

Abstract
For scaling drug plasma clearance (CLp) from adults to children, extrapolations of population pharmacokinetic (PopPK) covariate models between drugs sharing an elimination pathway have enabled accelerated development of pediatric models and dosing recommendations. This study aims at identifying conditions for which this approach consistently leads to accurate pathway specific CLp scaling from adults to children for drugs undergoing hepatic metabolism. A physiologically based pharmacokinetic (PBPK) simulation workflow utilizing mechanistic equations defining hepatic metabolism was developed. We found that drugs eliminated via the same pathway require similar pediatric dose adjustments only in specific cases, depending on drugs extraction ratio, unbound fraction, type of binding plasma protein, and the fraction metabolized by the isoenzyme pathway for which CLp is scaled. Overall, between‐drug extrapolation of pediatric covariate functions for CLp is mostly applicable to low and intermediate extraction ratio drugs eliminated by one isoenzyme and binding to human serum albumin in children older than 1 month.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Proofs of concept for extrapolation of pediatric covariate functions for CLp between drugs sharing the same elimination pathway have been published for a limited number of drugs eliminated through glucuronidation and renal excretion.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ The study identifies the conditions upon which between‐drug extrapolation of isoenzyme‐specific pediatric covariate functions consistently leads to accurate CLp scaling from adults to pediatric ages for drugs metabolized by one or multiple hepatic isoenzymes.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Between‐drug extrapolation of pediatric covariate functions for CLp is mostly applicable to low and intermediate extraction ratio drugs eliminated by one isoenzyme and binding to human serum albumin in children older than 1 month.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ We now have a tool available that can establish a priori whether two drugs metabolized by the same isoenzyme will require the same or different dose adjustments in pediatrics.
Accurate scaling of plasma clearance (CLp) of drugs from adults to the pediatric population is crucial for the definition of first‐in‐child doses and for the development of pediatric dose recommendations.1, 2, 3, 4 As illustrated in Figure 1, for drugs undergoing hepatic metabolism, CLp values are driven by the complex interplay between drug‐specific and system‐specific properties. Relevant parameters to describe hepatic clearance are hepatic blood flow (Qh), the unbound drug fraction in plasma, the blood to plasma ratio (B:P), and the intrinsic metabolic clearance in the liver based on unbound drug concentration (CLint).5 The Qh is a purely system‐specific parameter, whereas unbound fraction, B:P, and CLint are derived from both system‐specific and drug‐specific parameters. Moreover, in children, system‐specific parameters vary with age due to ontogenic processes, which in turn drive CLp changes across the pediatric age range (Figure 1).6 Because physiologically based pharmacokinetic (PBPK) modeling integrates all the above‐mentioned information,6, 7 it can provide a yardstick for scaling drug CLp from adults to various pediatric ages in the absence of pediatric clinical data,6, 8, 9, 10, 11 which is relevant for first‐in‐child dose definition. In addition to PBPK modeling, there is a need for model‐based scaling methods that aggregate the influence of ontogeny of the system‐specific parameters in a smaller set of equations, thereby facilitating scaling of pediatric CLp in drug development and clinical practice.
Figure 1.
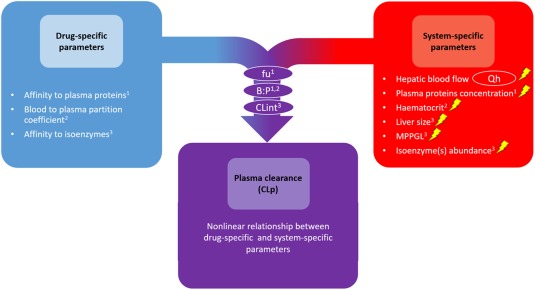
Schematic representation of the complex interplay between drug‐specific and system‐specific parameters driving hepatic CLp values. Parameters within circles are directly used in the physiologically based pharmacokinetic hepatic clearance model (e.g., dispersion model). Parameters in the purple circles represent composite parameters that are derived from the system‐specific parameters and the drug‐specific parameters indicated by the numbers in the subscripts. In children, each of the system‐specific parameters change with age, each represented by a lightning bolt. B:P, blood to plasma ratio; CLint, intrinsic metabolic clearance in the liver based on unbound drug concentrations; fu, unbound drug fraction in plasma; MPPGL, microsomal protein per gram of liver.
In pediatric population pharmacokinetic (PopPK) models, the net influence of ontogeny of system‐specific parameters on CLp of a drug is described using empirical covariate models derived from clinical data (i.e., concentration‐time profiles). Although these covariate models can be directly used as the basis for pediatric dose adjustments,12, 13, 14 this approach requires clinical data obtained upon the administration of every single drug of interest in every pediatric subpopulation.
Semimechanistic or semiphysiological PopPK pediatric scaling approaches have been proposed to bridge the gap between PBPK and PopPK methodologies, allowing for accelerated development of pediatric PopPK models and subsequent dosing recommendations. One of these approaches relies on extrapolations of PopPK covariate models between drugs that share an elimination pathway.15, 16, 17, 18, 19 Because PopPK covariate models are the basis of dose recommendations, this approach would also allow for extrapolation of pediatric dosing recommendations from a drug of which the changes in clearance have been quantified to other drugs for which no pediatric pharmacokinetic (PK) studies have been undertaken provided these drugs share the same elimination pathway. To date, proofs of concept for this method have been published for a limited number of drugs eliminated through glucuronidation or renal excretion.15, 16, 17, 18, 19 However, as illustrated in Figure 2, with this method, the between drug differences in CLp of drugs sharing the same elimination pathway are solely accounted for by the absolute value of the scaled CLp (e.g., adult CLp values), whereas CLp ontogeny is assumed to be purely system‐specific and, therefore, drug independent. This assumption is challenged by the fact that CLp of drugs with different properties might be impacted differently by the various ontogenic changes in system‐specific parameters (Figure 1).
Figure 2.

Illustration of between‐drug extrapolation of pediatric covariate functions to scale hepatic plasma clearance (CLp) from adults to pediatric patients for drugs eliminated by the same isoenzyme. The black dot in both graphs shows the adult hepatic CLp value for the model drug (“true” adult CLp_M) and the test drug (“true” adult CLp_T). The solid black line represents the change in CLp of the model drug throughout the pediatric age range, which is described by a pediatric covariate function based, in this example, on bodyweight (CLp ontogeny_M (bodyweight)). The dashed black line represents the scaled pediatric CLp predicted for the test drug by between‐drug extrapolation of the pediatric covariate function obtained for the model drug (CLp ontogeny_M (bodyweight)).
Therefore, the aim of the current study is to identify the conditions for which between‐drug extrapolation of pediatric covariate functions for hepatic metabolic CLp consistently leads to accurate pathway‐specific CLp scaling from adults to children of various ages (absolute prediction error ≤30%). We developed a PBPK‐based simulation workflow utilizing mechanistic equations defining hepatic metabolism to systematically screen a wide parameter space for both system‐specific and drug‐specific variables impacting hepatic CLp by one specific isoenzyme. Additionally, we investigated the impact of multiple elimination pathways on the between‐drug extrapolation potential of pediatric covariate functions for hepatic metabolic CLp. This allowed us to define a decision tree to identify the conditions leading to consistently accurate pediatric CLp scaling using between‐drug extrapolations of pathway‐specific pediatric covariate models.
METHODS
Model drug and test drug
Total (i.e., bound and unbound) hepatic metabolic plasma clearance will be referred to as CLp in this paper. We investigate the extrapolation potential of pediatric covariate functions scaling CLp from adults to pediatric patients between a model drug and a test drug both exclusively eliminated by the same hepatic isoenzyme. This method is illustrated in Figure 2. The impact of elimination by multiple isoenzymes is investigated as described under the section “Multiple elimination pathways.”
Model drug and determination of the pediatric covariate function: A pediatric covariate function is developed to describe the ontogeny of CLp of what will be referred to as the model drug (M).
Test drug and between‐drug extrapolation of the pediatric covariate function: The pediatric covariate function developed based on the model drug (M) is used to scale CLp for what will be referred to as the test drug (T) from adults to various pediatric ages.
PBPK‐based simulation workflow
A four‐step PBPK‐based simulation workflow (Figure 3) was developed in R software20 version 3.3.1 with R studio interface version 0.99.902 following a similar approach as previously published.21 This workflow investigates the impact of two main variables on the accuracy of pediatric CLp predictions based on extrapolation of pediatric covariate functions from a model drug to a test drug. These variables are the drug‐specific parameters of both the model drug and test drug and the ontogeny of the system‐specific parameters (Figure 1). This investigation was undertaken for 15 different elimination pathways. These elimination pathways correspond to elimination by the following isoenzymes: CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C18_19, CYP2D6, CYP2E1, CYP3A4, UGT1A1, UGT1A4, UGT1A6, UGT1A9, UGT2B7, and SULT1A1. Ultimately, conditions for which extrapolations consistently led to accurate CLp predictions were identified.
Figure 3.
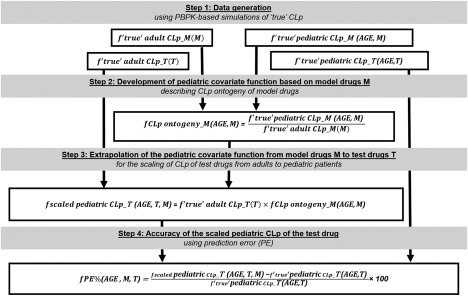
Physiologically based pharmacokinetic (PBPK)‐based simulation workflow used to investigate the between‐drug extrapolation potential of pediatric covariate models when scaling total plasma clearance (CLp) from adults to pediatric patients. The model drug is denoted with M and the test drug with T. AGE stands for pediatric postnatal age. All steps are performed for model drugs and test drugs binding to the same plasma proteins and eliminated by the same isoenzyme and repeated for each of the 15 isoenzymes and each of the 2 binding plasma proteins investigated.
Variables in the PBPK‐based simulation workflow
For a systematic investigation of the impact of each variable, we chose a factorial design (i.e., a global sensitivity analysis approach) with a wide yet realistic parameter space for each individual variable. For this purpose, continuous variables were transformed into categorical variables using a number of intermediate values within the defined parameter space allowing for the generation of single point estimates of the different functions of the PBPK‐based simulation workflow. To enable the computation across a wide parameter space and interpretability of the results, variability and uncertainty in both demographics and model parameters were not accounted for.
-
Ontogeny of system‐specific parameters:
To unravel the impact of ontogeny of the system‐specific parameters (see Figure 1) on the extrapolation potential of pediatric covariate functions between drugs sharing the same elimination pathway, simulations were performed for eight typical individuals for whom demographic characteristics and system‐specific parameters are specified in Table 1. The PBPK model parameters for typical individuals were computed as average values of men and women. Ontogeny functions for each system‐specific model parameter were taken from the Simcyp software (Simcyp, Sheffield, UK) V15.R1 library, except for the isoenzyme ontogeny of SULT1A1. For SULT1A1, maturity was taken to have been reached at birth,22 and, therefore, isoenzyme ontogeny of SULT1A1, defined as the pediatric to adult ratio of microsomal intrinsic clearance, was set to 1. More information can be found in Supplementary Material S1.
-
Drug‐specific properties of the model drugs and test drugs:
To unravel the impact of drug‐specific properties on the between‐drug extrapolation potential of pediatric covariate functions, a total of 7,560 hypothetical drugs with unique combinations of drug‐specific properties were generated and used as model drugs, as well as test drugs. This set of hypothetical drugs was generated using all possible combinations of different values of the following three drug‐specific properties: adult unbound fraction in plasma (range: 0.01–1; n = 10), blood‐to‐plasma partition coefficient or Kp (range: 0.35–40; n = 9), and adult total unbound intrinsic clearance value of one microgram of liver microsomes or adult CLint, mic, total (range: 0.56·10−6 to 0.209.10−3 L.min−1.mg−1 microsomal protein; n = 42). The hypothetical drugs were assumed to be exclusively bound to either human serum albumin (HSA) or alpha‐1 acid glycoprotein (AAG). More details can be found in Supplementary Material S1.
Table 1.
Demographic characteristics of the eight typical individuals implemented in the PBPK‐based simulation workflow and their corresponding system‐specific parameters values
| Demographic values | ||||||||
|---|---|---|---|---|---|---|---|---|
| Age | 1 day | 1 month | 6 months | 1 year | 2 years | 5 years | 15 years | 25 years |
| Bodyweight, kg | 3.45 | 4.30 | 7.55 | 9.90 | 12.35 | 18.25 | 54.25 | 72.65 |
| Height, cm | 49.75 | 54.25 | 66.00 | 74.75 | 86.00 | 108.25 | 166.00 | 172.30 |
| BSA, m2 | 0.22 | 0.26 | 0.38 | 0.46 | 0.55 | 0.74 | 1.60 | 1.86 |
| System‐specific parameters | ||||||||
| Qh, L/h | 6.55 | 7.83 | 12.95 | 17.65 | 24.65 | 41.14 | 89.75 | 87.92 |
| HSA, g/L | 35.78 | 39.94 | 42.07 | 42.90 | 43.73 | 44.82 | 44.68 | 43.94 |
| AAG, g/L | 0.2678 | 0.5497 | 0.6774 | 0.7172 | 0.7512 | 0.7877 | 0.6711 | 0.6847 |
| Hematocrit, % | 51.93 | 38.14 | 35.11 | 35.78 | 36.79 | 38.29 | 40.01 | 40.74 |
| Liver size, g | 133 | 159 | 249 | 313 | 385 | 544 | 1351 | 1614 |
| MPPGL | 25.53 | 25.60 | 25.99 | 26.45 | 27.36 | 29.97 | 36.80 | 39.79 |
| IO CYP1A2, % | 24 | 35 | 118 | 150 | 164 | 161 | 126 | 100 |
| IO CYP2A6, % | 2.10−9 | 0.48 | 99 | 100 | 100 | 100 | 100 | 100 |
| IO CYP2B6, % | 15 | 19 | 34 | 47 | 62 | 83 | 99 | 100 |
| IO CYP2C8, % | 38 | 86 | 97 | 99 | 99 | 100 | 100 | 100 |
| IO CYP2C9, % | 40 | 74 | 87 | 90 | 92 | 100 | 100 | 100 |
| IO CYP2C18–19, % | 30 | 33 | 84 | 95 | 97 | 98 | 100 | 100 |
| IO CYP2D6, % | 6 | 47 | 84 | 91 | 95 | 98 | 100 | 100 |
| IO CYP2E1, % | 10 | 37 | 59 | 67 | 74 | 82 | 88 | 100 |
| IO CYP3A4, % | 11 | 13 | 48 | 78 | 96 | 104 | 106 | 100 |
| IO UGT1A1, % | 0.2 | 23 | 98 | 104 | 100 | 100 | 100 | 100 |
| IO UGT1A4, % | 74 | 74 | 74 | 75 | 77 | 81 | 97 | 100 |
| IO UGT1A6, % | 15 | 30 | 63 | 76 | 87 | 95 | 100 | 100 |
| IO UGT1A9, % | 9 | 12 | 34 | 52 | 71 | 90 | 100 | 100 |
| IO UGT2B7, % | 8 | 9 | 11 | 13 | 18 | 32 | 79 | 100 |
| IO SULT1A1, % | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
AAG, alpha‐1 acid glycoprotein; BSA, body surface area; HSA, human serum albumin; MPPGL, milligram microsomal protein per gram of liver; IO, isoenzyme ontogeny expressed as percentage of adult microsomal intrinsic clearance; Qh, hepatic blood flow.
Steps of the PBPK‐based simulation workflow
Between‐drug extrapolation of covariate models was studied for model and test drugs sharing the same elimination pathway and binding to the same plasma protein. For each of the 15 elimination pathways investigated and each type of binding plasma protein, the following steps were undertaken, as also illustrated in Figure 3:
-
Step 1: Data generation
Using the dispersion model (Eqs. (1), (2), (3), (4), (5), (6)), PBPK‐based simulations of “true” CLp were performed for all hypothetical model drugs M and test drugs T in adults (“true” adult CLp_M and “true” adult CLp_T, respectively) as well as in each investigated pediatric age (“true” pediatric CLp_M and “true” pediatric CLp_T, respectively).(1) (2) (3) (4) (5) (6)
In these equations, is the overall total (i.e., bound and unbound) hepatic plasma clearance, CLB is the total whole blood clearance, B:P is the blood to plasma ratio, Qh is the hepatic blood flow, ER is the hepatic extraction ratio, fu is the unbound drug fraction in plasma, CLint is the total hepatic intrinsic clearance, RN is the efficiency number, and DN is the axial dispersion number. For the axial dispersion number (DN), a value of 0.17 was used.23 See Supplementary Material S1 for more details.
-
Step 2: Development of pediatric covariate function based on model drugs M
The pediatric covariate function (f CLp ontogeny_M) describes the ontogenic changes in “true” CLp from adults to pediatric patients and is derived from the model drug M. For each model drug M and investigated pediatric postnatal age (AGE), single point estimates of f CLp ontogeny_M were computed (see equation in Figure 3).
-
Step 3: Extrapolation of the pediatric covariate function from model drugs M to test drugs T
Analogous to Figure 2, scaled pediatric CLp of the test drugs (f scaled pediatric CLp_T) was computed by scaling the “true” adult CLp of each test drug T (“true” adult CLp_T) to each pediatric age. This scaling was performed by extrapolating the pediatric covariate function (f CLp ontogeny_M) from all model drugs to each test drug following the equation in Figure 3. This led, for each pediatric age and test drug, to as many scaled pediatric CLps as the number of model drugs.
-
Step 4: Accuracy of the scaled pediatric CLp of the test drug
The prediction errors (PEs) of scaled pediatric CLps obtained in step 3 was calculated by comparing, for each test drug and each age, the scaled pediatric CLps with the “true” CLp value obtained in step 1. This led, for each pediatric age and test drug, to as many PEs as the number of model drugs. Predictions were considered to be accurate when the absolute PE was 30% or lower.
Conditions leading to accurate CLp predictions
First, for the diversity of investigated ages and isoenzymes, trends in PEs with all drug properties of the model and the test drugs were separately assessed. These drug properties were the type of binding plasma protein, Kp, and unbound fraction in plasma B:P, CLint, and extraction ratio in adults, as well as the difference in the latter parameters between the model drug and test drug.
In order to define scenarios consistently leading to accurate CLp scaling from adults to the different pediatric ages using between‐drug extrapolation of pediatric covariate functions, drug properties allowing to best discriminate between accurate and inaccurate scaled pediatric CLp were identified by a hierarchical tree analysis (see Supplementary Material S1).24
All model‐test drug combinations were grouped into scenarios based on the most discriminative drug properties. Within each of these scenarios, multiple model‐test drug combinations are included, and for all combinations, both model and test drugs are eliminated by the same isoenzyme and binding to the same plasma protein. Overall accuracy of the test drug CLp predictions for each model‐test drug scenario was summarized per pediatric age group as follows:
Model‐test drug scenario systematically leading to accurate CLp scaling of the test drug: All CLp predictions within the defined scenario are accurate (absolute PE ≤30%).
Model‐test drug scenario not systematically leading to accurate CLp scaling of the test drug: At least one of the CLp predictions within the defined scenario is inaccurate (absolute PE >30%).
Multiple elimination pathways
Because many drugs are metabolized by multiple isoenzymes, we also evaluated the situation in which the model drug and/or the test drug are metabolized by two isoenzymes, namely IA and INA. The IA is the isoenzyme representing the elimination pathway accounted for by the covariate function, whereas INA is the isoenzyme representing the elimination pathway not accounted for by the covariate function. In order to include model drugs and/or test drugs undergoing metabolism through multiple hepatic isoenzymes, adaptations of the PBPK‐based simulation workflow earlier described were performed. Details on these alterations can be found in Supplementary Material S1.
RESULTS
Results for scenarios in which model and test drugs are metabolized by one isoenzyme
Visual inspection of the PEs revealed trends with age, type of metabolizing isoenzyme, the type of binding plasma protein, and extraction ratio in adults of the model drug and test drug. Figure 4 displays these trends in PEs for all hypothetical drugs that are exclusively metabolized by one isoenzyme and that are either exclusively binding to HSA (Figure 4 a) or to AAG (Figure 4 b). Results are categorized by age and by adult extraction ratio category of the model and test drugs, with increasing absolute PE values indicating increasing between‐drug differences in CLp ontogeny, with CLp standing for total (i.e., bound and unbound) hepatic metabolic plasma clearance. For each age, results are reported for the lowest, intermediate, and highest isoenzyme ontogeny values of the 15 isoenzymes included in the analysis.
Figure 4.

The prediction error (PE) of the total (bound and unbound) drug plasma clearance (CLp) predictions for scenarios in which model and test drugs are exclusively metabolized by one isoenzyme (fmA_adult = 100%) and exclusively binding to human serum albumin (a) or to alpha‐1 acid glycoprotein (b). The boxplots represent the minimum, firth quartile, median, third quartile, and maximum PE and are categorized by low (green), intermediate (blue), and high (pink) adult extraction ratio (ER) of the model drug and low (light color), intermediate (intermediate color), and high (dark color) adult extraction ratio of the test drug. For each age, the lowest, intermediate, and highest isoenzyme ontogeny values (percentage of adult CLint,mic) reported for the 15 isoenzymes are shown. The intermediate isoenzyme ontogeny was defined as the isoenzyme ontogeny value the closest to the mean of the lowest and highest isoenzyme ontogeny values for a specific age. Low, intermediate, and high extraction ratio correspond to extraction ratio ≤30%, 30% < extraction ratio ≤70% and extraction ratio >70%, respectively. The vertical solid black line indicates a PE of 0. The dotted black and dotted red lines indicate PE intervals of +/‐ 30% and +/‐ 50%, respectively. Note that the x‐axes are different for different ages.
Figure 4 shows similar trends in PEs between drugs binding to HSA and drugs binding to AAG, except in neonates of 1 day of age. However, for AAG bound drugs, the ranges in PE were either similar or higher than PE ranges for HSA bound drugs. For both HSA and AAG bound drugs, the range of absolute PE values tend to decrease with increasing age. Decreased isoenzyme ontogeny value tends to increase the range of the absolute PEs, with the highest absolute PEs being >1,000% in neonates of 1 day (for isoenzyme ontogeny ∼0%) and the highest absolute PE value in adolescents being only about 70% (for isoenzyme ontogeny = 79%).
Generally, within each age category and isoenzyme ontogeny category, the median PE can be seen to be closest to 0 when extrapolating pediatric covariate models between drugs of the same extraction ratio category, although even for these drugs the range in PE values shows a number of scenarios in which absolute PE values are higher than 30%. The absolute median PE was the highest when the model and test drugs belonged to extreme extraction ratio categories. Except for drugs binding to AAG in neonates of 1 day old, extrapolating pediatric covariate functions to a test drug of a lower extraction ratio category than the model drug systematically yields a positive median PE, indicating a bias toward overprediction of CLp of the test drug. The reverse trend is observed when extrapolating pediatric covariate functions to a test drug of a higher extraction ratio category than the model drug. These trends increase as the isoenzyme ontogeny decreases to values below 100%.
Overall, the hierarchical tree analysis revealed that the most discriminating drug properties for identifying systematically accurate CLp predictions across the pediatric age range were the extraction ratio of the test drug and/or the model drug, and the difference in extraction ratio and in unbound fraction in plasma between model drug and test drug (extraction ratio and unbound fraction defined in adults, see Supplementary Material S1). The additional influence of the difference in unbound fraction between the model drug and test drug on the PEs was not observed upon visual inspection of the trends in PEs, likely because trends in PE with unbound fraction are smaller than trends with extraction ratio and they depend on the extraction ratio of the model drug and test drug. As the extrapolations to the test drug require the use of a pediatric covariate model that is defined for the model drug, scenarios leading to accurate between‐drug extrapolations were defined using the extraction ratio of the model drug.
These most discriminative drug properties (i.e., extraction ratio of the model drug, and the difference in extraction ratio and in unbound fraction between the model drug and test drug) were used to define model‐test drug scenarios systematically leading to accurate CLp predictions in different age ranges, as displayed in the first row of Figure 5 a,b and of the figures in Supplementary Material S2 and S3. The first row in Figure 5 presents the results for drugs that are exclusively metabolized by CYP3A4 and that bind to HSA (Figure 5 a) or AAG (Figure 5 b), whereas Supplementary Material S2 and S3 show the results for substrates of all investigated isoenzymes binding to HSA and AAG, respectively. Although the results differ between the different isoenzymes and plasma proteins, they do reveal similar trends.
Figure 5.
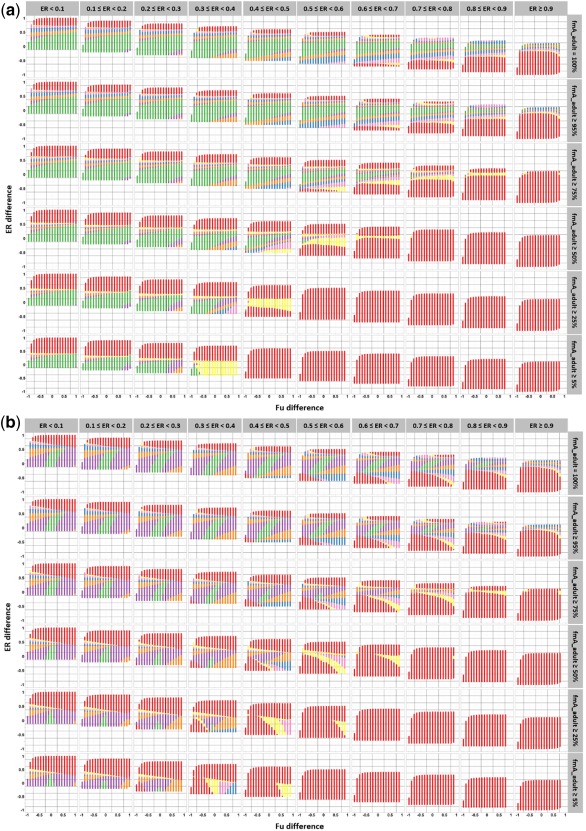
Illustration of model‐test drug scenarios that lead to accurate pathway‐specific drug plasma clearance (CLp) predictions for a test drug after between‐drug extrapolation of a pathway‐specific pediatric covariate function. Results are presented for drugs that are metabolized by CYP3A4 and that bind to human serum albumin (a) or alpha‐1 acid glycoprotein (b). Each column correspond to a range of extraction ratio (ER) values for the model drug in adults and each row to a specific range of fraction of drug (model and test drug) that is metabolized by CYP3A4 in adults. For each graph, the y‐axis represents the difference in extraction ratio between the test drug and the model drug (extraction ratio test drug – extraction ratio model drug) in adults, and the x‐axis represents the difference in unbound fraction in plasma between the test drugs and the model drug in adults (unbound fraction test drug – unbound fraction model drug). Each dot represents a model‐test drug scenario, including multiple model‐test drug combinations. A color code is used to indicate systematically accurate CLp predictions for all model‐test drug combinations within a model‐test drug scenario, for children ≥5 years (yellow), ≥2 years (pink), ≥1 year (blue), ≥6 months (orange), ≥1 month (purple), and ≥1 day term neonates (green). Red dots indicate model‐test drug scenarios leading to inaccurate predictions in children older than 5 years for at least one model‐test drug combination within the model‐test drug scenario. As an example, systematically accurate CLp scaling in children of 6 months and older is represented by the combination of the green, purple, and orange dots.
The between‐drug extrapolation potential of pathway‐specific pediatric covariate functions generally decreases with decreasing age, with patterns in model‐test drug scenarios systematically leading to accurately scaled pediatric CLp being highly dependent on both ontogeny of the system‐specific parameters and drug properties. For all ages, the between‐drug extrapolation potential increases with decreasing extraction ratio values of the model drug, with this effect being most pronounced in younger children (see Supplementary Material S2 and S3). Moreover, the extrapolation potential increases when the difference in extraction ratio between the model and test drugs decreases. Regarding the plasma protein binding, it can be seen that between‐drug extrapolation of pathway‐specific pediatric covariate functions generally yields more accurately scaled pediatric CLp for drugs binding to HSA compared with drugs binding to AAG. Additionally, the difference in plasma protein binding between the model and test drugs was found to mostly impact the method applicability in infants of 1 month or younger.
Results for model drugs and/or test drugs metabolized by several isoenzymes
When the model drug and/or the test drug are metabolized by two isoenzymes, the PE increases or decreases, as compared with drugs eliminated by one isoenzyme depending on the ontogeny of the isoenzyme representing the elimination pathway not accounted for by the covariate model (INA) and the fmA_adult values of the test drug and model drug. For the specific case in which the isoenzyme representing the elimination pathway accounted for by the covariate model (IA) and INA have similar ontogeny, the PEs are similar to the PE for drugs exclusively metabolized by one isoenzyme IA.
For model drugs and/or test drugs metabolized by several isoenzymes, the most discriminating drug properties to identify systematically accurate CLp predictions were the same as those found when both model and test drugs are metabolized by one unique isoenzyme. However, the larger the contribution of alternative metabolic pathways to the overall CLp in adults the lower the extrapolation potential of the pathway‐specific covariate function will be, as can be seen in the bottom rows of Figure 5 and Supplementary Material S2 and S3.
DISCUSSION
In this work, we believe for the first time, we systematically investigated the applicability of between‐drug extrapolation of pathway‐specific pediatric covariate functions for CLp of drugs undergoing hepatic metabolism in which CLp stands for total (i.e., bound and unbound) hepatic metabolic plasma clearance. Our results show that pediatric changes in CLp of drugs that are eliminated by the same hepatic elimination pathway will not always follow similar patterns and, therefore, in specific cases, these drugs will require a different pediatric PK‐based dose adjustment.
The CLp ontogeny of a specific elimination pathway was found to mainly depend on the following drug properties: the type of binding plasma protein, the adult unbound fraction, and extraction ratio of the drugs, and, in case of multiple elimination pathways, the number and type of isoenzymes responsible for the drug metabolism. This finding disproves the often implicitly made assumption that the ontogeny of CLp is drug independent.25, 26 Additionally, it highlights the importance of ontogeny processes other than isoenzyme ontogeny alone on CLp scaling from adults to children for drugs undergoing hepatic metabolism (Figure 1). Therefore, the identified drug properties should be taken into account when extrapolating pathway‐specific pediatric covariate functions between drugs. Figure 5 and Supplementary Material S2 and S3 were developed to guide the selection of scenarios that will systematically lead to accurate between‐drug extrapolation of pediatric covariate functions. These guides can also be interpreted as defining scenarios for which between‐drug extrapolation of pediatric covariate functions will approximate PBPK‐based predictions with an accepted error of +/‐ 30%. In these scenarios, the use of this semimechanistic scaling method can be encouraged to expedite pediatric CLp scaling and the development of dosing recommendations.
To illustrate the use of these guides, we can take the example of midazolam (extraction ratio = 0.44,27 unbound fraction in plasma = 0.02228 with binding to HSA,9 and fm_A ∼93%28, 29), sildenafil (extraction ratio = 0.45,30 unbound fraction in plasma ∼0.04 with binding to HSA,31 and fm_A = 79%31), and simvastatin (extraction ratio = 0.97,32 unbound fraction in plasma = 0.0233 with binding to HSA,34 and fm_A = 92%29), all mainly metabolized by CYP3A4. Based on the results for CYP3A4, we can anticipate that the PopPK covariate model describing CYP3A4‐mediated clearance ontogeny from adults to neonates of 1 day of midazolam could be extrapolated to sildenafil, because the extraction ratio model = 0.44, unbound fraction difference = 0.018, extraction ratio difference = 0.01, and fm_A ≥75% corresponds to a green area in Figure 5 a. The same plot also shows that this extrapolation from midazolam to simvastatin cannot be performed because the extraction ratio model = 0.44, unbound fraction difference = −0.002, extraction ratio difference = 0.53, and fm_A ≥75% corresponds to a red area. Importantly, this scaling method is isoenzyme‐specific, and one should be careful not to overlook minor pathways in adults that can become major pathways in children when aiming at scaling total clearance.
It should be noted that model‐test drug scenarios leading to systematically accurate CLp scaling were defined according to very strict criteria. Each dot in Figure 5 and the accompanying Supplementary Material S2–S3 represents a scenario that summarizes the PEs for many different model‐test drug combinations. The CLp predictions were defined as not systematically accurate for a scenario if only one model‐test drug combination within this scenario and, for drugs metabolized by several isoenzymes, if at least one specific INA (isoenzyme not accounted for by the covariate model) led to scaled CLp deviating >30% from its “true” value. However, it is likely for CLp scaling to be accurate for a (large) number of model‐test drug combinations within such scenarios, but in these scenarios the accuracy of the scaling method cannot be easily predicted a priori without PBPK modeling.
Overall, our results show that the between‐drug extrapolation potential of pathway‐specific pediatric covariate functions for CLp increases when the extraction ratio of the model drug in adults decreases. The applicability of this method decreases with age and with a decreased adult fraction of the test drug and/or model drug being metabolized by the isoenzyme pathway accounted for by the covariate model (fmA_adult). Moreover, these trends increase with increased extraction ratio of the model drug. Plasma protein binding to AAG also limits the between‐drug extrapolation potential of pediatric covariate functions for CLp, especially in young children, which can be explained by the more pronounced ontogeny pattern of AAG compared to HSA for these ages.35
In this work, we discovered that, for drugs metabolized by several isoenzymes, the ontogeny of CLp due to one specific isoenzyme (IA) is influenced by the ontogeny of the other isoenzymes responsible for the drug clearance (INA). We also found that this impact increases with increased adult extraction ratio and decreased fmA_adult, with fmA_adult standing for adult fraction of drug CLp due to IA. This is due to changes in extraction ratio with age contributed by the ontogeny of all isoenzymes involved in the drug CLp,36 changes, which, in turn, modify each isoenzyme‐specific CLp ontogenies. This was shown by the reduced applicability of the between‐drug extrapolation potential of pathway‐specific covariate models for CLp with decreased fmA_adult. This is further supported by the increase of these trends with increased extraction ratio of the model drug (see Figure 5 and Supplementary Material S2 and S3).
Although this workflow only investigated drugs undergoing hepatic metabolic CLp, the results on multiple elimination routes also apply to drugs undergoing hepatic metabolism in combination with renal clearance, as renal clearance does not impact the hepatic extraction ratio or the ontogeny of hepatic CLp. For these drugs, fmA_adult should be interpreted as the fraction of the total hepatic metabolic CLp in adults due to the isoenzyme pathway accounted for by the covariate model (IA). Additionally, this workflow can be used to develop guidance on the need for PK‐based dose adjustments in clinical situations of reduced plasma protein binding, for instance, uremia and hypoalbuminemia, or drug‐drug interactions. In this situation, the model drug properties correspond to the drug properties in normal clinical situations and the test drug properties are changed to unbound fraction and extraction ratio values adapted to the calculated values for plasma protein binding in adults. If such a model‐test drug combination yields an accurate CLp prediction for the test drug, the pediatric dose can be derived by applying the same dose adjustment factor used for maintenance doses in adults with a similar clinical condition.
An important limitation of the PBPK‐based simulation workflow is that active influx or efflux of drugs into or out of hepatocytes by transporters is not included, thereby implicitly assuming this process to be passive. The impact of active drug transport in the membranes of hepatocytes on the between‐drug extrapolation of pathway‐specific pediatric covariate functions requires further investigation.
In conclusion, the developed PBPK‐based simulation workflow utilizing mechanistic equations defining hepatic metabolism allowed, we believe for the first time, to unravel the variables most impacting CLp ontogeny and to define scenarios for which extrapolation of pediatric covariate functions from one drug to another systematically leads to accurate isoenzyme‐specific CLp scaling from adults to pediatric patients for 15 hepatic isoenzymes.
Source of Funding
This study was supported by the Innovational Research Incentives Scheme (Vidi grant, June 2013) of the Netherlands Organization for Scientific Research (NWO) to Catherijne A. J. Knibbe (2013).
Conflict of Interest
Trevor Johnson is a paid employee of Simcyp Limited (a Certara company). Amin Rostami‐Hodjegan holds shares in Certara, a company focusing on model‐informed drug development and also has shares in Diurnal, which focuses on developing high quality products for the life‐long treatment of chronic endocrine conditions. As Editor‐in‐Chief for CPT: Pharmacometrics & Systems Pharmacology, Piet H. van der Graaf was not involved in the review or decision process for this article.
Author Contributions
E.A.M.C., E.H.J.K., and C.A.J.K. wrote the manuscript. E.A.M.C., E.H.J.K., H.Y., P.V., T.J., A.R‐H., D.T., P.H.vdG., M.D., and C.A.J.K. designed the research. E.A.M.C. performed the research. E.A.M.C. and H.Y. analyzed the data.
Supporting information
Supplementary Material S1 Methodology
Supplementary Material S2 Model‐test drug scenarios, for drugs binding to human serum albumin that lead to accurate pathway specific CLp predictions for a test drug after between‐drug extrapolation of a pathway specific pediatric covariate function.
Supplementary Material S3 Model‐test drug scenarios, for drugs binding to alpha‐1 acid glycoprotein that lead to accurate pathway specific CLp predictions for a test drug after between‐drug extrapolation of a pathway specific pediatric covariate function.
Supplementary Material S4 R model code
References
- 1. Manolis, E. , Rohou, S. , Hemmings, R. , Salmonson, T. , Karlsson, M. & Milligan, P.A. The role of modeling and simulation in development and registration of medicinal products: output from the EFPIA/EMA Modeling and Simulation Workshop. CPT Pharmacometrics Syst. Pharmacol. 2, e31 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brussee, J.M. et al Children in clinical trials: towards evidence‐based pediatric pharmacotherapy using pharmacokinetic‐pharmacodynamic modeling. Expert Rev. Clin. Pharmacol. 9, 1235–1244 (2016). [DOI] [PubMed] [Google Scholar]
- 3. US Department of Health and Human Services, Food and Drug Administration Center for Drug Evaluation and Research (CDER) . General considerations for pediatric pharmacokinetic studies for drugs and biological products. <https://www.fda.gov/ohrms/dockets/ac/03/briefing/3927B1_04_GFI-Pharmacokinetic Guidance.pdf> (1998). Accessed 27 September 2017.
- 4. Zisowsky, J. , Krause, A. & Dingemanse, J. Drug development for pediatric populations: regulatory aspects. Pharmaceutics 2, 364–388 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Espié, P. , Tytgat, D. , Sargentini‐Maier, M.L. , Poggesi, I. & Watelet, J.B. Physiologically based pharmacokinetics (PBPK). Drug Metab. Rev. 41, 391–407 (2009). [DOI] [PubMed] [Google Scholar]
- 6. Maharaj, A.R. , Barrett, J.S. & Edginton, A.N. A workflow example of PBPK modeling to support pediatric research and development: case study with lorazepam. AAPS J. 15, 455–464 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maharaj, A.R. & Edginton, A.N. Physiologically based pharmacokinetic modeling and simulation in pediatric drug development. CPT Pharmacometrics Syst. Pharmacol. 3, 1–13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barrett, J.S. , Della Casa Alberighi, O. , Läer, S. & Meibohm, B. Physiologically based pharmacokinetic (PBPK) modeling in children. Clin. Pharmacol. Ther. 92, 40–49 (2012). [DOI] [PubMed] [Google Scholar]
- 9. Johnson, T.N. , Rostami‐Hodjegan, A. & Tucker, G.T. Prediction of the clearance of eleven drugs and associated variability in neonates, infants and children. Clin. Pharmacokinet. 45, 931–956 (2006). [DOI] [PubMed] [Google Scholar]
- 10. Björkman, S. Prediction of drug disposition in infants and children by means of physiologically based pharmacokinetic (PBPK) modelling: theophylline and midazolam as model drugs. Br. J. Clin. Pharmacol. 59, 691–704 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Johnson, T.N. & Rostami‐Hodjegan, A. Resurgence in the use of physiologically based pharmacokinetic models in pediatric clinical pharmacology: parallel shift in incorporating the knowledge of biological elements and increased applicability to drug development and clinical practice. Pediatr. Anesth. 21, 291–301 (2011). [DOI] [PubMed] [Google Scholar]
- 12. Joerger, M. Covariate pharmacokinetic model building in oncology and its potential clinical relevance. AAPS J. 14, 119–132 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Patel, K. & Kirkpatrick, C.M. Pharmacokinetic concepts revisited–basic and applied. Curr. Pharm. Biotechnol. 12, 1983–1990 (2011). [DOI] [PubMed] [Google Scholar]
- 14. Knibbe, C.A. , Tibboel, D. , de Wildt, S.N. , de Hoog, M. , Tjoeng, M.M. & Danhof, M. [Individualized dosing guidelines for children]. Ned. Tijdschr. Geneeskd. 157, A4214 (2013). [PubMed] [Google Scholar]
- 15. Krekels, E.H. et al From pediatric covariate model to semiphysiological function for maturation: part I–extrapolation of a covariate model from morphine to Zidovudine. CPT Pharmacometrics Syst. Pharmacol. 1, e9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krekels, E.H. et al From pediatric covariate model to semiphysiological function for maturation: part II–sensitivity to physiological and physicochemical properties. CPT Pharmacometrics Syst. Pharmacol. 1, e10 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao, W. , Biran, V. & Jacqz‐Aigrain, E. Amikacin maturation model as a marker of renal maturation to predict glomerular filtration rate and vancomycin clearance in neonates. Clin. Pharmacokinet. 52, 1127–1134 (2013). [DOI] [PubMed] [Google Scholar]
- 18. De Cock, R.F. et al Simultaneous pharmacokinetic modeling of gentamicin, tobramycin and vancomycin clearance from neonates to adults: towards a semi‐physiological function for maturation in glomerular filtration. Pharm. Res. 31, 2643–2654 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. De Cock, R.F. et al A neonatal amikacin covariate model can be used to predict ontogeny of other drugs eliminated through glomerular filtration in neonates. Pharm. Res. 31, 754–767 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. R Development Core Team . R: A language and environment for statistical computing version 3.4.3. <https://cran.r-project.org/doc/manuals/fullrefman.pdf> (2017).
- 21. Calvier, E.A. et al Allometric scaling of clearance in paediatric patients: when does the magic of 0.75 fade? Clin. Pharmacokinet. 56, 273–285 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hines, R.N. The ontogeny of drug metabolism enzymes and implications for adverse drug events. Pharmacol. Ther. 118, 250–267 (2008). [DOI] [PubMed] [Google Scholar]
- 23. Naritomi, Y. , Terashita, S. , Kimura, S. , Suzuki, A. , Kagayama, A. & Sugiyama, Y. Prediction of human hepatic clearance from in vivo animal experiments and in vitro metabolic studies with liver microsomes from animals and humans. Drug Metab. Dispos. 29, 1316–1324 (2001). [PubMed] [Google Scholar]
- 24. Izenman, A.J. Cluster analysis In: Modern Multivariate Statistical Techniques. Regression, Classification, and Manifold Learning. 407–462 (Springer‐Verlag, New York, NY: 2013). [Google Scholar]
- 25. Fernandez, E. , Perez, R. , Hernandez, A. , Tejada, P. , Arteta, M. & Ramos, J.T. Factors and mechanisms for pharmacokinetic differences between pediatric population and adults. Pharmaceutics 3, 53–72 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krekels, E.H. , Danhof, M. , Tibboel, D. & Knibbe, C.A. Ontogeny of hepatic glucuronidation; methods and results. Curr. Drug Metab. 13, 728–743 (2012). [DOI] [PubMed] [Google Scholar]
- 27. Hohmann, N. , Kocheise, F. , Carls, A. , Burhenne, J. , Haefeli, W.E. & Mikus, G. Midazolam microdose to determine systemic and pre‐systemic metabolic CYP3A activity in humans. Br. J. Clin. Pharmacol. 79, 278–285 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lown, K.S. et al The erythromycin breath test predicts the clearance of midazolam. Clin. Pharmacol. Ther. 57, 16–24 (1995). [DOI] [PubMed] [Google Scholar]
- 29. Venkatakrishnan, K. , Obach, R.S. & Rostami‐Hodjegan, A. Mechanism‐based inactivation of human cytochrome P450 enzymes: strategies for diagnosis and drug‐drug interaction risk assessment. Xenobiotica 37, 1225–1256 (2007). [DOI] [PubMed] [Google Scholar]
- 30. Zhao, P. et al Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin. Pharmacol. Ther. 89, 259–267 (2011). [DOI] [PubMed] [Google Scholar]
- 31. Mehrotra, N. , Gupta, M. , Kovar, A. & Meibohm, B. The role of pharmacokinetics and pharmacodynamics in phosphodiesterase‐5 inhibitor therapy. Int. J. Impot. Res. 19, 253–264 (2007). [DOI] [PubMed] [Google Scholar]
- 32. Fenneteau, F. , Poulin, P. & Nekka, F. Physiologically based predictions of the impact of inhibition of intestinal and hepatic metabolism on human pharmacokinetics of CYP3A substrates. J. Pharm. Sci. 99, 486–514 (2010). [DOI] [PubMed] [Google Scholar]
- 33. Vickers, S. , Duncan, C.A. , Chen, I.W. , Rosegay, A. & Duggan, D.E. Metabolic disposition studies on simvastatin, a cholesterol‐lowering prodrug. Drug Metab. Dispos. 18, 138–145 (1990). [PubMed] [Google Scholar]
- 34. Lennernäs, H. & Fager, G. Pharmacodynamics and pharmacokinetics of the HMG‐CoA reductase inhibitors. Similarities and differences. Clin. Pharmacokinet. 32, 403–425 (1997). [DOI] [PubMed] [Google Scholar]
- 35. McNamara, P.J. & Alcorn, J. Protein binding predictions in infants. AAPS PharmSci. 4, E4 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Salem, F. , Abduljalil, K. , Kamiyama, Y. & Rostami‐Hodjegan, A. Considering age variation when coining drugs as high versus low hepatic extraction ratio. Drug Metab. Dispos. 44, 1099–1102 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material S1 Methodology
Supplementary Material S2 Model‐test drug scenarios, for drugs binding to human serum albumin that lead to accurate pathway specific CLp predictions for a test drug after between‐drug extrapolation of a pathway specific pediatric covariate function.
Supplementary Material S3 Model‐test drug scenarios, for drugs binding to alpha‐1 acid glycoprotein that lead to accurate pathway specific CLp predictions for a test drug after between‐drug extrapolation of a pathway specific pediatric covariate function.
Supplementary Material S4 R model code