

Abstract
Translational pharmacokinetic (PK) models are needed to describe and predict drug concentration‐time profiles in lung tissue at the site of action to enable animal‐to‐man translation and prediction of efficacy in humans for inhaled medicines. Current pulmonary PK models are generally descriptive rather than predictive, drug/compound specific, and fail to show successful cross‐species translation. The objective of this work was to develop a robust compartmental modeling approach that captures key features of lung and systemic PK after pulmonary administration of a set of 12 soluble drugs containing single basic, dibasic, or cationic functional groups. The model is shown to allow translation between animal species and predicts drug concentrations in human lungs that correlate with the forced expiratory volume for different classes of bronchodilators. Thus, the pulmonary modeling approach has potential to be a key component in the prediction of human PK, efficacy, and safety for future inhaled medicines.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Most pulmonary PK models are descriptive rather than predictive, often single‐drug specific and have not been qualified by across animal scaling to show adequate predictions of preclinical lung profiles after pulmonary administration.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ The need of a modeling approach for soluble inhaled drugs capable of capturing PKs in the lungs and blood, allowing translation across species and prediction of clinical efficacy and safety in man.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Demonstrates utility of a compartmental model that describes key features of preclinical lung and systemic PK after pulmonary drug administration. The model‐predicted drug levels in human lungs were shown to correlate with efficacy, which supports the use of the model for estimating the human therapeutic dose.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ The translational modeling approach is shown to be valuable in rationalizing clinical lung function efficacy data for a range of inhaled bronchodilators, and provides drug discovery and development with a tool to assess and select future inhaled medicines.
Inhaled drug therapy is used for targeting the lung while minimizing systemic exposure in order to improve the benefit/risk margin for treatments of respiratory disease.1 A translational modeling approach capturing the pharmacokinetics (PKs) in both blood and lung after pulmonary delivery across species would be a valuable tool in the pursuit of novel inhaled therapeutics. To our knowledge, most of the available empirical compartmental or more mechanistic physiologically based pharmacokinetic models2 have not been evaluated to characterize lung PKs and efficacy. A notable exception is the work of Ericsson et al.,3 which provides a framework for prediction of the human therapeutic inhaled dose, and, although reasonably successful for bronchodilators, it delivers in contrast to the present work no further evaluation for the cross‐species translatability of an underlying lung PK model and no attempt was made to further quantitatively link PK to a measure of clinical efficacy. There is a wealth of preclinical and clinical efficacy data available for bronchodilators, with clinical efficacy measured by forced expiratory volume in 1 second (FEV1) in patients with respiratory diseases. Combining the efficacy data with lung PK modeling presents a unique opportunity to establish the first bronchodilator lung pharmacokinetic/pharmacodynamic (PK/PD) relationship which, in contrast to existing lung PK/PD models,4, 5, 6, 7 is driven by predicted lung concentrations from a validated translational PK model instead of retrospectively being optimized to best fit the measured effect.
Inhaled bronchodilators display pulmonary PK behavior associated with their physicochemical properties, which drive lung tissue retention and half‐lives, ranging from the rapidly absorbed single basic/quaternary amine drugs (e.g., terbutaline, salbutamol, and ipratropium) to recently developed dibasic amines (e.g., batefenterol and AZD3199) with lung half lives in the order of days.8, 9 In order to successfully capture these diverse lung retention behaviors, a compartmental model is required, where some of its parameters can be linked to actual physiology (e.g., lung weight, protein, and lung tissue binding) in order to allow for a successful cross‐species translation. Modeling preclinical data as a starting point offers the advantage of being data rich (lung and plasma PK) for many different inhaled pharmacological agents as well as facilitating the evaluation of cross‐species translation (e.g., rat to dog) via an appropriate scaling of the modeled PK parameters. The first original work directed toward the scaling of a preclinical model developed to predict events after pulmonary drug delivery was that of Jones et al.10 Their preclinical compartmental rat model was scaled to make reasonable predictions for the plasma exposure after inhalation of various neutral and basic drugs in humans. However, it only allowed for a mono‐exponential decline in pulmonary drug levels, which is an oversimplification, in particular for basic compounds.9, 11 In addition, no further attempt was made to predict the actual lung concentrations, a consequence of the absence of any lung PK data in the model development.
The objective of the current study was to develop a multicompartmental model to capture key features of both plasma and lung PK profiles after pulmonary and i.v. administration of a wide range of soluble bronchodilator drugs (chemically classified as either amines or quaternary amines) to rats and to assess the cross‐species translatability of the model using available plasma and lung PK data from intra‐tracheal (i.t.) dosed dogs. Finally, the ability of using the model to predict human plasma profiles for 12 inhaled drugs (primarily bronchodilators) and explore their pulmonary PK/PD relationships was investigated.
METHODS
Study design for animal experiments
All experimental procedures were performed in accordance with UK Home Office regulations under the Animals (Scientific Procedures) Act 1986. The in vivo PK profiles, including pulmonary PK, for all investigated bases and quaternary amines were acquired in male Sprague‐Dawley rats and, for a selection of compounds, also in male Beagle dogs after both bolus i.v. and i.t. administration of solutions (Table 1). To allow for a more peripheral deposition, typical for inhaled dosing, 100–200 µl air was injected behind 0.5 mL/kg of the i.t. dosed phosphate buffered (pH 7.4) saline solution (more information about analytics etc. is available in the Supplementary Material). The preclinical efficacy studies, from which also pulmonary PK was acquired, were carried out in male Dunkin Hartley guinea pigs, a well characterized species for studying pulmonary pharmacology15 the i.t. dose response for the compounds (except AZD4818 and terbutaline) was established via measurement of the inhibition of histamine (beta‐2 agonists) or methacholine (muscarinic antagonists) induced bronchoconstriction given 2 hours after a compound dose or in case of dual muscarinic antagonist/beta‐2 agonists (MABAs) after both histamine and methacholine challenge (for a detailed description see Supplementary Material). In addition, total lung concentrations for all 12 compounds were measured (see Bioanalytical Method) and combined with the other data in one joint dataset. To these data, a sigmoidal maximum effect (Emax) model was fit with the maximum effect fixed at 100% (Eq. 1) and the determined half‐maximal inhibitory lung concentration (IC50) values used to normalize the predicted lung concentrations in the following lung PK/PD analyses, in which the predicted lung concentrations were divided by the corresponding IC50.
| (1) |
Table 1.
Physicochemical properties and in vivo doses for selected bases and quaternary amines.
| Drug | Ion class | Solubilitya, µM | LogD7.4 b | pKa c | i.v./i.t. doses in the rat, µg/kg | i.v./i.t. doses in the dog, µg/kg |
|---|---|---|---|---|---|---|
| Salmeterol | Base | >380 | 2.2 | 9.1 | 14/18 | – |
| Formoterol | Base | 600 | 0.49 | 8.4 | 15/14 | 10/5 |
| Salbutamol | Base | >2,900 | −1.9 | 9.2 | 1,064/15 | – |
| Terbutaline | Base | 6,830 | −1.5 | 9.3 | 1,038/1080 | 500/50 |
| Indacaterol | Base | 25 | 2.8 | 8.3 | 1,000/15 | – |
| Tiotropium | QA | >2,800 | <‐1.3 | NA | 10,000/53 | 80/0.7 |
| Ipratropium | QA | >9,140 | −1.0 | NA | 1,000/1,002 | – |
| Glycopyrronium | QA | >4,050 | 0.12d | NA | 282/8.1 | – |
| AZD2115 | Dibase | 650 | 2.1 | 8.7/7.6 | 769/6.6 | – |
| Batefenterol | Dibase | 28 | 3.1 | 10/7.3 | 597/1.2 | – |
| AZD4818 | Dibase | >2,500 | 0.90 | 8.4/6.2 | 258/10.8 | – |
| AZD3199 | Dibase | 2,510 | 2.3 | 9.5/7.1 | 1,000/15.7 | – |
Determination of unbound fraction in plasma and lung tissue homogenate
The unbound drug fractions in either plasma or homogenized lung tissue were obtained from in vitro equilibrium dialyses experiments16 (see Supplementary Table S1).
BIOANALYTICAL METHOD
Compartmental model structure
The key features of pulmonary PK that were revealed by experimental determination of lung and plasma concentrations after both i.t. and i.v. dosing led to the conception of the compartmental model structure shown in Figure 1 a. The systemic PK is described by a serial three‐compartmental model (compartments 1, 4, and 5) linked to two serial lung compartments (compartments 2 and 3; Figure 1 b). Measured plasma concentration of drugs is represented by compartment 1. Measured lung concentration of drugs is represented by the sum of amounts in both lung compartments (compartments 2 and 3) divided by the physiological lung volume. Omission of a model description of dissolution of lung deposited material (after i.t.) is justified by the high solubility of the compounds. Equally, a model description of a mucociliary clearance route for solid particles was not included. The central lung compartment (compartment 2) is the dosing compartment for i.t. administration wherein the unbound drug is instantly available for further distribution. The lung compartment 3, which is lacking a defined volume, describes immobilized drug contained in a deep compartment of an as of yet undefined physiological identity (see Discussion).
Figure 1.
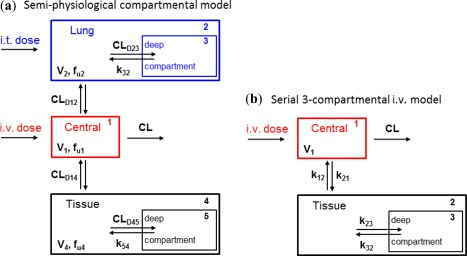
(a) Structure of the compartmental model for simultaneous fitting to lung and plasma concentration time profiles of i.v. and i.t. dosed rats. (b) Serial three‐compartmental model for fitting to plasma concentration time profiles of i.v. dosed rats. CL, clearance.
Compartments 4 and 5 represent drug distribution in all other tissues, and are identical in structure to compartments for the lung tissue. The distribution between the compartments is governed by unbound distributional clearances, with the exception of drug transfer out of the deep compartments 3 and 5, in which rate constants k32 and k54 are used because these compartments represent subcellular compartments that are not assigned specified volumes. All the unbound drug distribution clearances in the final model operate on unbound drug concentrations, parameterized as the product of the drug amounts in compartments 1, 2, and 4 and relevant unbound fractions, and divided by the compartmental volumes. Rate constants k32 and k54 were assumed to have equal values because it was assumed that the distribution from these deep tissue compartments is governed by similar physiological processes in all tissues (including the lungs). Finally, drug elimination occurs from compartment 1 and is parameterized by the plasma clearance (CL). The complete five‐compartmental model included the physiological lung volume (V2) and measured unbound fractions in plasma (fu1) and lung tissue homogenate (fu2) as fixed constants and is represented by the following five differential equations:
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
Thus, the fu2 constant simply describes nonspecific binding in the central lung compartment 2, as drugs in the so‐called deep compartment 3 was assumed to be liberated during the homogenization of lung tissue. Strong covariance between V4 and fu4 was observed in earlier modeling and was shown to be a result of the model being structurally unidentifiable (i.e., only the fraction of fu4/V4 could be determined). To render the final model at least structurally locally identifiable, this fraction was lumped into one parameter denoted with “fu4/V4” as the two parameters always appeared as a fraction in the model (for details see Supplementary Material and Supplementary Model Code).
Constants and initial estimates in a three‐step approach
For robust model fitting, some model parameters were fixed based on prior information. Physiological parameters were fixed based on literature and in‐house values; the physiological lung volume (V2) was set to the constant value of 6 mL/kg body weight for the rat, based on in‐house experimental rat data, which is consistent with literature values.17 Drug parameters, including systemic plasma CL, unbound fractions in both plasma and lung homogenate (fu1 and fu2, respectively) were fixed to their experimentally measured values. All other parameters were fitted using initial estimates obtained via a three step approach; step 1: analysis of plasma PK after i.v. dosing via fitting to a serial three‐compartmental model to obtain micro‐constants k12, k21, k23, and k32 (Figure 1 b); step 2: determination of the initial slope, terminal slope, and intercept from an analysis of the bi‐phasic lung profiles after i.t. dosing (Supplementary Table S2); and step 3: derivation of initial parameter estimates from equations involving parameter values obtained from previous steps (Supplementary Material and Table S3).
Modeling and software
For each compound, the compartmental model was fitted simultaneously to available individual and naively pooled drug concentration data from both plasma and lung samples obtained after i.t. and i.v. dosing, using the constants and initial estimates as defined above (assumed that all data came from one, the typical, individual; hence, the analysis is naïve with respect to the information regarding to which data belong to which individual). The weighting scheme was of a multiplicative nature in all cases, because measured plasma and lung concentration data are both believed to have a constant coefficient of variation. Phoenix 64 WinNonlin (Certara, NJ) was used for all the performed modeling.
Cross‐species scaling; simulation of dog and human PK
Scaling of the rat model to the dog and human compartmental models was performed by substituting measured dog and human values for clearance, unbound fractions in blood plasma, and the physiological lung volume. Rate constants k32 and k54, were assumed to be conserved across species equally so the volume V1 and “fu4/V4” and the latter is in agreement with allometric rules, considering that the exponent to scale a volume of distribution is typically assumed to be unity. CLD12 and CLD23 were recalculated in relation to literature values of the physiological lung volume, in which underlying rate constants were considered constant. Unbound distribution clearances CLD14 and CLD45 were allometrically scaled from the rat10, 18 (Eq. 7; Supplementary Table S3):
| (7) |
where the bodyweights of 0.25, 15, or 70 kg were used for rat, dog, and human, respectively. In the dog, with the i.t. dose input, the complete dose was modeled as being administered to the lung with no swallowed component and the model was used to simulate the drug concentration‐time profiles for both plasma and lungs. In contrast to i.t. dosing, following inhaled dosing in a human, a fraction of the total inhaled dose may be swallowed and absorbed from the gastro‐intestinal tract, which was accounted for in the model (Supplementary Material and Table S4).
Human pulmonary efficacy of bronchodilators
The clinical effect of bronchodilator drugs on lung function (forced expiratory volume in 1 second, FEV1 determined by spirometry) in patients with chronic obstructive pulmonary disease for a variety of dose regimens were obtained from published studies (Supplementary Table S5) or in‐house in case of AZD2115 (clinicaltrials.gov no. NCT01498081 and NCT02109406) and AZD4818. These latter studies were performed in accordance with the ethical principles that have their origin in the Declaration of Helsinki and that are consistent with the International Conference on Harmonisation/Good Clinical Practice and applicable regulatory requirements and the AstraZeneca policy on Bioethics. The reported mean values for the placebo‐corrected changes in trough FEV1 from baseline were directly linked to the predicted total lung concentrations at trough obtained via simulation of relevant dose regimens with the human compartmental model. Potency‐normalization was achieved through division of predicted total lung concentrations by the pharmacological total lung IC50 values obtained from the aforementioned in vivo guinea pig model. In order to establish sufficient ground to perform such a direct interspecies scaling, human and guinea pig in vitro potencies were obtained from published literature values. These experiments involved the measurement of relaxation of acetylcholine or carbachol precontracted isolated bronchial rings or lung slices (Table 2, 19, 20, 21, 22, 23, 24, 25, 26). Finally, pulmonary PK/PD relationships were obtained by fitting simple Emax models, for each bronchodilator class, to the normalized predicted lung concentration and trough efficacy data, which for many compounds included efficacy data after both single and multiple administrations and at various dose levels.
Table 2.
In vitro and in vivo potencies for bronchodilators.
| Drug | Drug class | In vivo guinea pig total lung IC50, nM (%CV)a | In vitro guinea pig trachea pIC50 | In vitro human bronchi pIC50 |
|---|---|---|---|---|
| Salmeterol | BA | 36 (48) | 7.719/7.520 | 7.321/8.122 |
| Formoterol | BA | 3.0 (37) | 8.819 | 9.121/8.721/8.823 |
| Salbutamol | BA | 90 (34) | 6.719 | 6.421/6.922 |
| Indacaterol | BA | 66 (75) | 7.719 | 6.622/7.424 |
| Tiotropium | MA | 4.7 (14) | 9.125 | 9.525 |
| Ipratropium | MA | 0.84 (25) | 8.625 | 9.525 |
| Glycopyrronium | MA | 10.4 (18) | 9.025 | 8.424/10.425 |
| AZD2115 | MABA | 126 (24) | NV | NV |
| Batefenterol | MABA | 33 (32) | 8.026 | NV |
| AZD3199 | BA | 895 (39) | 8.019 | NV |
BA, beta‐2 agonist; CV, coefficient of variation; IC50, half‐maximal inhibitory concentration; MA, muscarinic antagonist; MABA, muscarinic antagonist/beta‐2 agonist; NV, no value; pIC50, negative logarithm of the IC50 value in molar.
Derived from fitting a sigmoid maximum effect (Emax) model to the lung efficacy ‐ concentration data with Emax fixed at 100%.
RESULTS
Modeling of rat plasma and lung profiles
The PK datasets generated in this study share some unique features identified for these types of soluble bases and quaternary amines: often biphasic profiles in both plasma and lung irrespective of dosing route (i.v. or i.t.) and a clear first‐pass loading of drugs in the lungs following i.t. dosing, which resulted in 10–400‐fold higher lung drug concentrations compared to systemic dosing at near equivalent plasma exposures (Supplementary Material Raw Data). The developed compartmental model captured all these characteristics after a simultaneous model fit and provided an acceptable description of the total plasma and lung concentrations for all compounds (Figure 2). The quality of the simultaneous model fits was supported by the low to medium values for the coefficient of variation (<35%) associated with the majority of the modeled parameters (Supplementary Table S6) and the absence of any discernible pattern in the obtained residuals (analysis not shown). In addition, a sensitivity analysis further confirmed that small perturbations in optimized parameter estimates were cause for significant changes in the objective function value as determined by the likelihood‐ratio test (significance level of 0.05).
Figure 2.
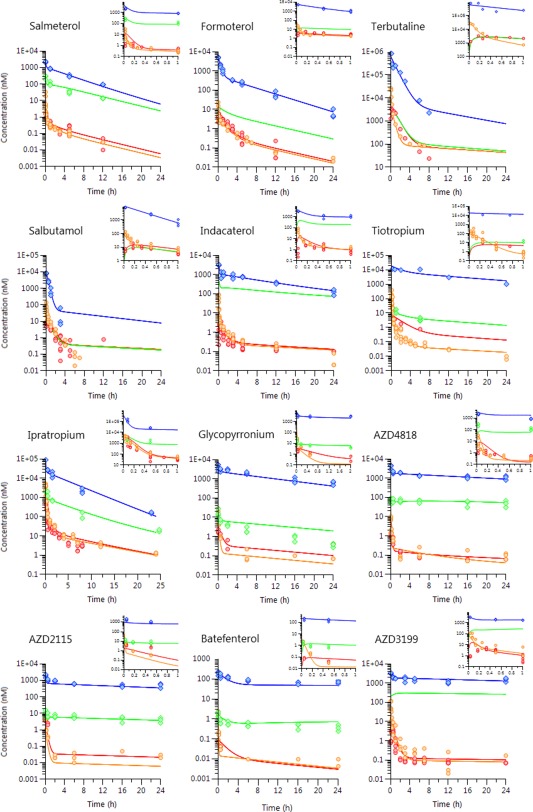
Observed individual plasma (circles) and lung concentrations (diamonds) with model‐fitted time profiles (solid lines) after i.t. and i.v. administration to rats. Plasma concentrations are colored red (after i.t.) or orange (after i.v.) and corresponding lung concentrations are either blue (after i.t.) or green (after i.v.).
Translation of rat model to dogs and humans
The translatability of the compartmental rat model to dogs was supported by agreement between observed and predicted PK profiles (Figure 3). The published and in‐house PK data in healthy volunteers for the 12 drugs demonstrates the ability of the rat model to predict human plasma PK profiles. Visual inspection of the predicted and observed PK data (Figure 4) shows an overall good performance in describing the human plasma profiles with predicted peak plasma concentration (Cmax) and last measured concentration (Clast) deviating less than twofold from the observed values in almost all of the cases without any outliers (Supplementary Table S7).
Figure 3.
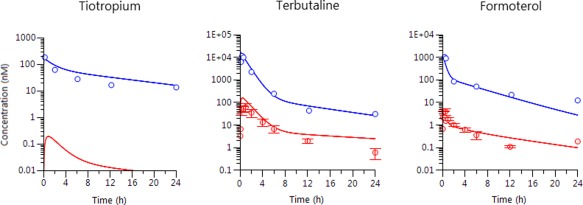
Observed averaged plasma (red) and individual lung (blue) concentrations (circles) with model‐simulated time profiles (solid lines) in dogs after i.t. dosing.
Figure 4.
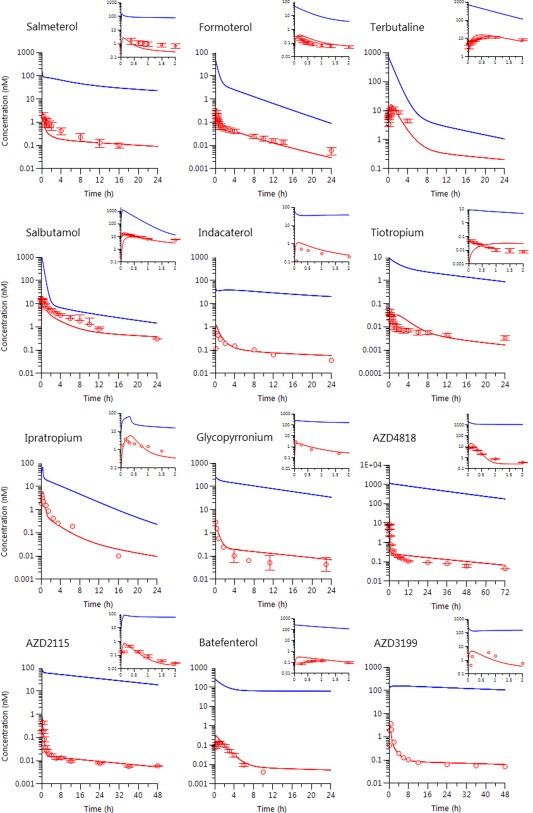
Observed human plasma concentrations (circles) vs. time after inhalation and simulated plasma and lung concentration time profiles (solid lines) based on the scaled compartmental model. Plasma concentrations are red colored and lung concentrations are blue.
Preclinical and clinical PK/PD assessment
The estimated in vivo potencies of the bronchodilator compounds in the guinea pig, in terms of the total lung concentrations required to cause 50% inhibition of bronchoconstriction (guinea pig IC50), are shown in Table 2 . 19, 20, 21, 22, 23, 24, 25, 26
The same table also highlights a relatively high degree of concordance between the reported guinea pig and human in vitro potencies, thus establishing the basis for the subsequent interspecies scaling of the in vivo total lung potency. The clinical pulmonary efficacy measures, the placebo corrected change in FEV1 from baseline at trough were plotted against the compartmental model predicted trough total lung concentrations. There is a correlation between efficacy and lung concentration for each individual drug (Figure 5 a) but the correlation for all the drugs was improved following correction for individual drug pharmacological in vivo potency measured in the guinea pig (Figure 5 b). The subsequent modeling of trough PK/PD relationships (Figure 5 c) resulted in good model fits for each pharmacological class and revealed that half‐maximal clinical response was associated with predicted lung concentrations that were 45–80% of the guinea pig model total lung IC50 in the case of muscarinic antagonists and MABAs (defined as ER50: the ratio of predicted human lung concentration/guinea pig IC50 that is associated with 50% of maximum improvement in trough FEV1; Figure 5 c). Interestingly, for beta‐2 agonists, the guinea pig model lung IC50 seemed to be ∼10‐fold higher than the lung concentration required to elicit half‐maximal response in patients with chronic obstructive pulmonary disease (Figure 5 c). The increased clinical effect for multiple dosing compared to single dosing observed for several of the drugs was closely matched by the predicted accumulation in lung concentrations. After once daily dosing, the predicted accumulation of lung trough levels after multiple dosing ranged from 1.3–2.6‐fold for glycopyrronium (25 µg) and indacaterol (300 µg), respectively, with an associated increase in FEV1 of around 40 mL and up to 15‐fold for batefenterol (800 µg) with an even larger increase in FEV1 at around 100 mL (see arrows in Figure 5 c and Supplementary Table S5).
Figure 5.
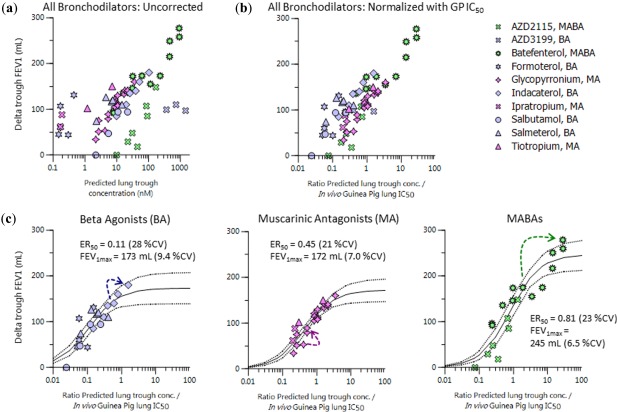
(a) Predicted trough lung concentrations vs. trough placebo corrected change in forced expiratory volume in 1 second (FEV1) from baseline in patients with chronic obstructive pulmonary disease for various inhaled doses and regimes of bronchodilators. (b) Similar to a but with division of concentration term by in vivo guinea pig (GP) lung half‐maximal inhibitory concentration (IC50). (c) Maximum effect (Emax) model fitting results (solid lines) of pulmonary pharmacokinetic/pharmacodynamic relationships at trough for various bronchodilator drug classes, taken from b, and dashed lines indicate 95% confidence intervals. The arrows indicate the degree of accumulation in going from single to multiple dosing (beta agonist (BA): indacaterol 300 µg; muscarinic antagonist (MA): glycopyrronium 25 µg; and muscarinic antagonist/beta‐2 agonist (MABA): batefenterol 800 µg). CV, coefficient of variation.
DISCUSSION
A compartmental modeling approach was shown to describe rat plasma and lung PK data for soluble drugs after pulmonary administration. Translatability of the model was demonstrated for dog and human data. The model adequately described the typical characteristics for a set of 12 soluble drugs belonging to different ion classes in which the majority of the i.t. dose was rapidly absorbed, occasionally within minutes and typically within a half an hour after administration. The model also captured the observed higher terminal phase lung concentrations after i.t. dosing (compared to i.v.) via a first pass loading of the deep lung compartment. Finally, also the large span in the observed terminal lung half‐lives was satisfactorily captured by the fitted lung model parameters. These results also provide a further rational/basis for the inclusion of multiple lung compartments in the population modeling of inhaled basic/cationic drugs, as already implemented for oladaterol27 and glycopyrronium.28 The presented pulmonary modeling approach should be used, not as a standalone, but combined with established methods10 for the prediction of clearance, oral bioavailability, and oral absorption rate constants to predict the human PK after a pulmonary delivery. With the integration of lung PK predictions and guinea pig pharmacology data, it was possible to model the clinical efficacy data for a broad range of bronchodilators. This suggests that the described modeling approach could have utility for the prediction of human inhaled dose and regimen for soluble molecules regardless of chemical class.
Compartmental model structure
The typical biphasic absorption from the lungs indicates the presence of a mechanism that retains part of the administered dose within the lung tissue. The model captured this through the use of a deep compartment from which drug release is slow relative to drug transfer into this compartment and between the lungs and the blood. The serial arrangement of these lung compartments and the similar setup for other tissues is essential in allowing an adequate description of the PK profiles and to describe a first pass loading of the lung tissue. This phenomenon is assumed to be the main driver for the increase in lung to plasma drug concentration ratio after lung delivery compared to i.v. dosing. Finally, parameters for capturing the unspecific binding in the lungs and the rest of the body, respectively, fu2 and fu4 (as part of “fu4/V4”), are likely correlated, and could even be presumed to be similar, although this would not affect the observed modeling performance and, in such a scenario, V4 would become a separate parameter.
The nature of the deep compartment
This preferential loading of a lung subcompartment could be due to lysosomal trapping as a result of the pH difference between cytosol (pH 7.2) and lysosomes (pH 5). This has previously been used as an explanation for increased tissue (including the lungs) concentration and consequently longer half‐lives for basic drugs2, 8 and, therefore, represents a plausible explanation for this reservoir. However, making physiological sense for basic drugs would not explain the behavior of the quaternary amines (tiotropium, ipratropium, and glycopyrronium), which are devoid of any ionizable centers and possess a permanent positive charge. These drugs might be retained due to a significantly slower passive cellular permeability or simply a result of active and drug concentration‐dependent epithelial cell uptake, as all investigated quaternary amines are OCT1 substrates,29 which is expressed in the lungs.30 This would have the effect of causing greater drug retention in the lung after pulmonary administration compared to when dosed systemically.
MODELING OUTCOME
Translation of the compartment model
The translation of the model from rat to dog involved direct scaling of the unbound lung clearances CLD12 and CLD23 in relation to the increased physiological lung volume. This resulted in an adequate prediction of both the plasma and lung profiles for two bases (terbutaline and formoterol) and the one quaternary amine (tiotropium) after i.t. dosing to dogs. Similarity between rat and dog in lung retention, as indicated by these results, provides further confidence in the translatability of the model to man, in particular in the prediction of pulmonary PK. Indeed, the simulated human plasma profiles after inhalation with the scaled model were generally similar to the observed data. The successful validation of the translatability of the model from rat to dog and the adequate prediction of human plasma profiles (see Supplementary Table S7) after inhalation builds confidence in using the predicted human lung PK in analyses of observed bronchodilator lung efficacy.
Predicting efficacy of the inhaled bronchodilators
The predicted potency‐normalized trough lung concentration was found to correlate with the placebo corrected change in FEV1 from baseline to a significant degree for the various bronchodilators, regardless of the pharmacological action (beta‐2 agonist or muscarinic antagonist). This positive result coming from the combination of guinea pig derived PD (total lung IC50s) with rat model derived PK (predicted trough lung concentrations) provided indirect evidence for the underlying assumption of a similar relative distribution of the compound in lung tissue between the rat and the guinea pig. In addition, the use of total levels was preferred over “free” in these PK/PD relationships, as the phenomenon of rebinding31, 32, 33 likely operated for these basic or cationic drugs and their membrane bound targets. Therefore, diffusion of drug molecules away from their unspecific binding sites (i.e., membranes), may allow them to consecutively bind (again) to nearby membrane bound receptors even when “bulk” unbound concentrations have dropped to insignificant levels. The PK/PD modeling of these relationships, separated out by pharmacological class, revealed that in order to affect a similar clinical response the beta‐2 agonists needed less coverage over the preclinical total lung IC50 compared to either the dual action MABAs or muscarinic antagonists. In the latter case, half‐maximal clinical response was associated with a predicted lung concentration that was similar to the guinea pig model lung IC50. This may be expected given the cross‐species similarity in potency and the anticipated direct PK/PD relationship for antagonists. On the other hand, the underprediction of the clinical effect from guinea pig in vivo potency (histamine challenge model) for beta‐2 agonists may be due to complexities of agonist PK/PD relationships or simply the fact that the size of the challenge in preclinical pharmacology models is manipulated to allow the largest window of effect to be observed. The translatability of dose is, therefore, generally uncertain and the better cross‐species relationship for muscarinic antagonists could simply be serendipitous.
The model was able to capture the accumulation in the effect observed following multiple dosing for some of the drugs, in particular for the dibasic MABAs, such as batefenterol with a predicted accumulation of lung trough concentration of 15‐fold. In contrast, drugs with only a minor observed accumulation in effect were also predicted to have a much lower accumulation of the lung trough concentration after multiple dosing. This builds confidence in the physiological relevance of the presented model. Last, the predicted terminal lung half‐lives for the various drugs are consistent with the optimal bronchodilators dosing regimen (e.g., the twice daily administered formoterol has a much shorter terminal lung half‐life (4 hours) compared to once daily administered indacaterol (14 hours)). Overall, these observations establish the translational value of the PK modeling approach and the preclinical PD models used to assess the bronchoprotective effect.
Generalization of the modeling approach
The successful extrapolation of the approach to predict the efficacy of inhaled bronchodilators to other target sites remains to be seen but we speculate that similar findings are anticipated when lung smooth muscle tissue is involved in combination with soluble compounds, as is the case for the studied bronchodilator drugs and recent pulmonary administered platelet‐derived growth factor receptor inhibitors.11 In cases of pulmonary administration of poorly soluble compounds, physiologically based pharmacokinetic approaches34 may offer a better alternative than the presented compartmental approach, because processes like mucociliary clearance are more easily modeled.
CONCLUSION
The modeling approach developed with the inclusion of a deep lung compartment was shown to allow translation of pulmonary PK across animal species to man for a large set of soluble inhaled bronchodilator drugs with a wide range of physicochemical, pharmacological, and PK properties. Furthermore, the observed correlation between predicted lung concentration and clinical lung function efficacy data suggests that the modeling approach has the potential to guide the development of novel inhaled soluble drugs and support the estimation of human inhaled therapeutic dose and dose regimen.
Conflict of Interest
The authors declared no competing interests for this work.
Supporting information
Supporting Information
Supporting Table S1
Supporting Table S2
Supporting Table S3
Supporting Table S4
Supporting Table S5
Supporting Table S6
Supporting Table S7
Supporting Information
Supporting Information
Acknowledgments
The authors wish to thank Britt‐Marie Fihn and Anders Lundqvist for their contributions to the in vivo studies and Erica Bäckström for providing measurements of the unbound fraction in homogenized lung tissue.
Author Contributions
R.H., M.F., U.E., and K.G. wrote the manuscript. K.G., R.H., and D.F. designed the research. D.F., R.H., and D.L.I.J. performed the research. D.F., R.H., and E.L.B. analyzed the data.
References
- 1. Tayab, Z.R. & Hochhaus, G. Pharmacokinetic/pharmacodynamic evaluation of inhalation drugs: application to targeted pulmonary delivery systems. Expert Opin. Drug Deliv. 2, 519–532 (2005). [DOI] [PubMed] [Google Scholar]
- 2. Borghardt, J.M. , Weber, B. , Staab, A. & Kloft, C. Pharmacometric models for characterizing the pharmacokinetics of orally inhaled drugs. AAPS J. 17, 853–870 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ericsson, T. , Fridén, M. , Kärrman‐Mårdh, C. , Dainty, I. & Grime, K. Benchmarking of human dose prediction for inhaled medicines from preclinical in vivo data. Pharm. Res. (2017). [DOI] [PubMed] [Google Scholar]
- 4. Wu, K. , Looby, M. , Pillai, G. , Pinault, G. , Drollman, A.F. & Pascoe, S. Population pharmacodynamic model of the longitudinal FEV1 response to an inhaled long‐acting anti‐muscarinic in COPD patients. J. Pharmacokinet. Pharmacodyn. 38, 105–119 (2011). [DOI] [PubMed] [Google Scholar]
- 5. Nielsen, J.C. , Hutmacher, M.M. , Cleton, A. , Martin, S.W. & Ribbing, J. Longitudinal FEV1 dose‐response model for inhaled PF‐00610355 and salmeterol in patients with chronic obstructive pulmonary disease. J. Pharmacokinet. Pharmacodyn. 39, 619–634 (2012). [DOI] [PubMed] [Google Scholar]
- 6. Gaz, C. , Cremona, G. , Panunzi, S. , Patterson, B. & De Gaetano, A. A geometrical approach to the PKPD modelling of inhaled bronchodilators. J. Pharmacokinet. Pharmacodyn. 39, 415–428 (2012). [DOI] [PubMed] [Google Scholar]
- 7. Agoram, B.M. , Milligan, P.A. & van der Graaf, P.H. A non‐parametric method to analyse time‐course of effect in the absence of pharmacokinetic data: application to inhaled bronchodilators. Eur. J. Pharm. Sci. 34, 250–256 (2008). [DOI] [PubMed] [Google Scholar]
- 8. Bäckström, E. , Boger, E. , Lundqvist, A. , Hammarlund‐Udenaes, M. & Fridén, M. Lung retention by lysosomal trapping of inhaled drugs can be predicted in vitro with lung slices. J. Pharm. Sci. 105, 3432–3439 (2016). [DOI] [PubMed] [Google Scholar]
- 9. Cooper, A.E. , Ferguson, D. & Grime, K. Optimisation of DMPK by the inhaled route: challenges and approaches. Curr. Drug Metab. 13, 457–473 (2012). [DOI] [PubMed] [Google Scholar]
- 10. Jones, R.M. & Harrison, A. A new methodology for predicting human pharmacokinetics for inhaled drugs from oratracheal pharmacokinetic data in rats. Xenobiotica 42, 75–85 (2012). [DOI] [PubMed] [Google Scholar]
- 11. Shaw, D.E. et al Optimization of platelet‐derived growth factor receptor (PDGFR) inhibitors for duration of action, as an inhaled therapy for lung remodeling in pulmonary arterial hypertension. J. Med. Chem. 59, 7901–7914 (2016). [DOI] [PubMed] [Google Scholar]
- 12. Wenlock, M.C. , Austin, R.P. , Potter, T. & Barton, P. A highly automated assay for determining the aqueous equilibrium solubility of drug discovery compounds. J. Lab. Autom. 16, 276–284 (2011). [DOI] [PubMed] [Google Scholar]
- 13. Wenlock, M.C. , Potter, T. , Barton, P. & Austin, R.P. A method for measuring the lipophilicity of compounds in mixtures of 10. J. Biomol. Screen. 16, 348–355 (2011). [DOI] [PubMed] [Google Scholar]
- 14. Wan, H. et al High‐throughput screening of pKa values of pharmaceuticals by pressure‐assisted capillary electrophoresis and mass spectrometry. Rapid Commun. Mass Spectrom. 17, 2639–2648 (2003). [DOI] [PubMed] [Google Scholar]
- 15. Walland, A. , Palluk, R. , Burkard, S. & Hammer, R. Compensation of muscarinic bronchial effects of talsaclidine by concomitant sympathetic activation in guinea pigs. Eur. J. Pharmacol. 330, 213–219 (1997). [DOI] [PubMed] [Google Scholar]
- 16. Bäckström, E. et al Development of a novel lung slice methodology for profiling of inhaled compounds. J. Pharm. Sci. 105, 838–845 (2016). [DOI] [PubMed] [Google Scholar]
- 17. Davies, B. & Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 10, 1093–1095 (1993). [DOI] [PubMed] [Google Scholar]
- 18. Gabrielsson, J. & Weiner, D. Pharmacokinetic and Pharmacodynamic Data Analysis, 5th edn. (Stockholm, Sweden, Swedish Pharmaceutical Press, 2017). [Google Scholar]
- 19. Stocks, M.J. et al Discovery of AZD3199, an inhaled ultralong acting β2 receptor agonist with rapid onset of action. ACS Med. Chem. Lett. 5, 416–421 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lindén, A. , Bergendal, A. , Ullman, A. , Skoogh, B.E. & Löfdahl, C.G. Salmeterol, formoterol, and salbutamol in the isolated guinea pig trachea: differences in maximum relaxant effect and potency but not in functional antagonism. Thorax 48, 547–553 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Molimard, M. , Naline, E. , Zhang, Y. , Le Gros, V. , Begaud, B. & Advenier, C . Long‐ and short‐acting beta2 adrenoceptor agonists: interactions in human contracted bronchi. Eur. Respir. J. 11, 583–588 (1998). [PubMed] [Google Scholar]
- 22. Naline, E. , Trifilieff, A. , Fairhurst, R.A. , Advenier, C. & Molimard, M. Effect of indacaterol, a novel long‐acting beta2‐agonist, on isolated human bronchi. Eur. Respir. J. 29, 575–581 (2007). [DOI] [PubMed] [Google Scholar]
- 23. Cazzola, M. et al Pharmacological characterization of the interaction between aclidinium bromide and formoterol fumarate on human isolated bronchi. Eur. J. Pharmacol. 745, 135–143 (2014). [DOI] [PubMed] [Google Scholar]
- 24. Cazzola, M. et al Pharmacological characterisation of the interaction between glycopyrronium bromide and indacaterol fumarate in human isolated bronchi, small airways and bronchial epithelial cells. Respir. Res. 17, 70 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Villetti, G. et al Pharmacological assessment of the duration of action of glycopyrrolate vs tiotropium and ipratropium in guinea‐pig and human airways. Br. J. Pharmacol. 148, 291–298 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hughes, A.D. et al Discovery of (R)‐1‐(3‐((2‐chloro‐4‐(((2‐hydroxy‐2‐(8‐hydroxy‐2‐oxo‐1,2‐dihydroquinolin‐5‐yl)ethyl)amino)methyl)‐5‐methoxyphenyl)amino)‐3‐oxopropyl)piperidin‐4‐yl [1,1'‐biphenyl]‐2‐ylcarbamate (TD‐5959, GSK961081, batefenterol): first‐in‐class dual pharmacology multivalent muscarinic antagonist and β2 agonist (MABA) for the treatment of chronic obstructive pulmonary disease (COPD). J. Med. Chem. 58, 2609–2622 (2015). [DOI] [PubMed] [Google Scholar]
- 27. Borghardt, J.M. , Weber, B. , Staab, A. , Kunz, C. & Kloft, C. Model‐based evaluation of pulmonary pharmacokinetics in asthmatic and COPD patients after oral olodaterol inhalation. Br. J. Clin. Pharmacol. 82, 739–753 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bartels, C. , Looby, M. , Sechaud, R. & Kaiser, G. Determination of the pharmacokinetics of glycopyrronium in the lung using a population pharmacokinetic modelling approach. Br. J. Clin. Pharmacol. 76, 868–879 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hendrickx, R. et al Identification of novel substrates and structure‐activity relationship of cellular uptake mediated by human organic cation transporters 1 and 2. J. Med. Chem. 56, 7232–7242 (2013). [DOI] [PubMed] [Google Scholar]
- 30. Koepsell, H. , Lips, K. & Volk, C. Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm. Res. 24, 1227–1251 (2007). [DOI] [PubMed] [Google Scholar]
- 31. Anderson, G.P. , Lindén, A. & Rabe, K.F. Why are long‐acting beta‐adrenoceptor agonists long‐acting? Eur. Respir. J. 7, 569–578 (1994). [DOI] [PubMed] [Google Scholar]
- 32. Jacobsen, J.R. Third‐generation long‐acting β2‐adrenoceptor agonists: medicinal chemistry strategies employed in the identification of once‐daily inhaled β2‐adrenoceptor agonists. Future Med. Chem. 3, 1607–1622 (2011). [DOI] [PubMed] [Google Scholar]
- 33. Vauquelin, G. & Charlton, S.J. Long‐lasting target binding and rebinding as mechanisms to prolong in vivo drug action. Br. J. Pharmacol. 161, 488–508 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Boger, E. et al Systems pharmacology approach for prediction of pulmonary and systemic pharmacokinetics and receptor occupancy of inhaled drugs. CPT Pharmacometrics Syst. Pharmacol. 5, 201–210 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Table S1
Supporting Table S2
Supporting Table S3
Supporting Table S4
Supporting Table S5
Supporting Table S6
Supporting Table S7
Supporting Information
Supporting Information