of repeats vary between the two strains, we used BCG as a surrogate for M. tuberculosis to determine the RNA levels. We first compared pre-crRNA levels in the WT and ΔcnpB strains. The results showed that pre-crRNAs were upregulated in the ΔcnpB strain, which was corrected by the complementation of the mutant with cnpB (Fig. 6).
FIG 5.

Comparison of reads of cas2 and its downstream RNA of M. tuberculosis WT and ΔcnpB analyzed using the Rockhopper program. Expression from both the positive strand (blue) and the negative strand (red) of the WT and ΔcnpB strains, respectively, is shown. The translational stop codon of cas2 is indicated with an arrow. The sequence of the first repeat in the complement orientation is indicated. DEG, differentially expressed genes; operons, multigene operons; UTRs, untranslated regions.
FIG 6.
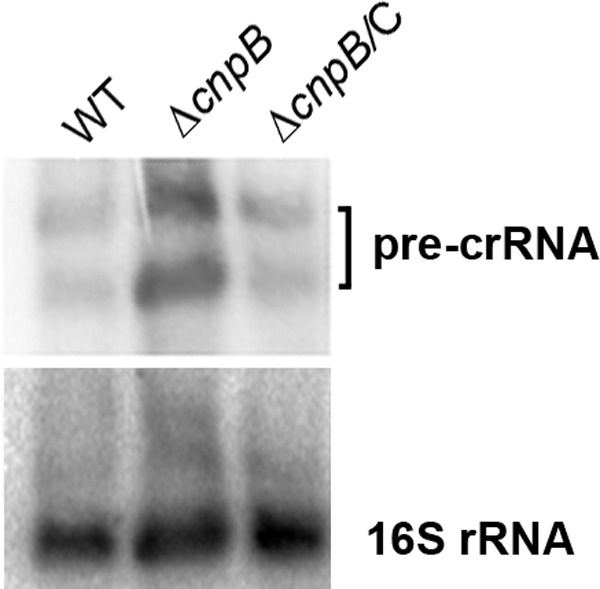
Northern blot analysis of pre-crRNAs. RNA samples of BCG WT, ΔcnpB, and complemented mutant strains were separated on 1% denatured agarose gels. After transfer onto a Hybond membrane, they were first hybridized with a probe specific to the M. tuberculosis CRISPR repeat sequence. The membrane was then stripped and reprobed with an oligonucleotide specific to M. tuberculosis 16S rRNA. The data shown are representative of three repeat experiments.
The size of the repeat sequence within the CRISPR arrays of both M. tuberculosis and BCG is 36 bp. The spacers in the M. tuberculosis CRISPR arrays are 25 to 41 bp, whereas they are 25 to 43 bp in BCG. Therefore, most of the processed crRNAs in BCG should be smaller than 70 nt. By using Northern blotting with a probe specific to the repeat sequence, we detected multiple RNA bands with sizes mostly smaller than that of the 73-nt His-tRNA control (Fig. 7). This result indicates that the CRISPR-Cas system in BCG is capable of processing pre-crRNAs into crRNAs. We also found levels of crRNAs in the ΔcnpB strain higher than those in the WT, which could be reduced by the complementation of the mutant with cnpB (Fig. 7; see Fig. S2 in the supplemental material). As expected, deletion of disA in the ΔcnpB genetic background did not alter the levels of crRNAs, indicating that the control of crRNAs by CnpB is c-di-AMP independent (Fig. 7). Interestingly, the elevated crRNA levels in the ΔcnpB strain were partially reduced by the expression of E. coli orn but not by the transformation of the control vector (Fig. 7), suggesting that the upregulation of the crRNAs by the deletion of cnpB is mediated by the loss of an Orn-like activity of CnpB. Taking the data together, we conclude that CnpB controls crRNA levels through an Orn-like activity, which is very likely mediated by nanoRNA, in TB complex mycobacteria.
FIG 7.
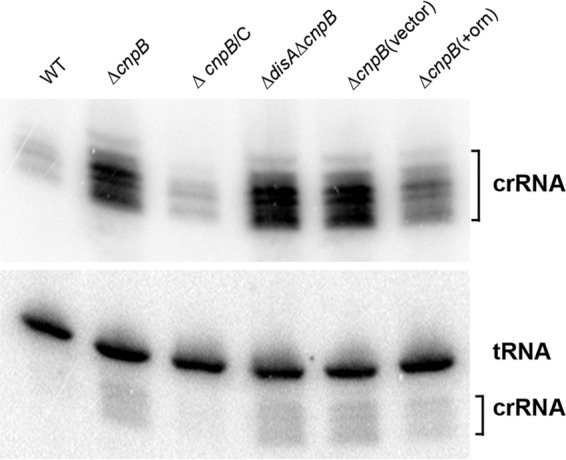
Northern blot analysis of crRNAs. RNA samples of BCG WT, ΔcnpB, complemented ΔcnpB, ΔdisA ΔcnpB, ΔcnpB expressing orn, and ΔcnpB bearing the control vector were separated in 10% polyacrylamide gels. After transfer onto a Hybond membrane, they were first hybridized with a probe specific to the M. tuberculosis CRISPR repeat sequence. The membrane was then stripped and reprobed with an oligonucleotide specific to M. tuberculosis His-tRNA. Note that the crRNA probes were not fully stripped off and residual signals remained in the blot with His-tRNA. The data shown are representative of three repeat experiments.
DISCUSSION
The CRISPR-Cas systems in TB complex mycobacteria have been recognized based on sequence analyses (16, 17) but have not been investigated experimentally. DHH superfamily proteins include RecJ, nanoRNases (NrnA), c-di-AMP phosphodiesterases, and pyrophosphatases (28). According to our current knowledge, M. tuberculosis CnpB is a DHH superfamily protein that cleaves c-di-AMP, c-di-GMP, and nanoRNA (19, 23). In this study, we found that CnpB controls the CRISPR-Cas systems in TB complex mycobacteria mediated by an Orn-like activity, possibly through nanoRNA rather than c-di-AMP or c-di-GMP (Fig. 8). Our study also shows for the first time that TB complex mycobacteria encode a functional system to process crRNAs.
FIG 8.

Hypothetical model of CnpB-controlled CRISPR-Cas system in TB complex mycobacteria. The meanings of the symbols in the ΔcnpB strain diagram are identical to those in the WT diagram. In this model, more nanoRNAs were accumulated in the ΔcnpB strain than in the WT, which induces the expression of both pre-crRNA and the cas genes. Eventually, more crRNAs were generated in the ΔcnpB strain than in the WT.
The components of CRISPR-Cas systems in some bacteria and their roles in gene editing have been well recognized. In contrast, our knowledge about regulation of CRISPR-Cas systems is still in an early stage. In several bacterial species, it has been shown that CRISPR-Cas systems are transcriptionally regulated by transcription factors, such as CRP, H-NS, LRP, and LeuO (9–15). In addition to these pleiotropic or global regulators, some dedicated regulators have also been shown to control the expression of CRISPR-Cas systems. These regulators include DevS of Myxococcus xanthus and Csa3 proteins of Sulfolobus islandicus (29).
The role of nanoRNAs in regulation of a CRISPR-Cas system has not been reported. It is possible that the regulation of the CRISPR-Cas system by CnpB that we found is orchestrated by a nanoRNA-responsive transcription factor. A CRP homolog (Rv3676) in M. tuberculosis has been well studied, but this transcription factor does not regulate the CRISPR-Cas system, according to multiple global gene expression analyses of M. tuberculosis Δcrp (30–32). Homologs of LRP (Rv3291c and Rv2779c) (33–36) and H-NS (Rv3597c) (37–40) have also been characterized in M. tuberculosis. However, it is unknown whether these homologs or other dedicated regulators regulate the expression of the cas genes, which warrants further investigation.
RNA turnover is an essential bioprocess in all organisms and is associated with the regulation of gene expression. RNA is degraded by RNases, but some RNases are unable to completely cleave the target RNAs. NanoRNAs are RNA degradation products of these RNases. For example, the end products of degradation catalyzed by RNase II and RNase R range from 1-mers to 6-mers, which vary in size with different RNases (41). M. tuberculosis CnpB and its homolog in Mycobacterium smegmatis have been shown to cleave 2-mer nanoRNAs (28). This process likely provides a source for nucleotide recycling. Bacterial nanoRNA levels in this study were not reported, as we had technical difficulty in examining nanoRNA by following a method used for Pseudomonas aeruginosa (42). Interestingly, in an orn deletion mutant of P. aeruginosa, nanoRNAs of 2-mers to 4-mers accumulate within bacteria and prime transcription initiation, which results in widespread transcription start site shifting (42). Additionally, the use of nanoRNAs to prime transcription initiation is also coupled with global alterations in gene expression, with a total of 1,158 genes differentially expressed in the orn mutant (42). Transcription start site shifting of cas6 is possible based on the assembled RNA reads. However, it is surprising that many fewer genes are differentially expressed in M. tuberculosis ΔcnpB than in the Δorn mutant of P. aeruginosa. Only the genes in the cas operon are highly expressed in M. tuberculosis ΔcnpB, suggesting that the upregulation of these genes is not due to transcription start site shifting caused by nanoRNA. The molecular basis of the CnpB-mediated gene regulation of M. tuberculosis will be further explored in our future studies.
crRNA is assembled into a ribonucleoprotein complex with Cas proteins. Six types of CRISPR-Cas systems are grouped into two classes: class 1 comprises multisubunit effector complexes, whereas class 2 possesses a single complex (6–8). Based on sequence analysis of the Cas proteins, M. tuberculosis possesses a class 1 type III CRISPR-Cas system (5). This type of CRISPR-Cas system typically targets single-stranded RNA (ssRNA) in a protospacer-adjacent motif (PAM)-independent manner (8). The target RNA-bound complex cleaves the target RNA transcript and, in addition, has a target RNA-stimulated nonspecific DNase activity that cleaves single-stranded DNA (8, 43–45). How the M. tuberculosis CRISPR-Cas system cleaves nucleic acids remains to be investigated.
The repeat sequences in the CRISPR arrays of TB complex mycobacteria are highly conserved, whereas the spacers vary slightly among different species (16). It is known that bacteria acquire spacer sequences from prior invasion of bacteriophages or plasmids. We compared all the M. tuberculosis spacer sequences with the Actinobacteriophage Database (http://www.phagesdb.org), which includes 1,435 currently sequenced mycobacteriophages. None of the spacer sequences matched any mycobacteriophage sequence. As pointed out by He et al., nearly all mycobacteriophages were initially isolated using M. smegmatis, in which no CRISPR locus can be found, as a host (16). Therefore, the targets of crRNAs of TB complex mycobacteria remain completely unknown, and exploration of these targets will provide more insights into the understanding of mycobacterial biology.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
M. tuberculosis H37Rv and BCG (strain Pasteur [Trudeau Institute]) and their derivatives were used in this study. ΔdisA, ΔcnpB, and ΔdisA ΔcnpB mutants of both M. tuberculosis and BCG were reported previously (19, 20). Both the ΔdisA and ΔcnpB mutants were complemented as described previously (19, 20). Bacteria were grown in mycomedium (Middlebrook 7H9 medium [BD] supplemented with 0.5% glycerol, 10% oleic acid-albumin-dextrose-catalase [OADC], and 0.05% Tween 80), Sauton's medium (46), or Middlebrook 7H10 agar (BD) supplemented with 10% OADC and 0.01% cycloheximide. Fresh cultures were inoculated from frozen stocks for every experiment. Bacteria were grown in tissue culture flasks standing with ambient air unless otherwise specified. E. coli strains were grown in Luria-Bertani (LB) broth or on LB agar plates. M. smegmatis mc2155 was grown in mycomedium. All cultures were grown at 37°C, except M. smegmatis, which was grown at 30°C in mycomedium. Kanamycin at 25 μg/ml, hygromycin at 50 μg/ml, or zeocin at 100 μg/ml was added when necessary.
Deletion of Rv1357c in M. tuberculosis.
All the plasmids used in this study are listed in Table 2. To construct strain ΔRv1357c of M. tuberculosis, an upstream fragment was amplified by PCR with primers JY007 and JY008 (see Table S1 in the supplemental material). This fragment was first cloned using a TA cloning kit (Invitrogen) and then subcloned into pJSC407 (kindly provided by Jeffery Cox [47]) between EcoRV and HindIII sites to generate pGB003. A downstream fragment of Rv1357c was amplified by PCR with primers JY009 and JY010 (see Table S1 in the supplemental material). The fragment was first cloned using the TA cloning kit (Invitrogen) and then subcloned into pGB003 between XbaI and KpnI sites to generate pGB004. Plasmid pGB004 was digested with PacI, ligated with PacI-digested phasmid phAE159 (generously provided by William Jacobs, Jr.) overnight, and then packaged using MaxPlax lambda packaging extracts (Epicentre Biotechnoloies) to generate phasmid pGB005 in E. coli HB101. The recombinant phasmid DNA was prepared from hygromycin-resistant colonies and transformed into M. smegmatis mc2155. Phages were prepared from a plaque derived from M. smegmatis by a method similar to that in previous reports (48, 49). M. tuberculosis bacteria grown to log phase were infected with high-titer phages. Hygromycin-resistant ΔRv1357c candidates were further screened by PCR using primers listed in Table S1 in the supplemental material.
TABLE 2.
Plasmids used in this study
| Plasmid | Descriptiona | Source or reference |
|---|---|---|
| pJSC407 | M. tuberculosis knockout plasmid; Hygr | 47 |
| phAE159 | Knockout phasmid; Carbr | 48 |
| pGB003 | pJSC407 carrying Rv1357c upstream fragment; Hygr | This study |
| pGB004 | pGB003 carrying Rv1357c downstream fragment; Hygr | This study |
| pGB005 | phAE159 carrying pGB004 plasmid DNA; Hygr | This study |
| pMBC1260 | pMBC304 carrying Rv0805 promoter; Kanr Apr | 19 |
| pGB228 | pMBC1260 carrying E. coli orn ORF; Kanr Apr | This study |
| pET28a(+) | Expression plasmid; Kanr | Novagen |
| pGB282 | pET28a(+) carrying E. coli orn ORF; Kanr | This study |
| pLACint | Promoter reporter vector with a promoterless lacZ; Kanr | 56 |
| pGB232 | pLacint carrying putative cas6 promoter (annotated); Kanr | This study |
| pGB248 | pLacint carrying putative cas6 promoter (identified); Kanr | This study |
Hygr, hygromycin resistance; Kanr, kanamycin resistance; Carbr, carbenicillin resistance; Apr, ampicillin resistance.
RNA extraction.
Total-RNA extraction from bacteria was carried out using the method reported by Mangan et al. (50). For RNA-Seq analysis and RT-PCR, the total RNA was treated twice with RNase-free DNase I on column with an RNeasy minikit (Qiagen) and an RNase-Free DNase set (Qiagen) following the manufacturer's instructions. For Northern blotting, RNA was treated twice with RNase-free DNase I in solution. The RNA concentration was determined spectrophotometrically using a Biophotometer (Eppendorf) at 260 nm. PCR of sigA was performed, using 0.1 μg of total RNA as a template, to ensure the absence of DNA contamination in the RNA samples.
RNA-Seq analysis.
For RNA-Seq analysis, RNA samples for three biological experiments were prepared. rRNA was removed from 2.5 μg of each total-RNA sample using the Ribo-Zero magnetic kit (Epicentre). Strand-specific DNA libraries for Illumina sequencing were prepared using the ScriptSeq complete kit (Epicentre) following the manufacturer's manual. Sequencing was performed using an Illumina HiSeq instrument at the University at Buffalo Next Generation Sequencing and Expression Analysis Core Facility. Sequencing files were uploaded to Galaxy of RNA-Rocket (http://rnaseq.pathogenportal.org), trimmed, aligned, and assembled according to the annotation of M. tuberculosis H37Rv (25). To compare RNA reads, RNA-Seq data were also uploaded to Rockhopper (https://cs.wellesley.edu/~btjaden/Rockhopper/) and analyzed using the program (51, 52).
Synthesis of cDNA and RT-PCR.
cDNA was prepared and amplified as previously described (53). Control reactions were performed using primers specific to 16S rRNA (54) or sigA (55) (see Table S1 in the supplemental material). PCR products were separated on 2% agarose gels. The mean intensity of each band was analyzed using ImageJ software (National Institutes of Health [NIH]). The relative expression was calculated by normalizing the intensity of the csm3 band with that of the sigA band amplified with the same cDNA sample.
Cloning of promoters.
The annotated intergenic region between Rv2824c and Rv2825c was PCR amplified using primers JY392 and JY393 (see Table S1 in the supplemental material). The promoter region that we predicted was amplified using primers JY432 and JY433. The PCR products were individually cloned into a single-copy integrative vector, pLACint, upstream of a promoterless lacZ gene, as previously reported (56).
β-Galactosidase activity assay.
Bacteria were grown to exponential phase by shaking at 37°C in mycomedium. A 300-μl portion of each strain was transferred to a fresh tube, followed by sonication as reported previously (57). The β-galactosidase activity was measured by a method similar to that in a previous report (58) using the fluorescent dye 5-acetylamino-fluorescein di-β-d-galactopyranoside (C2FDG) (Molecular Probes) and read in a CytoFluor multiwell plate reader (PerSpective Biosystems). Activity readings were normalized by the optical density of each culture at 650 nm (OD650), determined using a Thermomax microplate reader (Molecular Devices). The results were expressed as β-galactosidase activity divided by the OD.
Expression and purification of proteins.
Recombinant protein of M. tuberculosis CnpB was expressed and purified similarly to what we reported previously (19). The ORF of E. coli orn was amplified by PCR using primers JY518 and JY519 (see Table S1 in the supplemental material) and E. coli DH5α DNA as the template. The PCR product was digested with NdeI and HindIII and cloned into NdeI-HindIII-digested pET28a(+) (Novagen) to generate pGB282. This plasmid was transformed into E. coli BL21(DE3) to express Orn. The recombinant protein was purified by a method similar to what we reported previously (18, 59). Briefly, an overnight culture was inoculated in 500 ml LB broth containing 25 μg/ml kanamycin and incubated at 37°C with shaking to an OD600 of 0.6. The culture was then cooled to room temperature, followed by the addition of isopropyl-β-d-1-thiogalactopyranoside (IPTG) to a final concentration of 0.1 mM. After induction at room temperature for 4 h, the bacteria were harvested by centrifugation at 6,000 rpm for 10 min. The bacterial pellet was resuspended in 50 ml of buffer A (50 mM Tris-HCl, 150 mM NaCl, 10 mM imidazole, 10% glycerol) with 1% protease inhibitor (Roche) and sonicated on ice for 10 min, with 5-s pulses and 10-s intervals. After centrifugation at 12,000 rpm for 30 min at 4°C, the supernatant was loaded onto an Ni-nitrilotriacetic acid (NTA)-agarose column preequilibrated with buffer A at a flow rate of 0.5 ml/min. The column was subsequently washed with 50 ml of buffer B (50 mM Tris-HCl, 500 mM NaCl, 20 mM imidazole, pH 7.5) and 50 ml buffer C (50 mM Tris-HCl, 500 mM NaCl, 50 mM imidazole, pH 7.5) at 1 ml/min. The His-tagged proteins were eluted with 10 ml buffer D (50 mM Tris-HCl, 500 mM NaCl, 300 mM imidazole, pH 7.5) at 0.5 ml/min. All the eluted fractions were analyzed by SDS-PAGE, and collections were dialyzed against phosphate-buffered saline (PBS) at 4°C. The purified protein was stored in PBS with 10% glycerol at −80°C until it was used.
Expression of E. coli orn in mycobacteria.
The E. coli orn ORF was amplified by PCR using primers JY388 and JY389 (see Table S1 in the supplemental material) and E. coli DH5α DNA as the template. The PCR product was digested with KpnI and cloned into pMBC1260 at the KpnI site to generate pGB228. The inserted DNA was verified by PCR and sequencing. Plasmids pGB228 and, as the control plasmid, pMBC1260 were individually transformed into ΔcnpB mutants of M. tuberculosis and BCG, respectively, by electroporation. Transformants were selected on Middlebrook 7H10 plates containing 25 μg/ml kanamycin and were further verified by PCR prior to making stocks.
HPLC.
The reaction mixtures (10 μl) to determine the activity of Orn contained 50 mM Tris-HCl (pH 7.5), 1 mM MnCl2, and 0.5 mM the indicated nucleotide. The reaction was initiated by adding Orn or CnpB protein to 3 μM and was incubated for 1 h at 37°C. Subsequently, each reaction was terminated by adding 1 μl of 0.5 M EDTA, and the reaction mixture was diluted 1:5 with water. Finally, 20 μl of each sample was injected and separated by reverse-phase HPLC with a C18 column (250 by 4.6 mm; Vydac) using a Waters 625 LC system equipped with a 996 photodiode array detector and a 717 autosampler (Waters). The samples were eluted using the same buffers we reported previously (60). Nucleotides were monitored at 254 nm.
Detection of c-di-AMP.
Bacteria were grown in mycomedium for 7 days. After determination of the OD600 of each strain, the bacteria were harvested, and each bacterial pellet was resuspended in 0.5 ml 50 mM Tris-HCl (pH 8.0), heat killed, and disrupted with 1-mm beads using a bead beater (BioSpec). The lysate was collected, and the bacterial debris was removed by centrifugation for 10 min at 13,000 rpm. The supernatant was used as a bacterial c-di-AMP sample. c-di-AMP was then detected using an enzyme-linked immunosorbent assay (ELISA) as we described previously (61–63).
RACE.
The transcription start site was determined using 5′ RACE by following a previously reported method (64). Briefly, 5 μg total RNA of WT BCG was reverse transcribed using a cas6-specific primer, JY534 (see Table S1 in the supplemental material). cDNA was treated with an RNase H and RNase T1 mixture at 37°C for 30 min and was then purified with a S.N.A.P column (Invitrogen). A poly(C) tail was added to the cDNA at the 5′ end using terminal deoxynucleotidyl transferase (TdT). Subsequently, cDNA was amplified by PCR using primers JY532 and JY535 (see Table S1 in the supplemental material). The PCR product was cloned using a TA cloning kit (Invitrogen). The inserted DNA was sequenced with primer JY535 to determine the 5′ end of the transcript.
Northern blot analysis.
Northern blots were used to compare levels of both pre-crRNA and crRNA among different bacterial strains. DNA oligonucleotide probes specific for each RNA (see Table S1 in the supplemental material) were end labeled using 20 pmol of the oligonucleotide in a 10-μl reaction mixture containing 25 μM [γ-32P]ATP (MP Biomedicals) and 20 units T4 polynucleotide kinase (NEB) at 37°C for 1 h.
For Northern blotting of pre-crRNA, 5 μg total RNA was separated on a 1% denaturing formaldehyde agarose gel, which was capillary transferred onto a positively charged membrane (Hybond N+; GE Life Sciences) for blotting. Northern blotting of crRNA was performed by a method similar to that in a previous report (65). Briefly, 5 μg total RNA was separated on a 10% denaturing polyacrylamide gel, which was electronically transferred to a Hybond membrane for blotting. Hybridization was performed using Amersham Rapid-hyb buffer (GE Healthcare), following the recommended protocol for oligonucleotide probes, with a 6-h incubation at 42°C. The blotted membranes were washed once with 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) containing 0.1% SDS and twice with 0.2× SSC containing 0.1% SDS. The membranes were exposed to a PhosphorImager screen (Molecular Dynamics), scanned with a Storm 860 scanner (Molecular Dynamics), and analyzed with ImageQuant software (Molecular Dynamics). Subsequently, the hybridized membranes were stripped with a stripping solution (5 mM Tris-HCl, pH 8.0, 0.2 mM EDTA, 0.05% Na-pyrophosphate, 0.1× Denhardt's solution) for 30 min to 3 h at 80°C, followed by hybridization with different probes.
Statistical analysis.
All the data were analyzed by a two-tailed t test using Prism 5 (GraphPad Software), except that RNA-Seq data were exported from RNA-Rocket analysis. P values of <0.05 were considered to be statistically significant.
Accession number(s).
The RNA-Seq data have been deposited at NCBI Gene Expression Ominibus (GEO) under accession number GSE102816.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jeffery Cox for providing pJSC407 and pYO11; William Jacobs, Jr., for providing phAE159; Joseph Wade and Jing Wang for assistance with RNA-Seq analysis; and Kathleen McDonough for β-galactosidase activity assay. We are grateful to the Biochemistry Core of the Wadsworth Center for assistance with HPLC analysis and the Next Generation Sequencing Core Facility of the University at Buffalo for RNA-Seq.
This project is partly supported by a Scientist Development Grant from the American Heart Association 12SDG12080067 to G.B.
We have no conflict of interest to declare.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00743-17.
REFERENCES
- 1.Bhaya D, Davison M, Barrangou R. 2011. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet 45:273–297. doi: 10.1146/annurev-genet-110410-132430. [DOI] [PubMed] [Google Scholar]
- 2.Sorek R, Lawrence CM, Wiedenheft B. 2013. CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu Rev Biochem 82:237–266. doi: 10.1146/annurev-biochem-072911-172315. [DOI] [PubMed] [Google Scholar]
- 3.Westra ER, Buckling A, Fineran PC. 2014. CRISPR-Cas systems: beyond adaptive immunity. Nat Rev Microbiol 12:317–326. doi: 10.1038/nrmicro3241. [DOI] [PubMed] [Google Scholar]
- 4.Haft DH, Selengut J, Mongodin EF, Nelson KE. 2005. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput Biol 1:e60. doi: 10.1371/journal.pcbi.0010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, Horvath P, Moineau S, Mojica FJ, Wolf YI, Yakunin AF, van der Oost J, Koonin EV. 2011. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol 9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E, Haft DH, Horvath P, Moineau S, Mojica FJ, Terns RM, Terns MP, White MF, Yakunin AF, Garrett RA, van der Oost J, Backofen R, Koonin EV. 2015. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol 13:722–736. doi: 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mohanraju P, Makarova KS, Zetsche B, Zhang F, Koonin EV, van der Oost J. 2016. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science 353:aad5147. doi: 10.1126/science.aad5147. [DOI] [PubMed] [Google Scholar]
- 8.Tamulaitis G, Venclovas C, Siksnys V. 2017. Type III CRISPR-Cas immunity: major differences brushed aside. Trends Microbiol 25:49–61. doi: 10.1016/j.tim.2016.09.012. [DOI] [PubMed] [Google Scholar]
- 9.Agari Y, Sakamoto K, Tamakoshi M, Oshima T, Kuramitsu S, Shinkai A. 2010. Transcription profile of Thermus thermophilus CRISPR systems after phage infection. J Mol Biol 395:270–281. doi: 10.1016/j.jmb.2009.10.057. [DOI] [PubMed] [Google Scholar]
- 10.Medina-Aparicio L, Rebollar-Flores JE, Gallego-Hernandez AL, Vazquez A, Olvera L, Gutierrez-Rios RM, Calva E, Hernandez-Lucas I. 2011. The CRISPR/Cas immune system is an operon regulated by LeuO, H-NS, and leucine-responsive regulatory protein in Salmonella enterica serovar Typhi. J Bacteriol 193:2396–2407. doi: 10.1128/JB.01480-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patterson AG, Chang JT, Taylor C, Fineran PC. 2015. Regulation of the type I-F CRISPR-Cas system by CRP-cAMP and GalM controls spacer acquisition and interference. Nucleic Acids Res 43:6038–6048. doi: 10.1093/nar/gkv517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pul U, Wurm R, Arslan Z, Geissen R, Hofmann N, Wagner R. 2010. Identification and characterization of E. coli CRISPR-Cas promoters and their silencing by H-NS. Mol Microbiol 75:1495–1512. doi: 10.1111/j.1365-2958.2010.07073.x. [DOI] [PubMed] [Google Scholar]
- 13.Shinkai A, Kira S, Nakagawa N, Kashihara A, Kuramitsu S, Yokoyama S. 2007. Transcription activation mediated by a cyclic AMP receptor protein from Thermus thermophilus HB8. J Bacteriol 189:3891–3901. doi: 10.1128/JB.01739-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Westra ER, Pul U, Heidrich N, Jore MM, Lundgren M, Stratmann T, Wurm R, Raine A, Mescher M, Van Heereveld L, Mastop M, Wagner EG, Schnetz K, Van Der Oost J, Wagner R, Brouns SJ. 2010. H-NS-mediated repression of CRISPR-based immunity in Escherichia coli K12 can be relieved by the transcription activator LeuO. Mol Microbiol 77:1380–1393. doi: 10.1111/j.1365-2958.2010.07315.x. [DOI] [PubMed] [Google Scholar]
- 15.Yang CD, Chen YH, Huang HY, Huang HD, Tseng CP. 2014. CRP represses the CRISPR/Cas system in Escherichia coli: evidence that endogenous CRISPR spacers impede phage P1 replication. Mol Microbiol 92:1072–1091. doi: 10.1111/mmi.12614. [DOI] [PubMed] [Google Scholar]
- 16.He L, Fan X, Xie J. 2012. Comparative genomic structures of Mycobacterium CRISPR-Cas. J Cell Biochem 113:2464–2473. doi: 10.1002/jcb.24121. [DOI] [PubMed] [Google Scholar]
- 17.Supply P, Marceau M, Mangenot S, Roche D, Rouanet C, Khanna V, Majlessi L, Criscuolo A, Tap J, Pawlik A, Fiette L, Orgeur M, Fabre M, Parmentier C, Frigui W, Simeone R, Boritsch EC, Debrie AS, Willery E, Walker D, Quail MA, Ma L, Bouchier C, Salvignol G, Sayes F, Cascioferro A, Seemann T, Barbe V, Locht C, Gutierrez MC, Leclerc C, Bentley SD, Stinear TP, Brisse S, Medigue C, Parkhill J, Cruveiller S, Brosch R. 2013. Genomic analysis of smooth tubercle bacilli provides insights into ancestry and pathoadaptation of Mycobacterium tuberculosis. Nat Genet 45:172–179. doi: 10.1038/ng.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bai Y, Yang J, Zhou X, Ding X, Eisele LE, Bai G. 2012. Mycobacterium tuberculosis Rv3586 (DacA) is a diadenylate cyclase that converts ATP or ADP into c-di-AMP. PLoS One 7:e35206. doi: 10.1371/journal.pone.0035206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang J, Bai Y, Zhang Y, Gabrielle VD, Jin L, Bai G. 2014. Deletion of the cyclic di-AMP phosphodiesterase gene (cnpB) in Mycobacterium tuberculosis leads to reduced virulence in a mouse model of infection. Mol Microbiol 93:65–79. doi: 10.1111/mmi.12641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Yang J, Bai G. 31 January 2018. Cyclic di-AMP-mediated interaction between Mycobacterium tuberculosis ΔcnpB and macrophages implicates a novel strategy for improving BCG vaccination. Pathog Dis doi: 10.1093/femspd/fty008. [DOI] [PubMed] [Google Scholar]
- 21.Dey RJ, Dey B, Zheng Y, Cheung LS, Zhou J, Sayre D, Kumar P, Guo H, Lamichhane G, Sintim HO, Bishai WR. 2017. Inhibition of innate immune cytosolic surveillance by an M. tuberculosis phosphodiesterase. Nat Chem Biol 13:210–217. doi: 10.1038/nchembio.2254. [DOI] [PubMed] [Google Scholar]
- 22.Dey B, Dey RJ, Cheung LS, Pokkali S, Guo H, Lee JH, Bishai WR. 2015. A bacterial cyclic dinucleotide activates the cytosolic surveillance pathway and mediates innate resistance to tuberculosis. Nat Med 21:401–406. doi: 10.1038/nm.3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Postic G, Danchin A, Mechold U. 2012. Characterization of NrnA homologs from Mycobacterium tuberculosis and Mycoplasma pneumoniae. RNA 18:155–165. doi: 10.1261/rna.029132.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He Q, Wang F, Liu S, Zhu D, Cong H, Gao F, Li B, Wang H, Lin Z, Liao J, Gu L. 2016. Structural and biochemical insight into the mechanism of Rv2837c from Mycobacterium tuberculosis as a c-di-NMP phosphodiesterase. J Biol Chem 291:3668–3681. doi: 10.1074/jbc.M115.699801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE III, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 26.Gupta K, Kumar P, Chatterji D. 2010. Identification, activity and disulfide connectivity of C-di-GMP regulating proteins in Mycobacterium tuberculosis. PLoS One 5:e15072. doi: 10.1371/journal.pone.0015072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Botelho A, Canto A, Leao C, Cunha MV. 2015. Clustered regularly interspaced short palindromic repeats (CRISPRs) analysis of members of the Mycobacterium tuberculosis complex. Methods Mol Biol 1247:373–389. doi: 10.1007/978-1-4939-2004-4_27. [DOI] [PubMed] [Google Scholar]
- 28.Srivastav R, Kumar D, Grover A, Singh A, Manjasetty BA, Sharma R, Taneja B. 2014. Unique subunit packing in mycobacterial nanoRNase leads to alternate substrate recognitions in DHH phosphodiesterases. Nucleic Acids Res 42:7894–7910. doi: 10.1093/nar/gku425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patterson AG, Yevstigneyeva MS, Fineran PC. 2017. Regulation of CRISPR-Cas adaptive immune systems. Curr Opin Microbiol 37:1–7. doi: 10.1016/j.mib.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 30.Kahramanoglou C, Cortes T, Matange N, Hunt DM, Visweswariah SS, Young DB, Buxton RS. 2014. Genomic mapping of cAMP receptor protein (CRPMt) in Mycobacterium tuberculosis: relation to transcriptional start sites and the role of CRPMt as a transcription factor. Nucleic Acids Res 42:8320–8329. doi: 10.1093/nar/gku548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Knapp GS, Lyubetskaya A, Peterson MW, Gomes AL, Ma Z, Galagan JE, McDonough KA. 2015. Role of intragenic binding of cAMP responsive protein (CRP) in regulation of the succinate dehydrogenase genes Rv0249c-Rv0247c in TB complex mycobacteria. Nucleic Acids Res 43:5377–5393. doi: 10.1093/nar/gkv420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rickman L, Saldanha JW, Hunt DM, Hoar DN, Colston MJ, Millar JB, Buxton RS. 2004. A two-component signal transduction system with a PAS domain-containing sensor is required for virulence of Mycobacterium tuberculosis in mice. Biochem Biophys Res Commun 314:259–267. doi: 10.1016/j.bbrc.2003.12.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dey A, Shree S, Pandey SK, Tripathi RP, Ramachandran R. 2016. Crystal structure of Mycobacterium tuberculosis H37Rv AldR (Rv2779c), a regulator of the ald gene: DNA binding and identification of small molecule inhibitors. J Biol Chem 291:11967–11980. doi: 10.1074/jbc.M115.700484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parti RP, Shrivastava R, Srivastava S, Subramanian AR, Roy R, Srivastava BS, Srivastava R. 2008. A transposon insertion mutant of Mycobacterium fortuitum attenuated in virulence and persistence in a murine infection model that is complemented by Rv3291c of Mycobacterium tuberculosis. Microb Pathog 45:370–376. doi: 10.1016/j.micpath.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 35.Shrivastava T, Dey A, Ramachandran R. 2009. Ligand-induced structural transitions, mutational analysis, and ‘open’ quaternary structure of the M. tuberculosis feast/famine regulatory protein (Rv3291c). J Mol Biol 392:1007–1019. doi: 10.1016/j.jmb.2009.07.084. [DOI] [PubMed] [Google Scholar]
- 36.Song N, Cui Y, Li Z, Chen L, Liu S. 2016. New targets and cofactors for the transcription factor LrpA from Mycobacterium tuberculosis. DNA Cell Biol 35:167–176. doi: 10.1089/dna.2015.3040. [DOI] [PubMed] [Google Scholar]
- 37.Gordon BR, Imperial R, Wang L, Navarre WW, Liu J. 2008. Lsr2 of Mycobacterium represents a novel class of H-NS-like proteins. J Bacteriol 190:7052–7059. doi: 10.1128/JB.00733-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gordon BR, Li Y, Wang L, Sintsova A, van Bakel H, Tian S, Navarre WW, Xia B, Liu J. 2010. Lsr2 is a nucleoid-associated protein that targets AT-rich sequences and virulence genes in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 107:5154–5159. doi: 10.1073/pnas.0913551107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qu Y, Lim CJ, Whang YR, Liu J, Yan J. 2013. Mechanism of DNA organization by Mycobacterium tuberculosis protein Lsr2. Nucleic Acids Res 41:5263–5272. doi: 10.1093/nar/gkt249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sharadamma N, Harshavardhana Y, Singh P, Muniyappa K. 2010. Mycobacterium tuberculosis nucleoid-associated DNA-binding protein H-NS binds with high-affinity to the Holliday junction and inhibits strand exchange promoted by RecA protein. Nucleic Acids Res 38:3555–3569. doi: 10.1093/nar/gkq064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fang M, Zeisberg WM, Condon C, Ogryzko V, Danchin A, Mechold U. 2009. Degradation of nanoRNA is performed by multiple redundant RNases in Bacillus subtilis. Nucleic Acids Res 37:5114–5125. doi: 10.1093/nar/gkp527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goldman SR, Sharp JS, Vvedenskaya IO, Livny J, Dove SL, Nickels BE. 2011. NanoRNAs prime transcription initiation in vivo. Mol Cell 42:817–825. doi: 10.1016/j.molcel.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Elmore JR, Sheppard NF, Ramia N, Deighan T, Li H, Terns RM, Terns MP. 2016. Bipartite recognition of target RNAs activates DNA cleavage by the type III-B CRISPR-Cas system. Genes Dev 30:447–459. doi: 10.1101/gad.272153.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Estrella MA, Kuo FT, Bailey S. 2016. RNA-activated DNA cleavage by the type III-B CRISPR-Cas effector complex. Genes Dev 30:460–470. doi: 10.1101/gad.273722.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kazlauskiene M, Tamulaitis G, Kostiuk G, Venclovas C, Siksnys V. 2016. Spatiotemporal control of type III-A CRISPR-Cas immunity: coupling DNA degradation with the target RNA recognition. Mol Cell 62:295–306. doi: 10.1016/j.molcel.2016.03.024. [DOI] [PubMed] [Google Scholar]
- 46.Bai G, Schaak DD, McDonough KA. 2009. cAMP levels within Mycobacterium tuberculosis and Mycobacterium bovis BCG increase upon infection of macrophages. FEMS Immunol Med Microbiol 55:68–73. doi: 10.1111/j.1574-695X.2008.00500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grundner C, Cox JS, Alber T. 2008. Protein tyrosine phosphatase PtpA is not required for Mycobacterium tuberculosis growth in mice. FEMS Microbiol Lett 287:181–184. doi: 10.1111/j.1574-6968.2008.01309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bardarov S, Bardarov S Jr, Pavelka MS Jr, Sambandamurthy V, Larsen M, Tufariello J, Chan J, Hatfull G, Jacobs WR Jr. 2002. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148:3007–3017. doi: 10.1099/00221287-148-10-3007. [DOI] [PubMed] [Google Scholar]
- 49.Tufariello JM, Jacobs WR Jr, Chan J. 2004. Individual Mycobacterium tuberculosis resuscitation-promoting factor homologues are dispensable for growth in vitro and in vivo. Infect Immun 72:515–526. doi: 10.1128/IAI.72.1.515-526.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mangan JA, Sole KM, Mitchison DA, Butcher PD. 1997. An effective method of RNA extraction from bacteria refractory to disruption, including mycobacteria. Nucleic Acids Res 25:675–676. doi: 10.1093/nar/25.3.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McClure R, Balasubramanian D, Sun Y, Bobrovskyy M, Sumby P, Genco CA, Vanderpool CK, Tjaden B. 2013. Computational analysis of bacterial RNA-Seq data. Nucleic Acids Res 41:e140. doi: 10.1093/nar/gkt444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tjaden B. 2015. De novo assembly of bacterial transcriptomes from RNA-seq data. Genome Biol 16:1. doi: 10.1186/s13059-014-0572-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gazdik MA, McDonough KA. 2005. Identification of cyclic AMP-regulated genes in Mycobacterium tuberculosis complex bacteria under low-oxygen conditions. J Bacteriol 187:2681–2692. doi: 10.1128/JB.187.8.2681-2692.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alland D, Kramnik I, Weisbrod TR, Otsubo L, Cerny R, Miller LP, Jacobs WR Jr, Bloom BR. 1998. Identification of differentially expressed mRNA in prokaryotic organisms by customized amplification libraries (DECAL): the effect of isoniazid on gene expression in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 95:13227–13232. doi: 10.1073/pnas.95.22.13227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Manganelli R, Dubnau E, Tyagi S, Kramer FR, Smith I. 1999. Differential expression of 10 sigma factor genes in Mycobacterium tuberculosis. Mol Microbiol 31:715–724. doi: 10.1046/j.1365-2958.1999.01212.x. [DOI] [PubMed] [Google Scholar]
- 56.Purkayastha A, McCue LA, McDonough KA. 2002. Identification of a Mycobacterium tuberculosis putative classical nitroreductase gene whose expression is coregulated with that of the acr gene within macrophages, in standing versus shaking cultures, and under low oxygen conditions. Infect Immun 70:1518–1529. doi: 10.1128/IAI.70.3.1518-1529.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McDonough KA, Florczyk MA, Kress Y. 2000. Intracellular passage within macrophages affects the trafficking of virulent tubercle bacilli upon reinfection of other macrophages in a serum-dependent manner. Tuber Lung Dis 80:259–271. doi: 10.1054/tuld.2000.0268. [DOI] [PubMed] [Google Scholar]
- 58.Vasudeva-Rao HM, McDonough KA. 2008. Expression of the Mycobacterium tuberculosis acr-coregulated genes from the DevR (DosR) regulon is controlled by multiple levels of regulation. Infect Immun 76:2478–2489. doi: 10.1128/IAI.01443-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.El Qaidi S, Yang J, Zhang JR, Metzger DW, Bai G. 2013. The vitamin B6 biosynthesis pathway in Streptococcus pneumoniae is controlled by pyridoxal 5′-phosphate and the transcription factor PdxR and has an impact on ear infection. J Bacteriol 195:2187–2196. doi: 10.1128/JB.00041-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ryjenkov DA, Tarutina M, Moskvin OV, Gomelsky M. 2005. Cyclic diguanylate is a ubiquitous signaling molecule in bacteria: insights into biochemistry of the GGDEF protein domain. J Bacteriol 187:1792–1798. doi: 10.1128/JB.187.5.1792-1798.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bai Y, Yang J, Eisele LE, Underwood AJ, Koestler BJ, Waters CM, Metzger DW, Bai G. 2013. Two DHH subfamily 1 proteins in Streptococcus pneumoniae possess cyclic di-AMP phosphodiesterase activity and affect bacterial growth and virulence. J Bacteriol 195:5123–5132. doi: 10.1128/JB.00769-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bai Y, Yang J, Zarrella TM, Zhang Y, Metzger DW, Bai G. 2014. Cyclic di-AMP impairs potassium uptake mediated by a cyclic di-AMP binding protein in Streptococcus pneumoniae. J Bacteriol 196:614–623. doi: 10.1128/JB.01041-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Underwood AJ, Zhang Y, Metzger DW, Bai G. 2014. Detection of cyclic di-AMP using a competitive ELISA with a unique pneumococcal cyclic di-AMP binding protein. J Microbiol Methods 107:58–62. doi: 10.1016/j.mimet.2014.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miller E. 2016. Rapid amplification of cDNA ends for RNA transcript sequencing in Staphylococcus. Methods Mol Biol 1373:169–183. doi: 10.1007/7651_2015_282. [DOI] [PubMed] [Google Scholar]
- 65.DiChiara JM, Contreras-Martinez LM, Livny J, Smith D, McDonough KA, Belfort M. 2010. Multiple small RNAs identified in Mycobacterium bovis BCG are also expressed in Mycobacterium tuberculosis and Mycobacterium smegmatis. Nucleic Acids Res 38:4067–4078. doi: 10.1093/nar/gkq101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.