

Abstract
Drug‐induced cardiomyopathy contributes to drug attrition. We compared two pipelines of predictive modeling: (1) applying elastic net (EN) to differentially expressed genes (DEGs) of drugs; (2) applying integer linear programming (ILP) to construct each drug's signaling pathway starting from its targets to downstream proteins, to transcription factors, and to its DEGs in human cardiomyocytes, and then subjecting the genes/proteins in the drugs' signaling networks to EN regression. We classified 31 drugs with availability of DEGs into 13 toxic and 18 nontoxic drugs based on a clinical cardiomyopathy incidence cutoff of 0.1%. The ILP‐augmented modeling increased prediction accuracy from 79% to 88% (sensitivity: 88%; specificity: 89%) under leave‐one‐out cross validation. The ILP‐constructed signaling networks of drugs were better predictors than DEGs. Per literature, the microRNAs that reportedly regulate expression of our six top predictors are of diagnostic value for natural heart failure or doxorubicin‐induced cardiomyopathy. This translational predictive modeling might uncover potential biomarkers.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THIS TOPIC?
☑ There is no translational predictive modeling that integrates a drug's mode of action with clinical observation of toxicity.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study addresses the question of how to conduct systems pharmacology predictive modeling that integrates the modes of action of drugs and their clinically observed occurrence of treatment‐related cardiomyopathy.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ This study adds to the knowledge of (1) the proteins/genes that are top predictors of drug‐induced cardiomyopathy, and (2) utility of drugs' modes of action in the form of signaling pathways for predicting drug‐induced cardiomyopathy.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ This study enables pharmaceutical scientists to further translational system's pharmacology modeling to facilitate development of therapeutics.
Serious and life‐threatening drug‐induced adverse events cause drug attrition at various stages of drug development or modification of treatment regimens. For instance, anthracyclines, although effective to treat cancers, are known to cause irreversible, dose‐dependent cardiotoxicity (contractility‐related toxicity).1 Most recently, targeted therapy with tyrosine kinase inhibitors (TKIs) also cause such toxicity.1 The ability to predict drug‐induced cardiotoxicity may reduce drug attrition and advance precision medicine.
Predictive modeling of adverse drug reactions by integrating information across databases and knowledgebase of biological activities, chemistry, and adverse drug reactions has been undertaken.2, 3, 4 However, no predictive models of drug‐induced cardiomyopathy utilizing signaling network information have been constructed. Harpaz et al.4 stressed the importance of harnessing multiple sources of knowledge, biological information, and biomedical literature for predicting drug toxicity. In line with this notion, we reported herein predictive modeling by integrating prior knowledge, drug targets, and empirical data in order to enable the model to identify key predictors from a drug's mode of action, and to have the potential to inform lead identification and development.
To fill in the gap, we compiled a list of 31 toxic and nontoxic drugs that were transcriptomically profiled in human cardiomyocytes5, 6, 7; manually curated and compiled their clinical incidence of treatment‐related cardiomyopathy; and conducted predictive modeling of drug‐induced cardiomyopathy. Two predictive models were compared: (1) applying elastic net (EN) to gene expression data; and (2) applying integer linear programming (ILP) to construct a drug's signaling network to reflect its mechanism of action,8 and then subjecting the nodes in individual drugs' signaling networks to EN regression. The ILP formulation8 navigates a prior knowledge network of protein‐protein, protein‐transcription factor (TF), and TF‐gene interactions, and identifies the pathways that connect a drug's targets to its differentially expressed genes (DEGs). The ILP not only optimizes the solution of finding a drug's signaling pathways but also enhances performance of predictive modeling by enabling identification of the subset of DEGs that are functionally relevant to a drug's mode of action. We further referenced literature for the microRNAs, which are reportedly of diagnostic value for heart failure and for drug‐induced cardiomyopathy, as well as also regulating the expression of our predictors in hopes of shedding light on potential microRNAs as in vivo drug‐induced cardiomyopathy biomarkers.
METHODS
Compilation of drugs and their clinical incidence of drug‐induced cardiomyopathy
To compile the list of approved drugs that cause treatment‐related cardiomyopathy, we referenced the National Institutes of Health Common Terminology Criteria for Adverse Events (version 4.03)9 and the Medical Dictionary for Regulatory Activities10 for cardiomyopathy‐related terms to text‐mine approved drug labels. The terms used included cardiomyopathy, heart failure, congestive heart failure, cardiac failure, left ventricular dysfunction, left ventricular failure, and reduction in left ventricular ejection fraction. The current drug label PDF files (Drugs@FDA1) were processed using a text‐mining analysis pipeline, as published previously.11 Individual rates of occurrence for cardiomyopathy were extracted by manual curation of drug labels, published redacted new drug application reviews (Drugs@FDA), as well as published clinical studies.
Predictive modeling
Workflow and highlights of EN and ILP
As shown in Figure 1, we compared two pipelines of predictive modeling. For pipeline 1, we applied EN to DEGs of a drug. For pipeline 2, we applied ILP to construct each drug's signaling pathway, and then subjected the genes/proteins in each drug's signaling network to EN regression.
Figure 1.
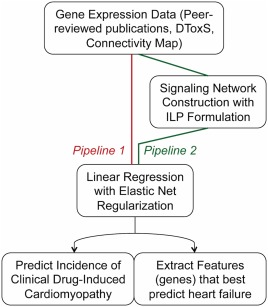
Workflow of predictive modeling. We built datasets using gene expression data and we compared two piplelines to predict clinical drug‐induced cardiomyopathy and extract features that best predict such toxicity. Running the Gene Expression Data at hand through a linear regression model with elastic net regularization or constructing signaling networks from the data before modeling using an integer linear programming (ILP) formulation. DToxS, Drug Toxicity Signature Generation Center.
The EN is useful for predictive modeling when predictors greatly outnumber observations while simultaneously being able to identify statistically significant predictors.12 The EN regularization is useful for analyzing genomics of drug sensitivity in cancer.13
We applied ILP to a drug's DEGs and protein targets to model its mode of action. These two levels of information are connected via signal transduction where the signal originates at drug targets, propagates intracellularly via a complex network of signaling cascades, passes through the layer of TFs, and finally reaches the transcriptomic level of DEGs. We modeled the interactions in the knowledge network by using the logic formalism,14 which identified the minimum subset of the network to achieve the desired connectivity. We constructed the specific signaling network for each drug using an ILP formulation, as published previously.8
The ILP will enhance predictive performance because it has the ability to capture cellular responses to a drug, to identify the subset of important functional DEGs, and to help differentiate between compounds and translate into improved performance.
Drug name normalization
Drug names were first normalized and identified by the PubChem compound identifier to ensure consistency when downloading data from Connectivity Map (CMap),15 Drug Toxicity Signature Generation Center (DToxS),5 Search Tool for Interactions of Chemicals (STITCH),16, 17 and literature.
Compilation of drug targets
We compiled the targets of individual drugs from STITCH,16, 17 and the “chemical‐protein links” database and selected only human proteins. The proteins were identified by the SwissProt/EnsEMBL‐identifier, and translated into HUGO Gene Nomenclature Committee gene symbols18 using the R biomaRt package, in order to match with the nodes in the prior‐knowledge network.19 We used STITCH's “interaction types for links” data file, from where we identified the drugs as activating or inhibiting individual target proteins. We used only those associating links between protein‐drug pairs with an evidence score of ≥ 0.7.
Gene expression data sources and handling
Wherever data were available in Affymetrix probe IDs, the probe IDs (Affymetrix GeneChip Human Genome U133A Array) were translated into HUGO Gene Nomenclature Committee gene symbols18 using the biomaRt package20 and hgu133a221 packages in R, an open source statistical computing graphics systems. Across all the gene lists, we kept only those genes with fold change > 2 and P value < 0.05 by a two‐tailed, two‐sample, unequal variance Student's t‐test, adjusted separately for the up and down gene lists with Bonferroni correction (P value adjusted for multiple comparisons).22
A list of 75 drugs with drug‐induced DEGs available from cancer cells15 in CMap were used for exploratory modeling (see Supplementary Table S1 in Supplementary Document‐CMap). To conduct robust predictive modeling, we exhausted literature and databases and found a list of 31 drugs of which drug‐induced DEGs in human cardiomyocytes5 and stem cells‐derived cardiomyocytes5, 6, 7 were available. The two data sources for drug‐induced perturbation of gene expression in cardiomyocytes were: (1) 30 drugs from DToxS5, where primary human adult cardiomyocytes were used; and (2) literature data of doxorubicin studied in human stem cell‐derived cardiomyocytes.6, 7 The size of each dataset was mainly constrained by the availability of DEGs data. For DToxS data, we downloaded the level two gene expression data, calculated the fold changes, kept only those DEGs with a P value < 0.05 and a fold change > 2, and merged them from different donors by averaging the fold changes while excluding any DEGs with opposite directions of fold change among donors.
Doxorubicin is widely studied for its dose‐dependent cardiac toxicity, and is commonly dosed at 40–60 mg/m.21 Following intravenous 60 mg/m2, its peak plasma concentration (Cmax) was 630 ng/mL (1,159 nM).23 See Supplementary Table S2 for a few studies of transcriptomic profiles of doxorubicin. For our modeling, we included the data from human‐induced pluripotent stem cells‐derived cardiomyocytes by Chaudhari et al.7 and Burridge et al.6 We included the gene expression data by Burridge et al.6 were at 100, 1,000, and 10,000 nM and those by Chaudhari et al.7 were at 156 nM (see Supplementary Table S3 for the rationale).
Identifying a drug's mode of action using ILP
We first built a prior‐knowledge network as a scaffold for constructing a drug's signaling network by downloading from Reactome19 the latest version (version 2015) of the “Functional interactions derived from Reactome.” As published previously,8 we merged those interactions with transcription factors and obtained a network across the protein, transcription factor, and gene levels, which contained 64,801 reactions, 2,585 signaling proteins, and 12,376 genes. We applied ILP to optimize a drug's signaling network by providing as the input the scaffold mentioned above and its targets.
The ILP formulation was solved using IBM ILOG CPLEX optimization studio8 for the objective of optimizing a drug's network. Based on the constraints that mimic signal transduction24 and adjustment to the specific case of very large (>10,000 nodes) networks,8 the algorithm minimized the mismatch between the data of gene expression measurements and the prior knowledge pathway topology. The output was the optimal signaling network of a drug, identifying the molecular interactions that seemed to be functional based on the input of DEGs and drug targets. We were able to select the minimum part of a prior‐knowledge network for each drug that could explain the data in hand. See Supplementary Document‐ILP for understanding the example of methotrexate signaling network captured by ILP (Supplementary Figure S1) and how the proposed ILP formulation works.
Comparing predictive modeling by applying EN to a drug's DEGs vs. to its ILP signaling network
To construct a matrix for EN regression, a drug was marked with 0 if classified as nontoxic and marked 1 if classified as toxic. We classified drugs by referencing approved labels for the criteria of “frequent adverse events being those occurring on one or more occasions in at least 1/100 patients; infrequent adverse events being those occurring in 1/100 to 1/1,000 patients; rare events being those occurring in less than 1/1,000 patients.” Referencing the definition of rare events used in drug labeling and considering the distribution of clinical incidence, the number of drugs with gene expression data available, and the heterogeneity of clinical studies, we classified drugs into two classes, toxic for those with incidence ≥ 0.1% and nontoxic for those with incidence < 0.1%.
A column of “cardiotoxicity” was created with the clinical incidence score: 1 for “toxic” and 0 for “nontoxic.” Each column corresponded to a single gene expressed in at least one of the DEGs signatures. Individual DEGs of a drug were assigned a value of 1, −1, or 0 to reflect upregulated, downregulated, or not reported, respectively (pipeline 1). The same assignments were applied to the nodes in each drug's ILP signaling network (pipeline 2).
In our modeling, we used EN regression,12 and more specifically a linear regression model with an EN penalty determined using the R package glmnet.25 The EN regularization is defined by two parameters, alpha and lambda. The EN regression is a mixing of LASSO and ridge regression and combines their two penalty terms for the alpha parameter. When alpha equals 0, EN performs as ridge regression and when alpha equals 1, EN performs as LASSO. In EN, the lambda parameter reflects shrinkage of the model's coefficients. When lambda equals 0, no shrinkage of the model's coefficients is performed but the coefficients decrease toward 0 (although not exactly equal 0) as its value increases. We tried a range of values for alpha from 0 to 1 by a 0.01 step and selected the one that minimized the mean squared error. For that alpha value, we selected the value of lambda that gave the minimum mean cross‐validated error.
To validate each model, we used leave‐one‐out cross validation (LOOCV) by leaving a drug's signature out one at a time (either DEGs or signaling network constructed from ILP) and did so across the whole list of drugs. Each time we calculated the accuracy, sensitivity, and specificity for a predictive model, and selected and reported the model with the highest accuracy along with its precision, sensitivity, and specificity. From the chosen predictive model, we extracted the predictors (genes/proteins) that best predicted drug‐induced cardiotoxicity. The receiver operating characteristic (ROC) and precision‐recall curves using the R package with the former plotted in smooth curve.
Pipeline 1 – applying EN to DEGs
The results of 75 drugs with DEGs from CMap are summarized in Supplementary Document‐CMap. Among these 75 drugs with their DEGs from CMap, 24 drugs were toxic and the remaining 51 drugs were nontoxic.
A model matrix was constructed using cardiomyocyte data, with the 34 observations (toxicity classification) as rows and 15,016 variables (gene expression) as columns. The predictive linear model was constructed by having as input all these variables for EN regularization. We tried all possible different cutoff scenarios (see the spreadsheet “summary” of Supplementary Table S4 for the results of the psccm_34_gen_heart trials and the detailed results of 18 models with different cutoffs in the spreadsheet “9”). For example, a cutoff of 10 meant that we ran the model by using only those genes that were expressed in at least 10 of the 34 signatures, meaning that the analysis started with 3,508 genes, whereas a cutoff of 15 started the analysis with the genes that appeared in at least 15 of the 34 signatures, meaning 464 genes were used as the cutoff.
Pipeline 2 – applying EN to gene/protein nodes in ILP‐constructed signaling networks
We first performed exploratory modeling using a list of 75 drugs with gene expression data available in CMap and concluded that signaling networks of drugs derived from ILP outperformed their DEGs when applying EN regularization (see Supplementary Figure S2 for ROC and precision‐recall curves in Supplementary Document‐CMap).
We were able to find the ILP solutions for drugs with gene expression data in cardiomyocytes (Supplementary Table S5) except cefuroxime, domperidone, and olmesartan. These three drugs were removed from this modeling exercise. At the end, we had 31 signaling networks from 28 drugs (15 nontoxic drugs and 13 toxic drugs). See Supplementary Table S5 for the gene/protein nodes in the signaling network of each individual drugs. We built a model matrix for the 31 signaling pathways/networks by using gene expression profiles from cardiomyocytes and by assigning 1 if a pathway node was upregulated, −1 if it was downregulated, and 0 if it was not present in a drug's optimized signaling network. See the spreadsheet “summary” of Supplementary Table S4 for the results of the psccm_34_ILP_heart trial and the detailed results of 31 models with different cutoffs in the spreadsheet “10.”
Biological context of predictors
To gain translational insight, we searched literature for microRNAs that have been shown to be diagnostic markers of heart failure and also involved in regulation of gene expression. We mined literature and MiRTarBase, a database of experimentally validated microRNA‐target interactions,26 for a list of microRNAs, which have been individually reported to regulate expression of our top gene/protein predictors, and also been reportedly detected in the circulation of patients with heart failure with a varying degree of severity27, 28 or of patients with doxorubicin‐induced cardiomyopathy.29
RESULTS
The list of drugs and toxicity profile
The list of 31 drugs with their clinical profiles of treatment‐related cardiomyopathy is summarized in Table 1. Literature search was also conducted to supplement clinical incidence of cardiomyopathy, if approved drug labels and published application reviews1 did not have such information. Among the 31 drugs, there were 13 toxic drugs (41.9%) and there were 18 nontoxic drugs (59.1%). For those drugs without mention of cardiomyopathy‐related toxicity described in their labels throughout the sections of clinical studies, postmarketing experiences, and warnings and precautions, we also searched literature and published reviews1 to reach the conclusion that they are nontoxic drugs.
Table 1.
The list of drugs with gene expression in cardiomyocytes and their cardiotoxicity classification
| Drug name | Classification | Referencea |
|---|---|---|
| Afatinib | 0 | Drugs@FDA and literature search |
| Alendronate | 0 | Drugs@FDA and literature search |
| Amiodarone | 1 | Drugs@FDA |
| Axitinib | 1 | Drugs@FDA |
| Bosutinib | 0 | Drugs@FDA and literature search |
| Cefuroxime | 0 | Drugs@FDA and literature search |
| Crizotinib | 0 | Drugs@FDA and literature search |
| Cyclosporine | 0 | Drugs@FDA and literature search |
| Cytarabine | 1 | NIH DailyMed |
| Dasatinib | 1 | Drugs@FDA |
| Diclofenac | 1 | Drugs@FDA |
| Domperidone | 0a | Not approved by FDA |
| Doxorubicin | 1 | Drugs@FDA |
| Diethylpropion | 0 | Drugs@FDA and literature search |
| Erlotinib | 0 | Drugs@FDA and literature search |
| Gefitinib | 0 | Drugs@FDA and literature search |
| Imatinib | 1 | Drugs@FDA |
| Lapatinib | 0 | Drugs@FDA |
| Methotrexate | 0 | Drugs@FDA and literature search |
| Olmesartan | 0 | Drugs@FDA and literature search |
| Paroxetine | 1 | Drugs@FDA |
| Ponatinib | 1 | Drugs@FDA |
| Regorafenib | 0 | Drugs@FDA and literature search |
| Ruxolitinib | 0 | Drugs@FDA and literature search |
| Sorafenib | 1 | Drugs@FDA |
| Sunitinib | 1 | Drugs@FDA |
| Tofacitinib | 0 | Drugs@FDA and literature search |
| Trametinib | 1 | Drugs@FDA and literature search |
| Ursodeoxycholic acid | 0 | Drugs@FDA and literature search |
| Vandetanib | 1 | Drugs@FDA |
| Vemurafenib | 0 | Drugs@FDA and literature search |
FDA, US Food and Drug Administration; NIH, National Institutes of Health.
Note: Toxic: 1 (clinical incidence ≥ 0.1%), and nontoxic: 0 (clinical incidence <0.1%). https://dailymed.nlm.nih.gov/dailymed/
Domperidone was profiled by Drug Toxicity Signature Generation Center (DtoxS) and toxicity information was from http://www.hc-sc.gc.ca/dhp-mps/medeff/reviews-examens/domperidone-eng.php.
Predictive modeling
Applying EN to DEGs (pipeline 1)
Using LOOCV across the whole list of 30 drugs and their gene expression signatures, we achieved 79% accuracy and 75% precision, with 80% sensitivity and 79% specificity when using those genes that were expressed in at least 11 of the 34 signatures (a cutoff of 11 in spreadsheet “9” of Supplementary Table S4). The results of EN regularization are shown in Figure 2 a,c, and the genes/proteins with non‐zero coefficients are PHF19, HSPA8, RIF1, CD46, MXRA7, RAB27A, TOMM20, MYO6, and CCNA2. The ROC curves and precision‐recall curves are shown in Figure 3 and Supplementary Figure S2 of Supplementary Document‐CMap), respectively.
Figure 2.
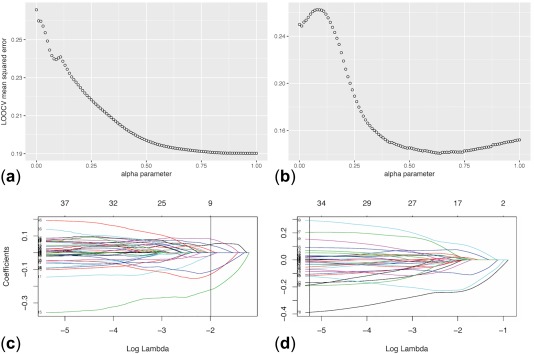
Plots of elastic net regularization results. (a and b) Show selection of the alpha parameter in the elastic net regularization by minimizing the leave‐one‐out cross validation (LOOCV) mean squared error to extract the features (genes) that best predict clinical incidence of cardiomyopathy. (c and d) Show the number of variables kept in the model, with a vertical line showing the optimal number for maximization of accuracy. a and c refer to the results of analyzing gene expression data only, whereas b and d correspond to the results of analyzing drugs' signaling networks obtained from integer linear programming formulation analysis. Each of the plotted lines in c and d corresponds to a variable (for example, a specific gene's expression) and shows how its coefficient changes with the log lambda parameter of elastic net. The vertical line shows the optimal number of parameters kept and their coefficients for maximization of accuracy.
Figure 3.
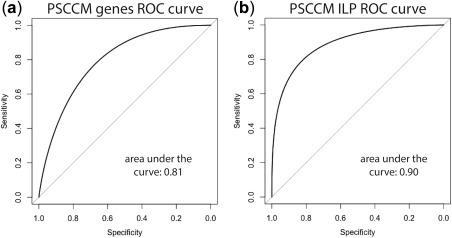
Receiver operating characteristic (ROC) curves. (a) ROC curve from modeling differentially expressed genes (DEGs) using elastic net (EN) and (b) ROC curve from modeling by subjecting these DEGs to integer linear programming (ILP) to construct their individual drugs' signaling networks and then subject these networks to EN. PSCCM, human cardiomyocytes.
Applying EN to the gene/protein nodes in ILP‐constructed signaling networks (pipeline 2)
By applying EN regression and LOOCV, we were able to increase both prediction accuracy and precision to 88%, with 88% sensitivity, and 89% specificity, compared with the results from EN regression of DEGs (Supplementary Table S4). The EN regularization is shown in Figure 2 b,d. The result for the psccm_34_ILP_heart trial is in the spreadsheet “summary” and the detailed results of 31 models with a cutoff ranging from 1 (5,012 genes/proteins in at least 1 drug) to 31 (5 genes/proteins in at least 31 network signatures) are in spreadsheet “10” of Supplementary Table S4. The highest accuracy, sensitivity, and specificity were achieved at cutoff of 10 with 189 genes/proteins from at least 10 drugs' signaling networks. The ROC and precision‐recall curves are shown in Figure 3 and Supplementary Figure S3, respectively.
We concluded that EN‐ILP (pipeline 2) outperformed EN alone (pipeline 1) when applied to the same set of DEGs.
Cardiac context of top predictors
Using EN regularization, we were able to extract the protein/gene predictors that best predict the toxicity classification of drug‐induced cardiotoxicity (either toxic for ≥ 0.1% clinical incidence or nontoxic for < 0.1%). The 33 protein/gene predictors along with their individual coefficients are summarized in Table 2. The network of the top 15 genes/proteins selected by the model is presented in Figure 4. Cardiac relevance of these predictors was reviewed and summarized in Supplementary Table S6. The protein and gene predictors identified by EN‐ILP reflected the key cellular biological factors for drug‐induced cardiotoxicity. The EN regularization in our predictive modeling selected the protein/gene predictors that best predicted drug‐induced cardiotoxicity.
Table 2.
Predictors with non‐zero coefficients from modeling/analysis of cardiomyocyte data
| Gene/protein | Coefficient | Gene/protein | Coefficient | Gene/protein | Coefficient | Gene/protein | Coefficient |
|---|---|---|---|---|---|---|---|
| CYP3A4 | −0.39 | TOP2A | −0.11 | FLI1 | −0.03 | H2AFX | −0.01 |
| ZNF823 | 0.29 | MAX | 0.09 | TCF12 | −0.03 | IRF1 | −0.011 |
| CASP3 | 0.20 | JUND | −0.08 | AHR | 0.03 | MAP3K5 | 0.01 |
| HJURP | −0.19 | MAPK12 | −0.07 | BCR | 0.03 | E2F1 | 0.01 |
| EPHA2 | −0.19 | RXRA | 0.07 | GATA3 | 0.03 | SMOC2 | 0.01 |
| STAT1 | −0.17 | HOXA5 | −0.07 | SMC3 | 0.02 | CYP2D6 | −0.01 |
| SP2 | 0.15 | STAT5A | −0.05 | EDN1 | 0.02 | ||
| PDGFR‐A | −0.12 | TCF7L2 | 0.05 | FOXF2 | −0.02 | ||
| TRIM28 | −0.12 | NR4A2 | −0.03 | CTCFL | −0.02 |
Nodes from drugs' signaling networks constructed using integer linear programming (ILP) included proteins (targets and protein‐protein interactions) and genes (differentially expressed). The gene/protein nodes from ILP were then subjected to elastic net regularization.
Figure 4.
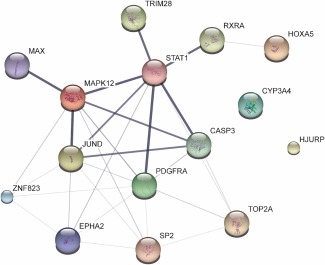
Interactions among the top 15 gene/protein predictors. Interactions among the top 15 genes/proteins selected by our model to best predict cardiomyopathy using cardiomyocytes data are depicted as a network using the STITCH website for visualization. Small nodes correspond to protein of unknown 3D structure and large nodes to known or predicted. Edges represent protein‐protein associations and the intensity of the line is proportional to the confidence score of each association. The confidence score is calculated by combining the probabilities from all evidence channels and is corrected for random observation probability.
We mined an evidence‐based database of microRNAs26 for those that reportedly regulate our top predictors, and also referenced literature to narrow the list to those that are reportedly of diagnostic value for heart failure. Summarized in Table 3 are our top 10 predictors and their individual regulating microRNAs that have reportedly been of diagnostic value for natural heart failure27, 28 or for doxorubicin‐induced cardiomyopathy.29
Table 3.
Top 10 predictors and their corresponding regulating microRNAs that are reportedly of diagnostic value for heart failure
| Predictors | Regulating microRNAsa that are of diagnostic value | References |
|---|---|---|
| CYP3A4 | No information | |
| ZNF823 | miR193‐3p (↓) | Schulte et al.27 |
| CASP3 | miR‐375b, miR‐26b‐5p (↓); miR‐30e‐5p (↓), let‐7a‐5p (↑) | Schulte et al.27; Marques et al.28 |
| HJURP | miR‐671‐5p (↑) | Schulte et al.27 |
| EPHA2 | miR‐26b‐5p (↓), miR‐193b‐3p (↓); miR‐16‐5p (↓) | Schulte et al.27; Marques et al.28 |
| STAT1 | miR 145‐5p (↓) | Schulte et al.27 |
| SP2 | miR‐29a‐3p (↓), miR‐638b | Schulte et al.27 |
| PDGFR‐A | miR‐140‐5p (↓); miR‐26b‐5p (↓); miR‐29b‐3p (↓); 181a‐5p (↑); miR‐1233 (↑) | Schulte et al.27; Marques et al.28 |
| TRIM28 | miR‐423‐5p (inconsistent reports), miR‐193b‐3p (↓), miR‐183‐3p (↓), miR‐92a‐3p (↓) | Schulte et al.27 |
| TOP2A | miR‐193b‐3p (↓), miR‐21‐5p (↑) | Schulte et al.27, Marques et al.28 |
Regulating microRNAs are from Chou et al.26 (http://mirtarbase.mbc.nctu.edu.tw).
Differentiating heart failure with reduced ejection fraction from heart failure with preserved ejection fraction. ↑and ↓ represent elevation and decrease, respectively, compared to healthy controls.
DISCUSSION
With the clinical incidence of drug‐induced cardiomyopathy as a dependent variable, ILP‐enhanced predictive modeling increased prediction accuracy from 79% to 88%, compared to modeling with EN and DEGs alone. This improved prediction signified the ability of ILP to computationally capture a drug's mode of action through constructing its signaling pathways for the purpose of predictive modeling. ILP offers the advantage of integrating our prior knowledge of biological protein interactions and drug targets (Reactome and STITCH), transcription factors, and DEGs into predictive modeling. ILP also optimizes the size of a drug's network signaure in addition to capturing the signaling pathways of a drug. Take lapatinib as an example, it had 2,265 DEGs from cardiomyocytes, whereas from this set of DEGs, its ILP network consisted of 1,923 nodes, including its targets, proteins involved in its signaling transduction, transcription factors, and functional DEGs.
The 33 gene/protein predictors along with their individual positive or negative coefficients could be used to predict “toxic” or “nontoxic” for a drug by linear summation using their individual levels of expression (either upregulation (+) or downregulation (‐)) from its ILP‐constructed signaling network. The predictive power of this system's pharmacology predictive model will increase with the amount of data in the training set.
Among the 31 drugs used to conduct predictive modeling, the distribution of toxic (n = 13) vs. nontoxic (n = 18) classification was acceptable, although not ideal. Among them, there were 18 kinase inhibitors (17 TKIs and 1 serine/threonine kinase inhibitor), which might seemingly be off‐balance from the perspective of the diversity of drug class. Vemurafenib is a serine/threonine kinase inhibitor and not toxic. The distribution of toxic (n = 8) and nontoxic (n = 9) drugs among the 17 TKIs was acceptable. TKIs, in general, lack target specificity, have multiple targets, and were designed to disrupt the signaling pathways that are vital to cancer cell survival.30 Unfortunately, several of these signaling pathways also play a critical role in cardiomyocyte biology31; consequently, several TKIs impair cardiac function. Within this context, our predictive modeling could be useful for predicting cardiac toxicity for future new chemical entities.
All top 15 gene/protein predictors have relevant cardiac functions except ZNF 823 (Supplementary Table S5). Interestingly, CYP3A4 was an important predictor. Although CYP3A4 does not have biological interactions with other predictors, as shown in Figure 4, it is a major drug metabolizing enzyme.1 Among the 31 drugs, 10 of 13 (85%) toxic drugs and 11 of 18 (61%) nontoxic drugs were metabolized by CYP3A4. The toxic drugs that are primarily or extensively metabolized by CYP3A4, included amiodarone, axitinib, cytrabine, dasatinib, doxorubicin, imatinib, ponatinib, sorafenib, sunitinib, and vandetanib.1, 32, 33, 34 For nontoxic drugs, they are bosutinib, crizotinib, cyclosporine, domperidone, erlotinib, gefitinib, lapatinib, regorafenib, ruxolitinib, tofacitinib, and ursodeoxycholic acid.1, 35
Some top predictors are biologically associated with focal adhesion kinase (FAK), a nonreceptor protein‐tyrosine kinase, which is involved in the cytoskeleton‐associated network of signaling proteins.36 Focal adhesion complexes play a critical role in how cultured cardiomyocytes respond to mechanical and neurohormonal stimuli, and in the development of heart failure.37 FAK activation plays a role in the adaptive response to cardiac afterload and in myocyte growth via the protein kinase B/mammalian target of rapamycin pathway.38 the FAK cleavage is mediated by CASP3 family during apoptosis of human normal cells,39 and occurs with activation of EPHA2 and p38 mitogen‐activated protein kinase during doxazosin‐induced apoptosis of a cardiac cell line.40 FAK activates STAT1 during cell attachment,41 and plays a role in cell migration with one of its actions being associated with platelet‐derived growth factor receptor (PDGFR) signaling complex.42 In short, the top predictors are important to maintain normal cardiac function.
Per literature, some microRNAs that reportedly regulated expression of our predictors have also been shown to be of diagnostic value for heart failure with a varying degree of severity (Table 3).27, 28 Among them, miR193‐3p and miR26b‐5p reportedly regulated more predictors than other microRNAs, and regulated four and three of our top predictors, respectively. It might be worthy of clinical studies to determine whether miR193‐3p and miR26b‐5p are useful in vivo biomarkers for drug‐induced cardiomyopathy. Literature search uncovered a recent study that investigated circulating microRNAs in children with anthracycline‐induced acute heart injury.29 Elevated miR‐29b and miR‐499 in the circulation seemed to correlate with troponin elevation in these children, and were identified as potential cardiomyopathy biomarkers.29 This observation of miR‐29b elevation in doxorubicin‐induced cardiomyopathy differed from an observation of decreased expression of miR‐29b‐3p in the coronary sinus blood of patients with heart failure.28 The MiR‐29b‐3p regulates expression of one of our top 10 predictors, PDGFR‐A. Further studies are needed to investigate the role of miR‐29b in drug‐induced cardiomyopathy or in natural heart failure. Even though miR‐27b reportedly regulated CYP3A4,26, 43 literature search did not uncover any reports that suggested miR‐27b to be of diagnostic value for drug‐induced cardiomyopathy.
Integrating clinical incidence with the modes of action of a drug, which is depicted as its signaling network, for predictive modeling is a strength of our study. There are, however, some limitations in our approach: (1) nontoxic slightly outnumbered toxic drugs; (2) limitation of ILP where no biological feedback controls were considered and assumptions adopted in ILP formulation; (3) DEGs of doxorubicin in cardiomyocytes were from different sources than the rest of 30 drugs; and (4) availability of transcriptomic profiling data in cardiomyocytes. Furthermore, our study inherited the shortcomings associated with the databases and knowledge base used for our modeling. The impact of disease indications on the incidence and severity of treatment‐related cardiomyopathy is not well characterized.
Our predictive modeling of integrating clinical incidence of drug‐induced cardiomyopathy with the signaling network of toxic and nontoxic drugs not only is useful for further improving its predictive power, but also identifies important gene/protein predictors that have relevant cardiac biological functions. Above all, the top genes/protein predictors are reportedly regulated by specific microRNAs that have been shown to be of diagnostic value for heart failure or drug‐induced cardiomyopathy. These predictors might be useful for shedding light on potential microRNAs as in vivo biomarkers of drug‐induced cardiomyopathy.
Conflict of Interest
The authors declared no conflict of interest.
Supporting information
Supplementary Document CMap Supplementary documentation of Connectivity Map (CMap) data: Predictive modeling using cancer cell lines data from CMap. This document contains Supplementary Table S1 (the list of 75 drugs) and Supplementary Figure S2.
Supplementary Document‐ILP A drug's mode of action illustrated by ILP. This document contains Supplementary Figure S1 (the methotrexate signaling network).
Supplementary Figure S3 (a and b) Precision/recall curves for the 34 differentially expressed genes modeled using elastic net (EN) and an integer linear programming‐EN combination, respectively.
Supplementary Table S2 A few studies of transcriptomic profiles of doxorubicin
Supplementary Table S3 Rationale for including 4 differentially expressed genes (DEGs) profiles of doxorubicin.
Supplementary Table S4 Detailed results of predictive modeling.
Supplementary Table S5 Integer linear programming (ILP) solutions of 34 differentially expressed genes (DEGs) profiles.
Supplementary Table S6 Biological functions of the top 15 gene/protein predictors.
Source of Funding
Dimitris Messinis was supported as ORISE fellow through a 2013 Medical Counter Measures Grant to J.P.F.B. Navya Varshney was supported as a pharmacy intern via fellowship from Office of Clinical Pharmacology. Assistance on using Betsy and computation efficiency by Dr Mike Mikailov (computer scientist, CDRH) is appreciated. This project was supported by an appointment to the Research Participation Program at CDER, administered by the Oak Ridge Institute for Science and Education (ORISE) through an interagency agreement between the US Department of Energy and the FDA. Helpful discussion about elastic net with Dr Hui Zou (Professor of Statistics, University of Minnesota) is appreciated. Comments on cardiac contractility and cardiomyopathy by Dr David Strauss (Division Director, Division of Applied Regulatory Science, Office of Clinical Pharmacology) are appreciated.
Author Contributions
D.E.M. and J.P.F.B. wrote the manuscript. D.E.M. and J.P.F.B. designed the research. D.E.M., J.G.H., and N.V. performed the research. I.N.M. and L.G.A. contributed new reagents/analytical tools.
Disclaimer
This article reflects the views of the authors and should not be construed to represent the FDA's views or policies.
References
- 1.Drugs@FDA: FDA Approved drug products. <http://www.https://www.accessdata.fda.gov/scripts/cder/daf/>. Accessed July 2016.
- 2. Pouliot, Y. , Chiang, A.P. & Butte, A.J. Predicting adverse drug reactions using publicly available PubChem BioAssay data. Clin. Pharmacol. Ther. 90, 90–99 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu, M. et al Large‐scale prediction of adverse drug reactions using chemical, biological, and phenotypic properties of drugs. J. Am. Med. Inform. Assoc. 19, e28–e35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harpaz, R. , DuMouchel, W. , Shah, N.H. , Madigan, D. , Ryan, P. & Friedman, C. Novel data‐mining methodologies for adverse drug event discovery and analysis. Clin. Pharmacol. Ther. 91, 1010–1021 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DToxS ‐ Drug Toxicity Signature Generation Center – Data and Resources. <https://martip03.u.hpc.mssm.edu/data.php>. Accessed February 2017.
- 6. Burridge, P.W. et al Human induced pluripotent stem cell‐derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin‐induced cardiotoxicity. Nat. Med. 22, 547–556 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chaudhari, U. et al Identification of genomic biomarkers for anthracycline‐induced cardiotoxicity in human iPSC‐derived cardiomyocytes: an in vitro repeated exposure toxicity approach for safety assessment. Arch. Toxicol. 90, 2763–2777 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Melas, I.N. et al Identification of drug‐specific pathways based on gene expression data: application to drug induced lung injury. Integr. Biol. (Camb). 7, 904–920 (2015). [DOI] [PubMed] [Google Scholar]
- 9. National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. <https://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctchtm>. Accessed July 2016.
- 10.Internatinal Council for Harmonisation of Technical Requirements for Pharmaceuticals (ICH). Medical Dictionary for Regulatory Activities (MeDRA). <http://www.meddra.com/>. Accessed July 2016.
- 11. Hur, J. , Guo, A.Y. , Loh, W.Y. , Feldman, E.L. & Bai, J.P. Integrated systems pharmacology analysis of clinical drug‐induced peripheral neuropathy. CPT Pharmacometrics Syst. Pharmacol. 3, e114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zou, H. & Hastie, T. Regularization and variable selection via the elastic net. J. R. Statist. Soc. B 67, 301–320 (2005). [Google Scholar]
- 13.Institute WTS. Genomics of Drug Sensitivity in Cancer. <http://www.cancer.rxgene.org/news>. Accessed August 2017.
- 14. Melas, I.N. , Samaga, R. , Alexopoulos, L.G. & Klamt, S. Detecting and removing inconsistencies between experimental data and signaling network topologies using integer linear programming on interaction graphs. PLoS Comput. Biol. 9, e1003204 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Connectivity Map (CMAP). <https://www.broadinstitute.org/connectivity-map-cmap>. Accessed August 2016.
- 16. Szklarczyk, D. , Santos, A. , von Mering, C. , Jensen, L.J. , Bork, P. & Kuhn, M. STITCH 5: augmenting protein‐chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 44, D380–D384 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Search Tool for Interactions of Chemicals (STITCH). Stitch‐European Molecular Biology Laboratory (EMBL). <http://stitch.embl.de/>. Accessed August 2016.
- 18.(EMBL‐EBI) EBI. HUGO Gene Nomenclature Committee. <http://www.genenames.org/>. Accessed August 2016.
- 19.Reactome. <http://www.reactome.org/>. Accessed August 2016.
- 20. Gentleman, R. Annotate: Annotation for microarrays. R package version 1.56.1. <https://www.bioconductor.org/packages/release/bioc/html/annotate.html>. Accessed August 2016.
- 21. Carlson, M. Affymetrix Human Genome U133A 2.0 Array annotation data (chip hgu133a2). R package version 3.2.3. <http://www.bioconductor.org/packages/release/data/annotation/html/hgu133a2.db.html>. Accessed August 2016.
- 22.The R Project for Statistical Computing. R: a language and environment for statistical computing. <https://www.r-project.org/> (2016).
- 23. Barpe, D.R. , Rosa, D.D. & Froehlich, P.E. Pharmacokinetic evaluation of doxorubicin plasma levels in normal and overweight patients with breast cancer and simulation of dose adjustment by different indexes of body mass. Eur. J. Pharm. Sci. 41, 458–463 (2010). [DOI] [PubMed] [Google Scholar]
- 24. Mitsos, A. , Melas, I.N. , Siminelakis, P. , Chairakaki, A.D. , Saez‐Rodriguez, J. & Alexopoulos, L.G. Identifying drug effects via pathway alterations using an integer linear programming optimization formulation on phosphoproteomic data. PLoS Comput. Biol. 5, e1000591 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Friedman, J. , Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1–22 (2010). [PMC free article] [PubMed] [Google Scholar]
- 26. Chou, C.H. et al miRTarBase 2016: updates to the experimentally validated miRNA‐target interactions database. Nucleic Acids Res. 44, D239–D247 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schulte, C. , Westermann, D. , Blankenberg, S. & Zeller, T. Diagnostic and prognostic value of circulating microRNAs in heart failure with preserved and reduced ejection fraction. World J. Cardiol. 7, 843–860 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marques, F.Z. , Vizi, D. , Khammy, O. , Mariani, J.A. & Kaye, D.M. The transcardiac gradient of cardio‐microRNAs in the failing heart. Eur. J. Heart Fail. 18, 1000–1008 (2016). [DOI] [PubMed] [Google Scholar]
- 29. Leger, K.J. , Leonard, D. , Nielson, D. , de Lemos, J.A. , Mammen, P.P. & Winick, N.J. Circulating microRNAs: potential markers of cardiotoxicity in children and young adults treated with anthracycline chemotherapy. J. Am. Heart Assoc. 6, e004653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Karaman, M.W. et al A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 26, 127–132 (2008). [DOI] [PubMed] [Google Scholar]
- 31. Force, T. , Krause, D.S. & Van Etten, R.A. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat. Rev. Cancer 7, 332–344 (2007). [DOI] [PubMed] [Google Scholar]
- 32. Fabre, G. , Julian, B. , Saint‐Aubert, B. , Joyeux, H. & Berger, Y. Evidence for CYP3A‐mediated N‐deethylation of amiodarone in human liver microsomal fractions. Drug Metab. Dispos. 21, 978–985 (1993). [PubMed] [Google Scholar]
- 33. Zientek, M.A. et al In vitro kinetic characterization of axitinib metabolism. Drug Metab. Dispos. 44, 102–114 (2016). [DOI] [PubMed] [Google Scholar]
- 34. Scripture, C.D. & Figg, W.D. Drug interactions in cancer therapy. Nat. Rev. Cancer 6, 546–558 (2006). [DOI] [PubMed] [Google Scholar]
- 35. Bodin, K. , Lindbom, U. & Diczfalusy, U. Novel pathways of bile acid metabolism involving CYP3A4. Biochim. Biophys. Acta 1687, 84–93 (2005). [DOI] [PubMed] [Google Scholar]
- 36. Schlaepfer, D.D. , Hauck, C.R. & Sieg, D.J. Signaling through focal adhesion kinase. Prog. Biophys. Mol. Biol. 71, 435–478 (1999). [DOI] [PubMed] [Google Scholar]
- 37. Samarel, A.M. Focal adhesion signaling in heart failure. Pflugers Arch. 466, 1101–1111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Clemente, C.F. et al Focal adhesion kinase governs cardiac concentric hypertrophic growth by activating the AKT and mTOR pathways. J. Mol. Cell. Cardiol. 52, 493–501 (2012). [DOI] [PubMed] [Google Scholar]
- 39. Grossmann, J. et al Hierarchical cleavage of focal adhesion kinase by caspases alters signal transduction during apoptosis of intestinal epithelial cells. Gastroenterology 120, 79–88 (2001). [DOI] [PubMed] [Google Scholar]
- 40. Jehle, J. et al Regulation of apoptosis in HL‐1 cardiomyocytes by phosphorylation of the receptor tyrosine kinase EphA2 and protection by lithocholic acid. Br. J. Pharmacol. 167, 1563–1572 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xie, B. et al Focal adhesion kinase activates Stat1 in integrin‐mediated cell migration and adhesion. J. Biol. Chem. 276, 19512–19523 (2001). [DOI] [PubMed] [Google Scholar]
- 42. Sieg, D.J. et al FAK integrates growth‐factor and integrin signals to promote cell migration. Nat. Cell. Biol. 2, 249–256 (2000). [DOI] [PubMed] [Google Scholar]
- 43. Pan, Y.Z. , Gao, W. & Yu, A.M. MicroRNAs regulate CYP3A4 expression via direct and indirect targeting. Drug Metab. Dispos. 37, 2112–2117 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Document CMap Supplementary documentation of Connectivity Map (CMap) data: Predictive modeling using cancer cell lines data from CMap. This document contains Supplementary Table S1 (the list of 75 drugs) and Supplementary Figure S2.
Supplementary Document‐ILP A drug's mode of action illustrated by ILP. This document contains Supplementary Figure S1 (the methotrexate signaling network).
Supplementary Figure S3 (a and b) Precision/recall curves for the 34 differentially expressed genes modeled using elastic net (EN) and an integer linear programming‐EN combination, respectively.
Supplementary Table S2 A few studies of transcriptomic profiles of doxorubicin
Supplementary Table S3 Rationale for including 4 differentially expressed genes (DEGs) profiles of doxorubicin.
Supplementary Table S4 Detailed results of predictive modeling.
Supplementary Table S5 Integer linear programming (ILP) solutions of 34 differentially expressed genes (DEGs) profiles.
Supplementary Table S6 Biological functions of the top 15 gene/protein predictors.