

Abstract
Rifampin is a pleiotropic inducer of multiple drug metabolizing enzymes and transporters. This work utilized a global approach to evaluate rifampin effects on conjugating enzyme gene expression with relevance to human xeno‐ and endo‐biotic metabolism. Primary human hepatocytes from 7 subjects were treated with rifampin (10 μmol/L, 24 hours). Standard methods for RNA‐seq library construction, EZBead preparation, and NextGen sequencing were used to measure UDP‐glucuronosyl transferase UGT, sulfonyltransferase SULT, N acetyltransferase NAT, and glutathione‐S‐transferase GST mRNA expression compared to vehicle control (0.01% MeOH). Rifampin‐induced (>1.25‐fold) mRNA expression of 13 clinically important phase II drug metabolizing genes and repressed (>1.25‐fold) the expression of 3 genes (P < .05). Rifampin‐induced miRNA expression changes correlated with mRNA changes and miRNAs were identified that may modulate conjugating enzyme expression. NAT2 gene expression was most strongly repressed (1.3‐fold) by rifampin while UGT1A4 and UGT1A1 genes were most strongly induced (7.9‐ and 4.8‐fold, respectively). Physiologically based pharmacokinetic modeling (PBPK) was used to simulate the clinical consequences of rifampin induction of CYP3A4‐ and UGT1A4‐mediated midazolam metabolism. Simulations evaluating isolated UGT1A4 induction predicted increased midazolam N‐glucuronide exposure (~4‐fold) with minimal reductions in parent midazolam exposure (~10%). Simulations accounting for simultaneous induction of both CYP3A4 and UGT1A4 predicted a ~10‐fold decrease in parent midazolam exposure with only a ~2‐fold decrease in midazolam N‐glucuronide metabolite exposure. These data reveal differential effects of rifampin on the human conjugating enzyme transcriptome and potential associations with miRNAs that form the basis for future mechanistic studies to elucidate the interplay of conjugating enzyme regulatory elements.
Keywords: drug metabolizing enzyme induction, miRNA modulation of mRNA, PBPK modeling, phase 2 enzyme induction, rifampin miRNA induction, rifampin mRNA repression
Abbreviations
- CYP
cytochrome P450
- DMSO
dimethyl sulfoxide
- GEO
Gene Expression Omnibus
- GST
glutathione‐S‐transferase
- miRNA
microRNA
- NAPQI
N‐acetyl‐p‐benzoquinone imine
- NAT
N acetyltransferase
- PBPK
physiologically based pharmacokinetic
- PXR
pregnane X receptor
- SULT
sulfonyltransferase
- TPMT
thiopurine S‐methyltransferase
- UGT
UDP‐glucuronosyl transferase
1. INTRODUCTION
Rifampin induction of cytochrome P450 is an extensively studied drug–drug interaction mechanism resulting in a substantial list of clinically important interactions that can lead to reduced drug efficacy or increased toxicity.1, 2 In contrast, relatively less is known about rifampin induction of human conjugating enzymes including uridine diphosphate glucuronosyltransferases (UGTs), sulfotransferases (SULTs), N‐acetyltransferases (NATs), thiopurine S‐methyltransferase (TPMT) and glutathione S‐transferases (GSTs).3 Rifampin is widely recognized as a pleiotropic but specific inducer of drug metabolizing enzymes and transporters with effects mediated mainly through activation of pregnane X receptor (PXR).4 The genes regulated by PXR include those encoding for human conjugating enzyme families (UGTs, SULTs, NATs, and GSTs). Previous studies demonstrated rifampin induction of miRNAs and association with repression of P450 genes, suggesting the possibility of additional epigenetic mechanisms underlying rifampin drug–drug interactions.5, 6 Epigenetic modulation of conjugating enzymes by miRNAs has also been demonstrated.7, 8, 9, 10 MiRNAs generally are thought to negatively regulate gene expression and reduce downstream protein translation via imperfect complementary binding with the 3′‐untranslated region. However, relatively little is known about the combined effects of rifampin‐induced changes in hepatic miRNA expression on the downstream expression of conjugating enzymes.
The UGT superfamily of conjugating enzymes contains 5 subfamilies (UGT1, UGT2A, UGT2B, UGT3, and UGT8). Three of these subfamilies (UGT1, UGT2A, and UGT2B) prominently contribute to the metabolism of drugs, dietary substances, toxicants, and endogenous substrates with broad and overlapping substrate specificities. These 3 subfamilies are encoded by 10 genes to generate 19 isoforms in humans.11 The UGT1A family shares a single chromosomal locus (band 2q37) with the 9 different functional isoforms being generated via splicing of shared exons 2‐5 to an isoform‐specific exon 1. Similarly, the UGT2A subfamily members share exons 2‐6 with an isoform‐specific exon 1. Conversely, the UGT2B family is composed of 7 functional enzymes encoded by individual genes. Each UGT possesses a unique 5′‐upstream promoter region that controls its transcription as well as more distant enhancer regions containing transcription factor‐binding sites that further control constitutive and inducible UGT expression. A wide variety of tissue‐specific and ligand‐activated transcription factors modulate the induction of UGT genes including PXR.12 In addition, epigenetic UGT regulation by miRNAs has recently been identified as another factor that modulates UGT expression and response to environmental exposures.7, 8, 9, 10, 13, 14 Taken together, evaluating the influence of rifampin on UGT mRNA expression and association with miRNA changes may help to unravel the complex regulatory network governing UGT expression and activity.
The cytosolic SULT family of enzymes contribute to the metabolism of several exogenous and endogenous substrates, including the clinically used drugs acetaminophen, minoxidil, and ethinyl estradiol. The SULT family is comprised of 13 members within 3 families (SULT1, SULT2, and SULT4). SULT activity varies widely among individuals due in part to genetic polymorphisms and susceptibility to induction via nuclear receptor activation.15, 16, 17 For women taking ethinyl estradiol, rifampin induction of SULTs may cause therapeutic failure of the oral contraceptive drug.18 Despite the clinical importance of SULT‐mediated xenobiotic metabolism, data describing mechanisms regulating SULT induction are rather sparse.
NATs, another family of conjugating enzymes, contribute to human xenobiotic and endogenous substrate metabolism. Two NATs, NAT1 and NAT2, are thought to be of primary importance to drug metabolism. Polymorphisms exist in both NAT1 and NAT2 genes with well‐established functional consequences in phenotypic slow acetylators. For example, slow acetylators are more susceptible to drug‐induced toxicities from hydralazine and isoniazid. Isoniazid and rifampin are also commonly coadministered for the treatment of latent tuberculosis, raising the potential for drug–drug interactions. Slow acetylators are also more prone to developing certain cancers.19 As a result, NAT modulation via small molecules and miRNAs has become a target of drug and biomarker development.20, 21 Considered together, understanding the rifampin‐induced changes in NAT expression and associated miRNAs may be of therapeutic and diagnostic value.
TPMT is the primary enzyme responsible for human metabolism of thiopurine drugs including azathioprine, thioguanine, and 6‐mercaptopurine. Genetic polymorphisms in TMPT can result in reduced enzyme activity leading to increased drug concentration and toxicities in certain patients. As a result, pharmacogenetics screening for TPMT deficiency is recommended prior to initiating thiopurine drug therapy. A previous report demonstrated no change in TPMT mRNA expression in human hepatocytes treated with rifampin3 but the potential influence of miRNAs has not been previously explored.
Human GSTs are a family of cytosolic enzymes that catalyze the transfer of the sulfhydryl group of glutathione to a large variety of electrophiles, including drug molecules such as busulfan and ethacrynic acid and reactive CYP450 metabolites such as N‐acetyl‐p‐benzoquinone imine (NAPQI). GST induction by drug molecules and dietary flavonoids has been previously reported22, 23 but the potential relationship with miRNA expression changes has not been evaluated.
The first aim of this report was to describe the effects of rifampin treatment on the regulation of hepatic conjugating enzyme mRNA expression and the relationships with regulation of miRNA expression in primary human hepatocytes. The second aim was to further assess the impact of rifampin modulation of UGT mRNA expression in human renal proximal tubule cells to evaluate the potential for tissue‐specific changes in enzyme regulation. Finally, based upon the in vitro and in silico study results, rifampin induction of UGT1A4‐mediated metabolism was selected for further evaluation via physiologically based pharmacokinetic (PBPK) modeling and simulation. The overarching goal of this work was to globally evaluate rifampin's effects on conjugating enzyme gene expression with relevance to human xeno‐ and endobiotic metabolism.
2. MATERIALS AND METHODS
2.1. Primary human hepatocytes and drug treatments
This study evaluated mRNA and miRNA expression data collected in a previously published human hepatocyte experiment.5, 6 In brief, freshly isolated human hepatocytes from 7 different donors were obtained from CellzDirect (Durham, NC) and were plated on 12‐well collagen‐coated plates cultured in Williams’ E medium without phenol red containing Primary Hepatocyte Maintenance Supplements (Life Technologies Corporation, Carlsbad, CA). Cultures from each donor were considered biological replicates (n = 7). All studies were performed within 72 and 120 hours following the time of hepatocyte isolation. Hepatocytes were treated with rifampin (10 μmol/L) or corresponding vehicle control (0.1% methanol) for 24 hours. The commercially obtained human hepatocytes were deidentified and specific demographic and/or clinical information were not available from the supplier.
2.2. MicroRNA expression profiling and bioinformatics analysis
Total RNA, including small RNAs, was isolated from the human hepatocytes following treatment, using the miRNeasy kit (Qiagen, Valencia, CA) with optional on‐column DNase treatment included in the purification. Expression of 754 miRNAs was measured using the Taqman OpenArray Human miRNA Panel with an NT Cycler (Applied Biosystems, Foster City, CA). Each subject's RNA was analyzed on 2 different OpenArrays to yield technical duplicates. Threshold cycles were set manually based on visual inspection of the real‐time amplification curves of each individual miRNA. Final analysis of technical duplicates was completed within a single project to ensure that the same adjusted threshold was applied to each pair. CT values were transformed to positive values (40‐CT) to ensure appropriate directionality of effect for the correlation analyses with RNA‐seq data. The remaining miRNA bioinformatics analyses mirrored that described in a previous analysis of this data set for evaluation of transport protein changes.5
2.3. Bioinformatic analysis of the RNA‐seq data
RNA‐seq library construction, EZBead preparation, and NextGen sequencing were performed using standard methods as described previously6 and used to measure UGT, SULT, NAT, TPMT, and GST mRNAs and compared to vehicle control (0.1% methanol). UGT1A genes were identified and quantified by unique exons 1 as exons 2‐5 are shared across this gene subfamily. The RNA‐Seq data analysis included quality assessment and sequence alignment prior to differential gene expression analysis as described previously.6 In brief, SOLiD Instrument Control Software and Experiment Tracking Software were used for read quality recalibration. Each sequence was scanned for low‐quality reads and any read length of less than 35 bases was discarded to effectively eliminate low‐quality reads while retaining high‐quality regions. BFAST was used as the primary sequence alignment algorithm employing a TopHat‐like strategy to align sequencing reads that crossed splicing junctions. Sequence reads were aligned to a filtering index to exclude sequences that were not of interest (eg, repeats and ribosomal RNA). Analyses were restricted to uniquely aligned sequences with 2 or less mismatches. Differentially expressed genes were identified using edgeR following exclusion of genes with less than 1 read per million mappable reads in more than half of samples. A generalized linear model considering the effects of individual donors as a random effect was used to identify gene expression levels directly affected by rifampin treatment. The P‐values were calculated for each gene and Benjaminin and Hochberg's algorithm was used to control the false discovery rate. Data reported in the primary tables and figures only for genes up‐ or downregulated >1.25‐fold by rifampin and P < .05. Clustering of mRNA expression changes and hepatocyte donors depicted in dendrograms were determined, using Euclidian distances and the complete linkage clustering method. Data visualization and hierarchical cluster analysis were performed with R software (build 3.2.3) and R Studio (v. 0.99.491), using the gplots and ggplots2 packages.
2.4. Rifampin treatment of human renal proximal tubule cells
Immortalized normal human proximal tubular kidney (NHPTK) cells24 were maintained in REGM media (Lonza, Basel, Switzerland) supplemented with 10% fetal bovine serum (Hyclone, GE Healthcare Bio‐Sciences, Pittsburgh, PA, USA) to maintain appropriate renal phenotype. NHPTK cells were maintained at 37°C in 95% humidified atmosphere (5% CO2). Studies were performed on cells in passages 6‐9 (corresponding to passage 3‐6 post‐immortalization) with individual passages considered a biological replicate (n = 4). NHPTK cells were treated with rifampin (10 μmol/L) or vehicle control (0.01% methanol) for 24 hours. Following treatment, ~1 million cells were washed in ice‐cold PBS, recovered via centrifugation, and the resultant pellet stored at −80°C pending RNA isolation.
2.5. Quantitative real‐time PCR of renal cells
Total RNA was extracted, using the miRNeasy Plus Mini Kit (Qiagen, Hilden, Germany) manufacturer protocol. UGT1A1, 1A6, 1A9, and 2B7, expression levels were determined via qRT‐PCR using GAPDH as an endogenous control. RNA quantification and quality were assessed, using optical spectrometry ratios (260/280 and 260/230 nm); mRNA was reverse transcribed to cDNA using the iScript Reverse Transcription Kit (Bio‐Rad, Hercules, CA) and diluted to obtain 25 ng/mL final cDNA concentration. Here, qRT‐PCR was performed on an Applied Biosystems Quantum Studio Viia 7 system with iTaq Universal SYBR Green (Bio‐Rad) and custom made primers (Life Technologies). The thermocycler parameters were 95°C for 30 seconds, then 40 cycles consisting of 95°C for 15 seconds followed by an annealing temperature for 30 seconds. Primer sequences and annealing temperatures are provided in Table S1. The delta–‐delta CT method was applied to determine the relative expression of each gene for rifampin and vehicle‐treated cells as previously described.5 The fold change in gene expression is represented as the mean ± SEM of the biological replicates (n = 4).
2.6. ChIP‐seq PXR‐binding site in silico analysis
The conjugating enzyme genes in this study were evaluated in silico for PXR‐binding sites using a publically available ChIP‐Seq database generated using HepG2 cells treated with vehicle (dimethyl sulfoxide, DMSO) or rifampin.25 The in silico ChIP‐Seq testing was conducted as described previously.5 Promoter regions were specified as ±2 kb based upon the coordinates of each transcription start site.
2.7. Accession numbers
Raw RNA‐seq data were made publicly available through the National Center for Biotechnology Information Gene Expression Omnibus (GEO) database and can be accessed, using GEO series accession number GSE799933 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE79933). OpenArray miRNA data were made publically available through the Indiana University Center for Computational Biology and Bioinformatics and can be accessed at http://compbio.iupui.edu/group/6/pages/rifampin.
2.8. Physiologically based modeling and simulation
The potential clinical impact of rifampin induction of UGT1A4‐mediated midazolam N‐glucuronidation was evaluated via PBPK modeling and simulation, using the SimCYP population‐based simulator (version 15.1; SimCYP Limited, Sheffield, UK). The midazolam SimCYP library file was modified to describe the clinically observed disposition of the UGT1A4‐mediated N‐glucuronide metabolite of midazolam. The midazolam N‐glucuronide compound file was linked to the parent compound and designated as “Primary Metabolite 1” within the software. Midazolam N‐glucuronide model development was accomplished, using clinical data previously acquired during the control phase of a healthy volunteer (n = 12) herbal product–drug interaction study.26 SimCYP model parameters are available in Table S2. Simulated pharmacokinetic outcomes within 30% of observed endpoints were deemed sufficiently accurate to proceed with interaction simulations. Drug–drug interactions resulting from coadministration of rifampin (600 mg/day orally for 3 days) with midazolam (5 mg orally on day 3) were simulated in 10 virtual trials of 10 healthy volunteers (ages 20–50 years, 50:50 male:female). Initial simulations evaluated only the impact of a fivefold increase in the hepatic UGT1A4 mediated metabolism of midazolam achieved semimechanistically using the “UGT scalar” option within SimCYP. Subsequent simulations incorporated the impact of a simultaneous fivefold increase in UGT1A4 metabolism along with mechanistic description of rifampin‐induced changes in CYP3A4 activity. The multiple dose rifampicin library file within the SimCYP model was used as provided to describe rifampin induction of CYP3A. The only exception was that the maximal fold induction (Indmax) for CYP3A4/5 was set to 22.7, the mean value observed in the current hepatocyte experiments. The directly observed fold mRNA change was utilized based upon the assertion that appropriate use of higher Indmax values improves model prediction accuracy of drug–drug interactions mediated via CYP3A4 induction.27
3. RESULTS
3.1. Rifampin regulation of hepatocyte drug metabolizing gene expression
The effects of rifampin on the hepatocyte expression of 53 phase II drug metabolizing enzyme enzymes was evaluated by differential mRNA expression. The effects of rifampin on selected enzymes (greater than 1.25‐fold change in mRNA expression and P < .05) are outlined in Table 1. Rifampin treatment significantly induced the expression of 13 genes and repressed the expression 3 genes. UGT1A5 expression was induced by ~twofold in agreement with previous reports of rifampin induction (3.5‐fold)28 in human hepatocytes. UGT family mRNA expression was isoform dependent and induced or not changed in response to rifampin treatment (Figure 1). However, multiple members of the UGT1A enzyme family consistently demonstrated induction in response to rifampin treatment across all 7 hepatocyte donors (Figure S1). Here, mRNA expression of 3 NAT isoforms appeared to be repressed in response to rifampin treatment with the remaining isoforms largely unchanged (Figure 1). SULT2A1 mRNA expression was induced while SULT1B1 and 1E1 expression was repressed (Figure 1). SULT1E1 and SULT1B1 mRNA expression were repressed to a similar extent, a result consistent with reported coregulation of these 2 genes.29 Changes in GST mRNA expression were modest with mixed induction and repression observed (Figure 1). Consistent with previous report,3 TPMT mRNA expression was unchanged by rifampin treatment. Observed changes in mRNA expression were largely consistent across biological replicates with the exception of strong induction of UGT2A1 and GSTO2 observed only in hepatocytes from donor 5 (Figure S1). UGT1A4 and UGT1A1 were most strongly induced suggesting the possibility of clinically relevant drug–drug interactions resulting from concomitant rifampin administration with drug substrates of these enzymes and prompting further evaluation via physiologically based modeling and simulation.
Table 1.
Effect of rifampin on the expression of selected conjugative drug metabolizing enzymes in human hepatocytes
| Gene | Fold changea | P‐value | FDR | Examples of substrates |
|---|---|---|---|---|
| Upregulated | ||||
| UGT1A4 | 4.93 | 9.85 × 10−113 | 1.14 × 10−109 | Amitriptyline, endoxifen, imipramine, midazolam |
| UGT1A1 | 3.19 | 2.82 × 10−70 | 1.64 × 10−67 | Acetaminophen, bilirubin, SN‐38, raltegravir |
| SULT2A1 | 2.44 | 2.78 × 10−44 | 9.59 × 10−42 | Androgens, dehydroepiandrosterone |
| UGT1A3 | 2.40 | 2.76 × 10−30 | 7.06 × 10−28 | Ezetimibe, naproxen, quercetin |
| UGT1A5 | 2.07 | 8.79 × 10−17 | 1.15 × 10−14 | 1‐hydroxypyrene, 4‐methylumbelliferone, scopoletin |
| GSTA1 | 1.92 | 1.51 × 10−21 | 2.72 × 10−19 | Busulfan, chlorambucil, thiotepa, androstene‐3,17‐dione |
| UGT2B4 | 1.89 | 3.54 × 10−24 | 7.30 × 10−22 | Lorazepam, bile acids, carvedilol |
| GSTA2 | 1.87 | 3.80 × 10−14 | 3.57 × 10−12 | Busulfan, dibenzopyrene diolepoxide |
| UGT2B11 | 1.76 | 1.27 × 10−3 | 1.32 × 10−2 | 12‐hydroxyeicosatetraenoic acid (HETE), 15‐HETE |
| GSTM2 | 1.69 | 6.63 × 10−3 | 4.71 × 10−2 | 1‐chloro‐2,4‐dinitrobenzene |
| GSTM1 | 1.68 | 1.65 × 10−7 | 6.08 × 10−6 | Artemisinin |
| SULT1A2 | 1.61 | 2.16 × 10−2 | 1.07 × 10−1 | Minoxidil, β‐napthol |
| UGT2B15 | 1.28 | 1.70 × 10−4 | 2.64 × 10−3 | Acetaminophen, (S)‐oxazepam, tolcapone |
| Downregulated | ||||
| SULT1B1 | −0.55 | 2.72 × 10−18 | 4.09 × 10−16 | 1‐napthol, 4‐nitrophenol, tri‐iodothyronine |
| SULT1E1 | −0.57 | 9.87 × 10−10 | 5.30 × 10−8 | Estrogen, naringenin, 4‐hydroxytamoxifen, curcumin |
| NAT2 | −0.75 | 5.06 × 10−5 | 9.41 × 10−4 | Dapsone, sulfasalazine, isoniazid |
FDR, false discovery rate.
Rifampin/control; reported only for genes up‐ or down‐regulated >1.25‐fold and P < 0.05.
Figure 1.
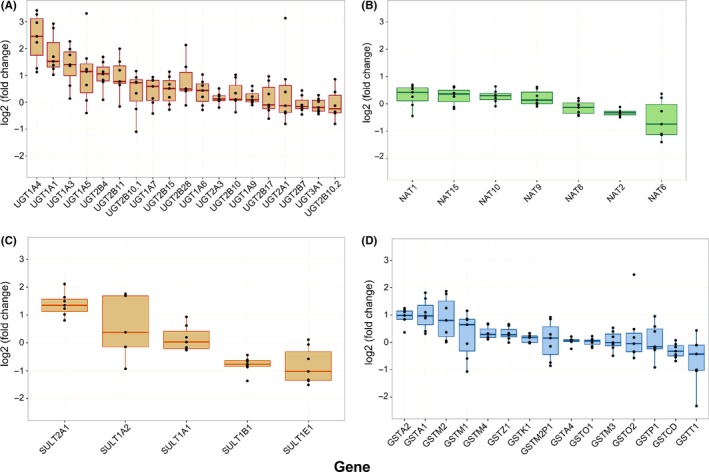
Individual changes in mRNA expression grouped by gene family; (A) UDP‐glucuronsyltransferases (UGTs), (B) N‐acetyltransferases (NATs), (C) sulfotransferases (SULTs), and (D) glutathione‐S‐transferases determined via RNA‐seq. Dots denote individual observed data points for each biological replicate (n = 7). Boxes denote observed median and interquartile range (IQR), whereas whiskers depict 1.5 times the IQR.
3.2. Conjugating enzymes and CYP450 isoforms appear to be coordinately regulated
Coordinate regulation of CYPs, UGTs, and transporters has been proposed as a defense mechanism providing protection against various chemical stressors.30 Correlation analysis suggests that several conjugating enzymes are coordinately regulated in response to rifampin treatment. As expected, multiple UGT genes displayed strong positive correlations in rifampin‐induced expression changes (Table 2). UGT1A4 and UGT2B15 expression changes positively correlated with changes in UGT2B4 expression. Our results are in agreement with the literature reported protein pairs of UGT1A4/2B4 (rs=.71, P < .0001, n = 82) and UGT2B4/2B15 (rs = .63, P < .0001, n = 83) from a recent meta‐analysis.31 Interestingly, expression changes of multiple GST enzymes were negatively correlated with changes in UGT1A and 2B expression (Table 2). Changes in UGT1A1 expression correlated positively with changes in CYP3A7 and CYP2B6 expression while UGT1A5 expression negatively correlated with changes in CYP3A5 and CYP2E1 (Table 3). Interestingly, significant correlations were not observed between the major CYPs (eg, CYP1A2, 2C8, 2C9, and 3A4/5) and UGTs (UGT1A1, 1A3, 1A4, 1A9, 2B7). These data provide further evidence for coordinate regulation of drug metabolizing enzymes in response to rifampin treatment.
Table 2.
Correlations of rifampin‐induced changes in the mRNA expression among the conjugating enzymes
| Gene 1 | Gene 2 | Correlation coefficienta | P‐value |
|---|---|---|---|
| Positive correlation | |||
| GSTA1 | SULT2A1 | .96 | .003 |
| UGT1A3 | UGT2B4 | .96 | .003 |
| GSTM2 | GSTM4 | .93 | .007 |
| SULT1B1 | UGT1A5 | .89 | .012 |
| UGT2B15 | UGT2B4 | .89 | .012 |
| SULT1E1 | UGT2B17 | .86 | .024 |
| SULT2A1 | UGT2B15 | .86 | .024 |
| UGT1A3 | UGT2B15 | .86 | .024 |
| UGT1A3 | UGT2B17 | .86 | .024 |
| GSTA1 | UGT2B15 | .82 | .034 |
| UGT1A3 | UGT1A5 | .82 | .034 |
| UGT1A4 | UGT2B4 | .82 | .034 |
| UGT1A5 | UGT2B17 | .82 | .034 |
| UGT1A9 | UGT2B11 | .82 | .034 |
| GSTA1 | SULT1A2 | .79 | .048 |
| NAT2 | SULT1A1 | .79 | .048 |
| Negative correlation | |||
| GSTT1 | UGT2B4 | −.96 | .003 |
| GSTT1 | UGT1A3 | −.93 | .007 |
| GSTA4 | UGT1A1 | −.86 | .024 |
| GSTM1 | UGT2B11 | −.86 | .024 |
| GSTA1 | GSTT1 | −.82 | .034 |
| GSTO1 | UGT1A4 | −.82 | .034 |
| GSTT1 | UGT2B15 | −.82 | .034 |
| GSTA1 | GSTM2 | −.79 | .048 |
| GSTZ1 | UGT1A4 | −.79 | .048 |
Spearman correlation as assumption of normal distribution could not be made. Data recovered via RNA‐seq.
Only significant correlations (P < 0.05 by Spearman correlation) are reported for those conjugating enzymes modulated by rifampin (>1.25 mean fold change, FDR <0.05).
Table 3.
CYP450 genes correlated with clinically relevant conjugating enzyme genes
| Gene 1 | Gene 2 | Correlation coefficienta | P‐value |
|---|---|---|---|
| Positive correlation | |||
| UGT1A1 | CYP3A7 | .96 | .003 |
| UGT1A5 | CYP1B1 | .86 | .024 |
| UGT1A9 | CYP1A1 | .86 | .024 |
| UGT2B4 | CYP2D6 | .86 | .024 |
| SULT1B1 | CYP1B1 | .82 | .034 |
| NAT2 | CYP2J2 | .82 | .034 |
| UGT1A1 | CYP2B6 | .79 | .048 |
| Negative correlation | |||
| GSTM1 | CYP4F2 | −.86 | .024 |
| UGT1A5 | CYP3A5 | −.79 | .048 |
| UGT1A5 | CYP2E1 | −.79 | .048 |
Spearman correlation as assumption of normal distribution could not be made. Data recovered via RNA‐seq. Only significant correlations reported (P < 0.05 by Spearman correlation).
3.3. Changes in miRNA and conjugating enzyme mRNA expression are highly correlated
Correlation analyses between changes in miRNA expression and mRNA changes were performed to identify miRNAs that may regulate conjugating enzyme expression. Typically, miRNAs are expected to downregulate target gene expression which would result in a negative correlation in this analysis. However, changes in miRNA expression were both positively and negatively correlated with conjugating enzyme mRNA expression (Table 4), similar to a previous report of miRNA and CYP450 correlations.6 Five of the miRNA/RNA pairs identified, using correlation analysis were also predicted via TargetScan (Release 7.1).32 hsa‐miR‐200b was negatively correlated with SULT1A1, SULT1A2, and NAT2 (Table 4); consistent with downregulation of those genes by the miRNA. Rifampin‐induced expression of hsa‐miR‐200b may underlie the observed repression of NAT2 (Figure 1B, Table 1). The hsa‐miR‐766 was previously predicted in silico to target the HNF4α nuclear receptor6 which may explain the observed correlations with changes in UGT1A3, UGT2B4, UGT2B15, GSTO1, and GSTT1 mRNA expression. The vast miRNA and transcription factor network that controls the expression of the various conjugating enzymes likely underlies the observed positive and negative correlations.
Table 4.
Conjugating enzyme‐miRNA pair correlations consistent with miRNA modulation of conjugating enzyme gene expression in response to rifampin treatment
| miRNA | Gene 2 | Correlation coefficienta | P‐value |
|---|---|---|---|
| Positive correlation | |||
| hsa‐miR‐638 | GSTT1 | .99 | .0004 |
| hsa‐miR‐766 | GSTT1 | .96 | .003 |
| hsa‐miR‐92a | UGT1A9 | .93 | .007 |
| hsa‐miR‐335 | GSTCD | .93 | .007 |
| hsa‐miR‐342‐3p | GSTA4 | .93 | .007 |
| hsa‐miR‐92a | UGT2B11 | .89 | .012 |
| hsa‐miR‐92a | UGT2B7 | .89 | .012 |
| hsa‐miR‐30dc | GSTM4b | .86 | .024 |
| hsa‐miR‐660 | GSTA4 | .86 | .024 |
| hsa‐miR‐320 | UGT3A1b | .86 | .024 |
| hsa‐miR‐616 | SULT1A1 | .86 | .024 |
| hsa‐miR‐200a | GSTZ1 | .86 | .024 |
| hsa‐miR‐200a | GSTO1 | .86 | .024 |
| hsa‐miR‐21 | GSTA4 | .86 | .024 |
| hsa‐miR‐886‐3p | GSTM2P1 | .82 | .034 |
| hsa‐miR‐92a | TPMTb | .82 | .034 |
| hsa‐miR‐320 | SULT1A1 | .82 | .034 |
| HSA‐MIR‐1180 | NAT15 | .82 | .034 |
| hsa‐miR‐361 | NAT1 | .79 | .048 |
| hsa‐miR‐92a | GSTCD | .79 | .048 |
| hsa‐miR‐30dc | GSTM2 | .79 | .048 |
| hsa‐miR‐660 | GSTT1 | .79 | .048 |
| hsa‐miR‐21 | GSTT1 | .79 | .048 |
| Negative correlation | |||
| hsa‐miR‐766 | UGT1A3 | −.96 | .003 |
| hsa‐miR‐148bc | GSTO1 | −.96 | .003 |
| hsa‐miR‐200bc | SULT1A1 | −.9 | .006 |
| hsa‐miR‐766 | UGT2B4b | −.93 | .007 |
| hsa‐miR‐18a | UGT2B17 | −.89 | .012 |
| hsa‐miR‐200bc | NAT2 | −.85 | .016 |
| hsa‐miR‐107 | NAT1 | −.86 | .024 |
| hsa‐miR‐660 | SULT1A2 | −.82 | .034 |
| hsa‐miR‐638 | GSTA1 | −.82 | .034 |
| hsa‐miR‐638 | UGT2B15 | −.82 | .034 |
| hsa‐miR‐25 | GSTZ1 | −.82 | .034 |
| hsa‐miR‐18a | UGT2B15 | −.82 | .034 |
| hsa‐miR‐23ac | UGT2B10b | −.82 | .034 |
| hsa‐miR‐744 | UGT2A3 | −.82 | .034 |
| hsa‐miR‐766 | GSTA1 | −.79 | .048 |
| hsa‐miR‐766 | UGT2B15 | −.79 | .048 |
| hsa‐miR‐218 | GSTZ1 | −.79 | .048 |
| hsa‐miR‐31 | GSTO1 | −.79 | .048 |
| hsa‐miR‐200bc | SULT1A2 | −.76 | .049 |
Spearman correlation as assumption of normal distribution could not be made. Only significant correlations reported (P < .05 by Spearman correlation). Only those genes and miRNAs significantly altered by rifampin treatment (up‐ or down‐regulated >1.25‐fold and P < .05) were included in the correlation analyses.
Denotes genes predicted via TargetScan to be targets of the correlated miRNA.
Correspond to the nonpredominantly expressed miRNA sequence. TPMT, thiopurine S‐methyltransferase.
3.4. Rifampin modulation of UGT gene expression is cell line specific and appears to be largely PXR dependent
Rifampin treatment did not significantly alter the expression of UGT1A1, 1A6, 1A9, or 2B7 in NHPTK cells. The observed fold changes ranged from 0.95 to 1.04, consistent with a previously reported lack of PXR expression in this cell line.5 In Silico ChIP‐Seq analysis of rifampin‐treated HepG2 cells found PXR peaks within the promoter regions of 4 clinically relevant conjugating enzyme genes: UGT1A4, UGT1A6, SULT2A1, and GSTO1. ChiP‐Seq analysis was in agreement with the RNA‐seq results demonstrating increased gene expression of UGT1A4 and SULT2A1, suggesting a PXR‐mediated induction process.
3.5. Physiologically based modeling and simulation suggests that UGT induction contributes to observed rifampin–drug interactions with dual CYP3A/UGT substrates
Simulated midazolam and midazolam N‐glucuronide concentration‐time profiles closely approximated clinically observed disposition and pharmacokinetic outcomes (Figure 2A and B, Table 5). Simulations evaluating the impact of rifampin‐induced UGT1A4 metabolism in isolation predicted markedly increased midazolam N‐glucuronide exposure (~fourfold) with minimal reductions in parent midazolam exposure (~10%), consistent with midazolam clearance‐mediated primarily by CYP3A4 (Figure 2C and D, Table 5). CYP3A4 induction only was predicted to reduce systemic midazolam exposure by nearly 10‐fold, in concordance with previous clinical and PBPK model‐predicted reports of hepatic CYP3A4 induction.27 Midazolam N‐glucuronide exposure was also predicted to be substantially reduced by rifampin, reflective of drastically reduced parent midazolam exposure leading to reduced substrate availability for UGT1A4‐mediated N‐glucuronidation. Simultaneous evaluation of UGT1A4 and CYP3A4 induction predicted >10‐fold mean reduction in plasma midazolam exposure but only ~2‐fold reduction in midazolam N‐glucuronide exposure, suggesting that the effects of limited substrate availability are partially overcome by simultaneous UGT1A4 induction.
Figure 2.
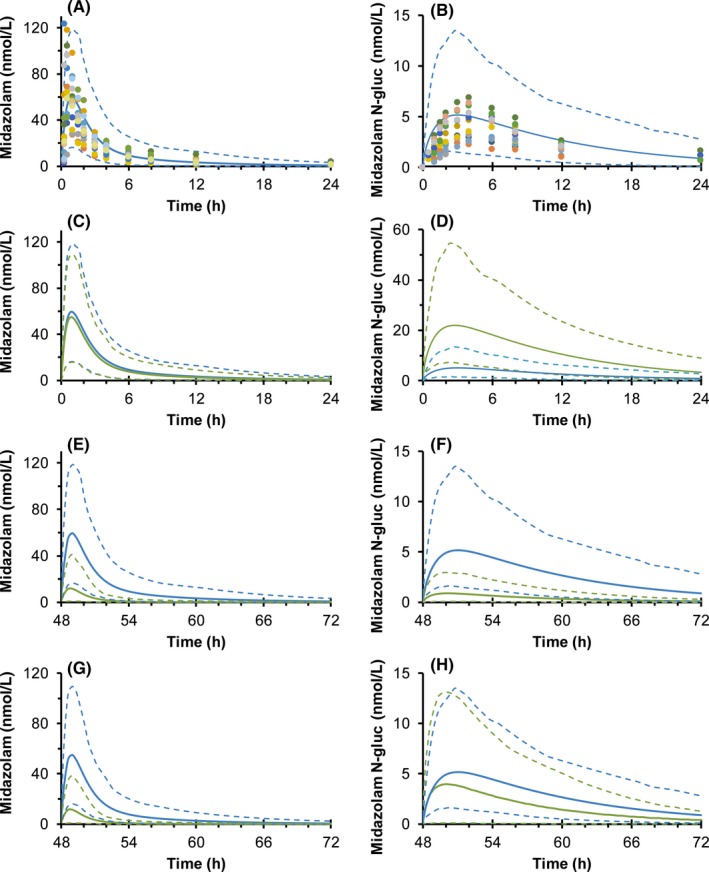
Physiologically based pharmacokinetic model predicted effects of rifampin‐induced midazolam metabolism. Observed and predicted midazolam (A) and midazolam N‐glucuronide (B) concentration‐time profiles following oral administration of midazolam (5 mg) to healthy volunteers (n = 12). Predicted midazolam and N‐glucuronide concentration‐time profiles considering the effects of rifampin coadministration (600 mg/day orally for 3 days) resulting in the following: UGT1A4 induction only (C and D), CYP3A induction only (E and F), and simultaneous induction of both UGT1A4 and CYP3A (G and H). Blue and green lines correspond to midazolam and N‐glucuronide disposition when taken alone or with rifampin, respectively. Solid and dashed lines denote the predicted arithmetic mean and 95% confidence intervals, respectively. Dots denote observed individual data; each color represents data from a single healthy volunteer (n = 12). PBPK, physiologically based pharmacokinetic
Table 5.
Model‐predicted rifampin mediated drug–drug interaction with midazolam
| Midazolam | Midazolam N‐glucuronide | ||||
|---|---|---|---|---|---|
| Control (no interaction) | |||||
| AUCobs | 209 (160‐274) | AUCobs | 54.7 (45.3‐66.0) | ||
| AUCpred | 185 (162‐213) | AUCpred | 51.7 (44.9‐59.7) | ||
|
|
74.2 (56.5‐97.5) |
|
4.17 (3.46‐5.03) | ||
|
|
50.0 (44.1‐56.6) |
|
4.25 (3.73‐4.84) | ||
| Rifampin UGT induction only | |||||
| AUCind | 165 (144‐188) | AUCind | 219 (192‐250) | ||
| 1/AUCratio | 1.12 (0.98‐1.28) | AUCratio | 4.24 (3.22‐5.57) | ||
|
|
46.7 (41.3‐52.8) |
|
18.5 (16.4‐20.9) | ||
| 1/ | 1.07 (0.95‐1.21) |
|
4.35 (3.39‐5.62) | ||
| Rifampin CYP3A induction only | |||||
| AUCind | 19.1 (15.4‐23.7) | AUCind | 5.43 (4.36‐6.77) | ||
| 1/AUCratio | 9.72 (8.48‐11.2) | 1/AUCratio | 9.52 (8.30‐10.9) | ||
|
|
7.44 (6.01‐9.22) |
|
0.54 (0.43‐0.66) | ||
| 1/ | 6.71 (5.87‐7.67) | 1/ | 7.94 (9.08‐6.93) | ||
| Rifampin CYP3A and UGT induction | |||||
| AUCind | 18.4 (14.9‐22.8) | AUCind | 24.8 (20.0‐30.78) | ||
| 1/AUCratio | 10.1 (9.32‐11.1) | 1/AUCratio | 2.08 (2.24‐1.94) | ||
|
|
7.23 (5.85‐8.94) |
|
2.48 (2.02‐3.05) | ||
| 1/ | 6.91 (6.33‐7.69) | 1/ | 1.71 (1.59‐1.85) | ||
Observed data recovered from a healthy volunteer (n = 12) study in which participants were administered a single oral dose (5 mg) of midazolam. AUCobs, observed area under the plasma concentration‐time curve (nmol/L × hours); AUCpred, predicted area under the plasma concentration‐time curve (nmol/L × hours); AUCInd, predicted area under the plasma concentration‐time curve following rifampin induction (nmol/L × hours); AUCratio, rifampin treatment:control ratio; , maximal observed plasma concentration (nmol/L); , maximal predicted concentration (nmol/L); , rifampin treatment:control ratio; , predicted maximal concentration following rifampin induction (nmol/L). Values denote geometric mean and 95% confidence intervals.
4. DISCUSSION
Successful mitigation of CYP450 metabolic liabilities during drug development has increased the importance of non‐CYP450 enzyme contributions to drug metabolism. However, evaluation of non‐CYP450 mediated xenobiotic metabolism continues to pose research, development, and regulatory challenges.33 Standardized in vitro models to assess conjugating enzyme contributions to xenobiotic clearance and approaches to predict clinical consequences are evolving. As a result, mechanistic understanding of these metabolic pathways and reports of the effects of modulators of conjugating enzyme activity are relatively limited compared to the CYP450 system. This report outlines the effects of rifampin induction of conjugating enzyme expression and potential modulation via induced miRNA expression. These data: (1) show that several clinically relevant conjugating enzymes are inducible by rifampin; (2) support the association of rifampin‐induced miRNA modulation of conjugating enzyme expression; (3) indicate that UGT induction is cell‐line dependent; and (4) suggest the potential clinical relevance of UGT induction by rifampin.
Of the non‐CYP450 enzymes that contribute to xenobiotic metabolism, UGTs are the most important in terms of abundance in hepatic and extrahepatic tissues as well as in the wide range of xenobiotics including many drugs and endobiotics they metabolize. Drug–drug interactions mediated via UGT induction are inherently difficult to predict from in vitro data.33 LC‐MS/MS approaches to directly quantify UGT protein content within in vitro systems and human tissues34, 35, 36, 37, 38, 39, 40 have enhanced in vitro–‐in vivo extrapolation of UGT mediated metabolism. Future studies of UGT induction could leverage combinatorial approaches (eg, RNAseq and LC‐MS/MS) to provide complementary information. UGT1A1 has been previously demonstrated to be induced by treatment with dietary polyphenols including resveratrol, curcumin, and chrysin in Caco‐2 cells,41, 42 human hepatocytes,43 and PXR reporter assays.44 However, rapid metabolism and minimal systemic exposure of many dietary polyphenols may limit their ability to induce hepatic UGTs in vivo.45 In contrast, rifampin and other prototypic drug inducers are expected to produce systemic exposure sufficient to result in clinically relevant enzyme induction.46 Rifampin induction of UGT1A5 has been reported previously in human hepatocytes. Detection of hepatic UGT1A5 protein in human livers not exposed to rifampin is limited, suggesting that hepatic UGT1A5 is expressed only in response to xenobiotic exposure.
In agreement with previous report, SULT2A1 mRNA expression was induced while SULT1A1 was not altered by treatment with rifampin.47 Consistent repression of SULT1E1 and SULT1B1 gene expression was not expected to result from rifampin treatment particularly when considering previous reports of the interaction between rifampin and ethinyl estradiol.18 However, repression of SULT1E1 and SULT1B1 may result from the complex interplay of multiple rifampin‐induced transcription factors, similar to the mechanism described for rifampin repression of CYP7A1 expression.48 This may suggest that rifampin induces regulatory elements that act to suppress SULT1E1 and SULT1B1 mRNA expression, such as miRNAs or transcription repressors, or mechanisms other than rifampin activation of PXR may underlie the observed changes.
Expression of NAT2 mRNA was the most strongly repressed gene by treatment with rifampin. The mechanistic basis for this observation remains to be elucidated. Nevertheless, this novel in vitro observation may provide an alternate explanation for the clinically observed increase in hepatotoxicity that results from coadministration of rifampin with the NAT2 substrate isoniazid.49, 50 The mechanistic underpinnings of this drug–drug interaction have been the source of some debate as it does not appear to be related directly to induction of CYP450‐mediated reactive metabolite concentrations in humans or PXR‐humanized mice.51, 52 Mouse models suggest that human PXR modulates hepatotoxicity associated with rifampin and isoniazid via increased accumulation of an endogenous hepatotoxin.51 However, human NAT2 genetic polymorphisms that result in a slow acetylator phenotype have been strongly associated with increased risk of isoniazid hepatotoxicity. It then leads that perhaps rifampin down‐regulation of NAT2 is creating a drug‐induced slow acetylator phenotype that leads to increased risk of isoniazid hepatotoxicity when administered with rifampin. Rifampin‐induced formation of hydrazine from isoniazid has been posited to underlie increased hydrazine plasma levels observed in patients taking rifampin and isoniazid as compared to those taking isoniazid alone.53 Alternatively, repressed NAT2 activity leading to impaired hydrazine elimination, or a combination of both increased formation and reduced elimination, may explain the apparent increase in hydrazine exposure caused by rifampin. Further reduction in limited NAT2 activity by rifampin could potentially explain reports of increased incidence of hepatotoxicity when slow acetylators take isoniazid and rifampin.54
The alpha‐class GSTs catalyze the GSH‐dependent detoxification of several alkylating chemotherapy agents and numerous environmental pollutants.55 GST induction has also been suggested, using high‐sensitivity real‐time PCR3 and likely represents another defense mechanism against xenobiotic exposure. The observed changes in GST expression measured via RNAseq are in alignment with previous reports using alternate quantification approaches.
Several miRNAs have been suggested to modulate conjugating enzyme expression and function. miR‐376c was identified as a modulator of UGT2B15 and UGT2B17 in prostate cancer cell lines.10 This observation was later confirmed and expanded to include miR‐331‐5p, miR‐376c‐3p (formerly miR‐368‐1) for UGT2B15 and miR‐376c, miR‐409, and miR‐494 for UGT2B17.9 miR‐216b has been identified in silico as a potential modulator of several UGT2B family members, including UGT2B15.7 Similarly, miR‐491‐3p is associated with UGT1A1 expression and activity changes in hepatic cell lines8 However, the expected inverse correlation between the levels of miR‐491‐3p and UGT1A1 mRNA were not demonstrated in a panel of 38 normal livers. More recently, a functional genomics approach assessed the complete compliment of miRNAs that could regulate UGT1A expression and identified 6 additional miRNAs (miR‐21‐3p, miR‐200a‐3p, miR‐103b, miR‐1286, miR‐376b‐3p, and miR‐141‐3p) that decrease UGT1A‐dependent activity.13 SULT1A1 expression in human liver has been associated with miR‐631 expression levels.56 miR‐133a has been associated with repression of GSTP1 mRNA and protein in lung and bladder cancer cell lines57, 58 while miR‐133b has been associated with repressed GSTP1 mRNA expression in prostate cancer cell lines.59 miR‐513a‐3p has also been associated with repressed GSTP1 expression in lung cancer cells.60 An inverse correlation between PXR translational efficiency and miR‐148a has also been reported.61 Interestingly, none of these miRNAs were revealed by our correlation analysis. This may be the result of both direct and indirect mechanisms mediated via rifampin induction. However, the miRNA‐mRNA pairs identified in Table 4, particularly those predicted via TargetScan, may represent a reasonable starting point to better understand the interplay of miRNA modulation and transcription factor activation via functional and mechanistic studies.
Induction of UGT1A4 observed in vitro prompted evaluation via PBPK modeling and simulation. An available dataset which included the UGT1A4 mediated N‐glucuronide metabolite of midazolam was utilized to evaluate the potential contribution of isolated and simultaneous CYP450 and UGT1A4 induction. It is acknowledged that induction of midazolam N‐glucuronidation is unlikely to be of clinical significance owing to the relatively minor contribution of UGT1A4 to overall midazolam clearance. However, this exemplar drug–drug interaction highlights the potential importance of considering simultaneous induction of alternate pathways, particularly for drugs where the fraction metabolized by CYP450 is relatively lower than that of midazolam. Including induction of all known metabolic pathways into predictive PBPK drug–drug interaction models may also help alleviate the systematic tendency to under predict the magnitude of drug–drug interactions resulting from induction.
These data reveal differential effects of rifampin on the human conjugating enzyme transcriptome and potential associations with miRNAs. The magnitude of phase 2 enzyme mRNA induction in response to rifampin was relatively lower than that observed for induction of CYP450 enzymes. This may be the result of a relatively lesser contribution of PXR‐mediated induction to the overall induction potential of the conjugating enzymes evaluated. We acknowledge that mRNA expression changes may not directly reflect changes in protein content and activity. Further studies are needed to evaluate the correlations between rifampin‐induced mRNA expression changes, miRNA modulation, and enzyme activity as posttranscriptional and posttranslation modifications may alter this relationship. However, this global expression approach was aimed at revealing additional factors that might contribute to regulation of important drug metabolizing enzymes. These findings should inform future studies to elucidate and quantitatively predict the impact of epigenetic regulation and conjugating enzyme induction on clinical drug disposition.
AUTHOR CONTRIBUTIONS
Participated in research design: Gufford, Liu, Desta, and Skaar; while Gufford and Eadon conducted experiments. Contributed new reagents or analytical tools: Eadon; performed data analysis: Gufford, Robarge, Lin, Gao, Liu, and Skaar. Lastly, Gufford, Eadon, Robarge, Liu, Desta, and Skaar wrote or contributed to writing of the manuscript.
DISCLOSURES
None declared.
Supporting information
Figure S1. Hierarchical clustering of rifampin‐induced changes in conjugating enzyme mRNA expression determined via RNA‐seq. Clustering of mRNA expression changes and hepatocyte donors depicted by the dendrograms were determined, using Euclidian distances and the complete linkage clustering method. Red = induced genes; blue = repressed genes.
Table S1. Primer sequences and annealing temperatures
Table S2. SimCYP model input parameters for midazolam and midazolam N‐glucuronide
ACKNOWLEDGEMENTS
We thank Mary F. Paine for kindly providing midazolam and midazolam‐N‐glucuronide clinical pharmacokinetic data used to support PBPK model development.
Gufford BT, Robarge JD, Eadon MT, et al. Rifampin modulation of xeno‐ and endobiotic conjugating enzyme mRNA expression and associated microRNAs in human hepatocytes. Pharmacol Res Perspect. 2018;e00386 https://doi.org/10.1002/prp2.386
Funding Information
This work was supported by the National Institutes of Health National Institute of General Medical Sciences (grant 1R01GM088076). B.T.G. and J.D.R. supported by the National Institute of General Medical Sciences (grant T32 GM008425). B.T.G. also supported by the Indiana University Center for AIDs Research. M.T.E. supported by the PhRMA Foundation Clinical Pharmacology Award.
REFERENCES
- 1. Niemi M, Backman JT, Fromm MF, Neuvonen PJ, Kivisto KT. Pharmacokinetic interactions with rifampicin: clinical relevance. Clin Pharmacokinet. 2003;42:819–850. [DOI] [PubMed] [Google Scholar]
- 2. Sousa M, Pozniak A, Boffito M. Pharmacokinetics and pharmacodynamics of drug interactions involving rifampicin, rifabutin and antimalarial drugs. J Antimicrob Chemother. 2008;62:872–878. [DOI] [PubMed] [Google Scholar]
- 3. Nishimura M, Yoshitsugu H, Naito S, Hiraoka I. Evaluation of gene induction of drug‐metabolizing enzymes and transporters in primary culture of human hepatocytes using high‐sensitivity real‐time reverse transcription PCR. Yakugaku Zasshi. 2002;122:339–361. [DOI] [PubMed] [Google Scholar]
- 4. Chen J, Raymond K. Roles of rifampicin in drug‐drug interactions: underlying molecular mechanisms involving the nuclear pregnane X receptor. Ann Clin Microbiol Antimicrob. 2006;5:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Benson EA, Eadon MT, Desta Z, et al. Rifampin regulation of drug transporters gene expression and the association of MicroRNAs in human hepatocytes. Front Pharmacol. 2016;7:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ramamoorthy A, Liu Y, Philips S, et al. Regulation of microRNA expression by rifampin in human hepatocytes. Drug Metab Dispos. 2013;41:1763–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dluzen DF, Lazarus P. MicroRNA regulation of the major drug‐metabolizing enzymes and related transcription factors. Drug Metab Rev. 2015;47(3):320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dluzen DF, Sun D, Salzberg AC, et al. Regulation of UDP‐glucuronosyltransferase 1A1 expression and activity by microRNA 491‐3p. J Pharmacol Exp Ther. 2014;348:465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Margaillan G, Levesque E, Guillemette C. Epigenetic regulation of steroid inactivating UDP‐glucuronosyltransferases by microRNAs in prostate cancer. J Steroid Biochem Mol Biol. 2016;155(Pt A):85–93. [DOI] [PubMed] [Google Scholar]
- 10. Wijayakumara DD, Hu DG, Meech R, McKinnon RA, Mackenzie PI. Regulation of Human UGT2B15 and UGT2B17 by miR‐376c in Prostate Cancer Cell Lines. J Pharmacol Exp Ther. 2015;354:417–425. [DOI] [PubMed] [Google Scholar]
- 11. Guillemette C, Levesque E, Harvey M, Bellemare J, Menard V. UGT genomic diversity: beyond gene duplication. Drug Metab Rev. 2010;42:24–44. [DOI] [PubMed] [Google Scholar]
- 12. Hu DG, Meech R, McKinnon RA, Mackenzie PI. Transcriptional regulation of human UDP‐glucuronosyltransferase genes. Drug Metab Rev. 2014;46:421–458. [DOI] [PubMed] [Google Scholar]
- 13. Papageorgiou I, Court MH. Identification and validation of microRNAs directly regulating the UDP‐glucuronosyltransferase 1A subfamily enzymes by a functional genomics approach. Biochem Pharmacol. 2017;137:93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Papageorgiou I, Freytsis M, Court MH. Transcriptome association analysis identifies miR‐375 as a major determinant of variable acetaminophen glucuronidation by human liver. Biochem Pharmacol. 2016;117:78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gamage N, Barnett A, Hempel N, et al. Human sulfotransferases and their role in chemical metabolism. Toxicol Sci. 2006;90:5–22. [DOI] [PubMed] [Google Scholar]
- 16. Lindsay J, Wang LL, Li Y, Zhou SF. Structure, function and polymorphism of human cytosolic sulfotransferases. Curr Drug Metab. 2008;9:99–105. [DOI] [PubMed] [Google Scholar]
- 17. Runge‐Morris M, Kocarek TA. Regulation of sulfotransferases by xenobiotic receptors. Curr Drug Metab. 2005;6:299–307. [DOI] [PubMed] [Google Scholar]
- 18. Li AP, Hartman NR, Lu C, Collins JM, Strong JM. Effects of cytochrome P450 inducers on 17alpha‐ethinyloestradiol (EE2) conjugation by primary human hepatocytes. Br J Clin Pharmacol. 1999;48:733–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sim E, Lack N, Wang CJ, et al. Arylamine N‐acetyltransferases: structural and functional implications of polymorphisms. Toxicology. 2008;254:170–183. [DOI] [PubMed] [Google Scholar]
- 20. Endo Y, Yamashita H, Takahashi S, et al. Immunohistochemical determination of the miR‐1290 target arylamine N‐acetyltransferase 1 (NAT1) as a prognostic biomarker in breast cancer. BMC Cancer. 2014;14:990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sim E, Abuhammad A, Ryan A. Arylamine N‐acetyltransferases: from drug metabolism and pharmacogenetics to drug discovery. Br J Pharmacol. 2014;171:2705–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moon YJ, Wang X, Morris ME. Dietary flavonoids: effects on xenobiotic and carcinogen metabolism. Toxicolo In Vitro. 2006;20:187–210. [DOI] [PubMed] [Google Scholar]
- 23. Rae JM, Johnson MD, Lippman ME, Flockhart DA. Rifampin is a selective, pleiotropic inducer of drug metabolism genes in human hepatocytes: studies with cDNA and oligonucleotide expression arrays. J Pharmacol Exp Ther. 2001;299:849–857. [PubMed] [Google Scholar]
- 24. Herbert BS, Grimes BR, Xu WM, et al. A telomerase immortalized human proximal tubule cell line with a truncation mutation (Q4004X) in polycystin‐1. PLoS ONE. 2013;8:e55191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smith RP, Eckalbar WL, Morrissey KM, et al. Genome‐wide discovery of drug‐dependent human liver regulatory elements. PLoS Genet. 2014;10:e1004648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brantley SJ, Gufford BT, Dua R, et al. Physiologically based pharmacokinetic modeling framework for quantitative prediction of an herb‐drug interaction. CPT Pharmacometrics Syst Pharmacol. 2014;3:e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Almond LM, Mukadam S, Gardner I, et al. Prediction of drug‐drug interactions arising from CYP3A induction using a physiologically based dynamic model. Drug Metab Dispos. 2016;44:821–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Finel M, Li X, Gardner‐Stephen D, Bratton S, Mackenzie PI, Radominska‐Pandya A. Human UDP‐glucuronosyltransferase 1A5: identification, expression, and activity. J Pharmacol Exp Ther. 2005;315:1143–1149. [DOI] [PubMed] [Google Scholar]
- 29. Meinl W, Glatt H. Structure and localization of the human SULT1B1 gene: neighborhood to SULT1E1 and a SULT1D pseudogene. Biochem Biophys Res Commun. 2001;288:855–862. [DOI] [PubMed] [Google Scholar]
- 30. Kohle C, Bock KW. Coordinate regulation of human drug‐metabolizing enzymes, and conjugate transporters by the Ah receptor, pregnane X receptor and constitutive androstane receptor. Biochem Pharmacol. 2009;77:689–699. [DOI] [PubMed] [Google Scholar]
- 31. Achour B, Rostami‐Hodjegan A, Barber J. Protein expression of various hepatic uridine 5′‐diphosphate glucuronosyltransferase (UGT) enzymes and their inter‐correlations: a meta‐analysis. Biopharm Drug Dispos. 2014;35:353–361. [DOI] [PubMed] [Google Scholar]
- 32. Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;2:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Argikar UA, Potter PM, Hutzler JM, Marathe PH. Challenges and opportunities with non‐CYP enzymes aldehyde oxidase, carboxylesterase, and UDP‐glucuronosyltransferase: focus on reaction phenotyping and prediction of human clearance. AAPS J. 2016;18:1391–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fallon JK, Neubert H, Goosen TC, Smith PC. Targeted precise quantification of 12 human recombinant uridine‐diphosphate glucuronosyl transferase 1A and 2B isoforms using nano‐ultra‐high‐performance liquid chromatography/tandem mass spectrometry with selected reaction monitoring. Drug Metab Dispos. 2013;41:2076–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fallon JK, Neubert H, Hyland R, Goosen TC, Smith PC. Targeted quantitative proteomics for the analysis of 14 UGT1As and ‐2Bs in human liver using NanoUPLC‐MS/MS with selected reaction monitoring. J Proteome Res. 2013;12:4402–4413. [DOI] [PubMed] [Google Scholar]
- 36. Groer C, Busch D, Patrzyk M, et al. Absolute protein quantification of clinically relevant cytochrome P450 enzymes and UDP‐glucuronosyltransferases by mass spectrometry‐based targeted proteomics. J Pharm Biomed Anal. 2014;100:393–401. [DOI] [PubMed] [Google Scholar]
- 37. Harbourt DE, Fallon JK, Ito S, et al. Quantification of human uridine‐diphosphate glucuronosyl transferase 1A isoforms in liver, intestine, and kidney using nanobore liquid chromatography‐tandem mass spectrometry. Anal Chem. 2012;84:98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Margaillan G, Rouleau M, Fallon JK, et al. Quantitative profiling of human renal UDP‐glucuronosyltransferases and glucuronidation activity: a comparison of normal and tumoral kidney tissues. Drug Metab Dispos. 2015;43:611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Margaillan G, Rouleau M, Klein K, et al. Multiplexed targeted quantitative proteomics predicts hepatic glucuronidation potential. Drug Metab Dispos. 2015c;43:1331–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sato Y, Nagata M, Tetsuka K, et al. Optimized methods for targeted peptide‐based quantification of human uridine 5′‐diphosphate‐glucuronosyltransferases in biological specimens using liquid chromatography‐tandem mass spectrometry. Drug Metab Dispos. 2014;42:885–889. [DOI] [PubMed] [Google Scholar]
- 41. Galijatovic A, Walle UK, Walle T. Induction of UDP‐glucuronosyltransferase by the flavonoids chrysin and quercetin in Caco‐2 cells. Pharm Res. 2000;17:21–26. [DOI] [PubMed] [Google Scholar]
- 42. Iwuchukwu OF, Tallarida RJ, Nagar S. Resveratrol in combination with other dietary polyphenols concomitantly enhances antiproliferation and UGT1A1 induction in Caco‐2 cells. Life Sci. 2011;88:1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Walle T, Otake Y, Galijatovic A, Ritter JK, Walle UK. Induction of UDP‐glucuronosyltransferase UGT1A1 by the flavonoid chrysin in the human hepatoma cell line hep G2. Drug Metab Dispos. 2000;28:1077–1082. [PubMed] [Google Scholar]
- 44. Wang J, Huang M, Hu H, Yu L, Zeng S. Pregnane X receptor‐mediated transcriptional activation of UDP‐glucuronosyltransferase 1A1 by natural constituents from foods and herbs. Food Chem. 2014;164:74–80. [DOI] [PubMed] [Google Scholar]
- 45. Smith CM, Graham RA, Krol WL, et al. Differential UGT1A1 induction by chrysin in primary human hepatocytes and HepG2 Cells. J Pharmacol Exp Ther. 2005;315:1256–1264. [DOI] [PubMed] [Google Scholar]
- 46. Smith CM, Faucette SR, Wang H, LeCluyse EL. Modulation of UDP‐glucuronosyltransferase 1A1 in primary human hepatocytes by prototypical inducers. J Biochem Mol Toxicol. 2005;19:96–108. [DOI] [PubMed] [Google Scholar]
- 47. Nishimura M, Koeda A, Shimizu T, et al. Comparison of inducibility of sulfotransferase and UDP‐glucuronosyltransferase mRNAs by prototypical microsomal enzyme inducers in primary cultures of human and cynomolgus monkey hepatocytes. Drug Metab Pharmacokinet. 2008;23:45–53. [DOI] [PubMed] [Google Scholar]
- 48. Li T, Chiang JY. Mechanism of rifampicin and pregnane X receptor inhibition of human cholesterol 7 alpha‐hydroxylase gene transcription. Am J Physiol Gastrointest Liver Physiol. 2005;288:G74–G84. [DOI] [PubMed] [Google Scholar]
- 49. Gangadharam PR. Isoniazid, rifampin, and hepatotoxicity. Am Rev Respir Dis. 1986;133:963–965. [DOI] [PubMed] [Google Scholar]
- 50. Skakun NP, Shman'ko VV. Synergistic effect of rifampicin on hepatotoxicity of isoniazid. Antibiot Med Biotekhnol. 1985;30:185–189. [PubMed] [Google Scholar]
- 51. Li F, Lu J, Cheng J, et al. Human PXR modulates hepatotoxicity associated with rifampicin and isoniazid co‐therapy. Nat Med. 2013;19:418–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Steele MA, Burk RF, DesPrez RM. Toxic hepatitis with isoniazid and rifampin. A meta‐analysis. Chest. 1991;99:465–471. [DOI] [PubMed] [Google Scholar]
- 53. Sarma GR, Immanuel C, Kailasam S, Narayana AS, Venkatesan P. Rifampin‐induced release of hydrazine from isoniazid. A possible cause of hepatitis during treatment of tuberculosis with regimens containing isoniazid and rifampin. Am Rev Respir Dis. 1986;133:1072–1075. [DOI] [PubMed] [Google Scholar]
- 54. Ohno M, Yamaguchi I, Yamamoto I, et al. Slow N‐acetyltransferase 2 genotype affects the incidence of isoniazid and rifampicin‐induced hepatotoxicity. Int J Tuberc Lung Dis. 2000;4:256–261. [PubMed] [Google Scholar]
- 55. Coles BF, Kadlubar FF. Human alpha class glutathione S‐transferases: genetic polymorphism, expression, and susceptibility to disease. Methods Enzymol. 2005;401:9–42. [DOI] [PubMed] [Google Scholar]
- 56. Yu X, Dhakal IB, Beggs M, et al. Functional genetic variants in the 3′‐untranslated region of sulfotransferase isoform 1A1 (SULT1A1) and their effect on enzymatic activity. Toxicol Sci. 2010;118:391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Moriya Y, Nohata N, Kinoshita T, et al. Tumor suppressive microRNA‐133a regulates novel molecular networks in lung squamous cell carcinoma. J Hum Genet. 2012;57:38–45. [DOI] [PubMed] [Google Scholar]
- 58. Uchida Y, Chiyomaru T, Enokida H, et al. MiR‐133a induces apoptosis through direct regulation of GSTP1 in bladder cancer cell lines. Urol Oncol. 2013;31:115–123. [DOI] [PubMed] [Google Scholar]
- 59. Patron JP, Fendler A, Bild M, et al. MiR‐133b targets antiapoptotic genes and enhances death receptor‐induced apoptosis. PLoS ONE. 2012;7:e35345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang X, Zhu J, Xing R, et al. miR‐513a‐3p sensitizes human lung adenocarcinoma cells to chemotherapy by targeting GSTP1. Lung Cancer. 2012;77:488–494. [DOI] [PubMed] [Google Scholar]
- 61. Takagi S, Nakajima M, Mohri T, Yokoi T. Post‐transcriptional regulation of human pregnane X receptor by micro‐RNA affects the expression of cytochrome P450 3A4. J Biol Chem. 2008;283:9674–9680. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Hierarchical clustering of rifampin‐induced changes in conjugating enzyme mRNA expression determined via RNA‐seq. Clustering of mRNA expression changes and hepatocyte donors depicted by the dendrograms were determined, using Euclidian distances and the complete linkage clustering method. Red = induced genes; blue = repressed genes.
Table S1. Primer sequences and annealing temperatures
Table S2. SimCYP model input parameters for midazolam and midazolam N‐glucuronide