

Abstract
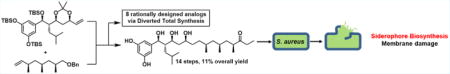
The baulamycins were identified as in vitro siderophore biosynthesis inhibitors. Diverted total synthesis was used to construct the natural products and eight strategic analogues, three of which had improved inhibitory activity. Biological testing then revealed that membrane damage is the predominant mode of action in Staphylococcus aureus cells.
Graphical abstract
The development of clinical antibiotics has revolutionized medicine and overall quality of life over the past century; however, current problems with resistance have made some in the field question whether we are at the brink of a “post-antibiotic era”.1 While antibiotic-resistant bacteria are not a newly discovered phenomenon, current mis- and overuse of antibiotics has exacerbated the problem.2 With the recent emergence of multidrug resistant “superbugs”, new antibiotics with novel mechanisms of action are urgently needed. The strategy of modifying existing scaffolds such as penicillin, erythromycin, and tetracycline has only delayed the development of resistance, as these compounds target essential metabolic pathways, thereby creating a significant selection pressure. New small molecule agents that attenuate virulence or target certain pathways that are upregulated during a pathogen’s host invasion would be a valuable addition to the current clinical arsenal as they do not generate such pressures.3
One such strategy is to interfere with microbes’ uptake of essential nutrients, for example, iron. Iron acquisition and utilization is energy-intensive and has therefore evolved to be a competitive process among microbes in Nature. In an aerobic human host environment, free iron is in the ferric (3+) state, which has poor aqueous solubility. Most of this iron is stored in hemoglobin and ferritin. Iron transport throughout the host is accomplished by high-affinity transferrin complexes. Bacteria compete with the host by secreting iron-chelating small molecules called siderophores that bind iron with even higher affinity than transferrins. The resultant siderophore–iron complexes are taken in by the bacterium for its essential metabolic processes.4 In contrast to “classical” antibiotics that nonselectively kill an entire population of bacteria, disruption of iron uptake has been shown to be an effective strategy to reduce a bacterial population’s growth and virulence.5
It is for these reasons that we were drawn to the baulamycins, compounds isolated in 2014 by Sherman and co-workers, that were shown to inhibit bacterial iron acquisition in vitro (Figure 1A).6 The compounds were identified as inhibitors of SbnE and were able to outcompete citric acid, presumably by mimicking its structure via the baulamycins’ 1,3,5-triol moiety. The initial isolation paper showed that the compounds displayed broad-spectrum whole cell inhibitory activity in both iron-rich media (IRM) and iron-depleted media (IDM). For Staphylococcus aureus in particular, the differences between IRM and IDM IC50 values were minimal (86 vs 69 μM for MSSA and 130 vs 130 μM for MRSA). This discrepancy coupled with ambiguity regarding the relative and absolute stereochemistry7 served as the ideal platform to leverage diverted total synthesis (DTS) toward better understanding the biological mode of action (Figure 1A).8,9
Figure 1.
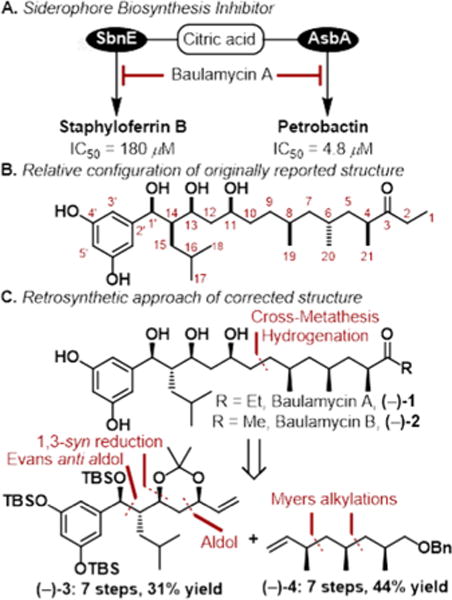
(a) Enzymes that the baulamycins were shown to inhibit in vitro and IC50 values. (b) Originally reported structure of baulamycin A (with numbering convention) (c) Correct structure as confirmed by Aggarwal and co-workers, retrosynthetic approach used herein.
Previous efforts from our laboratory have successfully utilized DTS to both identify modes of action of natural products and also uncover unique phenotypes.10–12 With these principles in mind, we designed a synthesis that would be both flexible, allowing for the construction of all possible stereoisomers, and strategic in order to answer specific hypothesis-driven inquiries. Herein we report the total syntheses and biological investigation of the baulamycins and eight rationally designed analogues in whole-cell assays. Our results provide evidence that membrane damage is the predominant mechanism of action of the natural products in contrast to previous reports.
Our retrosynthetic analysis led us to propose a late-stage crossmetathesis and hydrogenation providing fragments (−)-3 and (−)-4 (Figure 1C). We first began our work toward the originally proposed structure (Figure 1B) but were able to quickly pivot upon the disclosure of the correct stereoisomer (Figure 1C) during the course of this project (vida infra).13 In a forward sense, the synthesis of the originally reported left fragment began with known Evans syn aldol product (−)-7. The chiral oxazolidinone was then removed by transamidation to the corresponding Weinreb amide, followed by TBS protection yielding (−)-8 in 81% over two steps. Displacement of the amide with methylmagnesium bromide yielded methyl ketone (−)-9, which was then converted to the corresponding (−)-diisopino-campheylborane-derived enolate and treated with acrolein to yield the β-hydroxyketone (−)-10 as a single diastereomer resulting from synergistic 1,4-syn-1,5-syn induction. Subsequent diastereoselective 1,3-syn reduction by NaBH4 in the presence of Et2BOMe yielded the 1,3-diol, which was then converted to the corresponding acetonide (−)-11 (Scheme 1A).
Scheme 1.
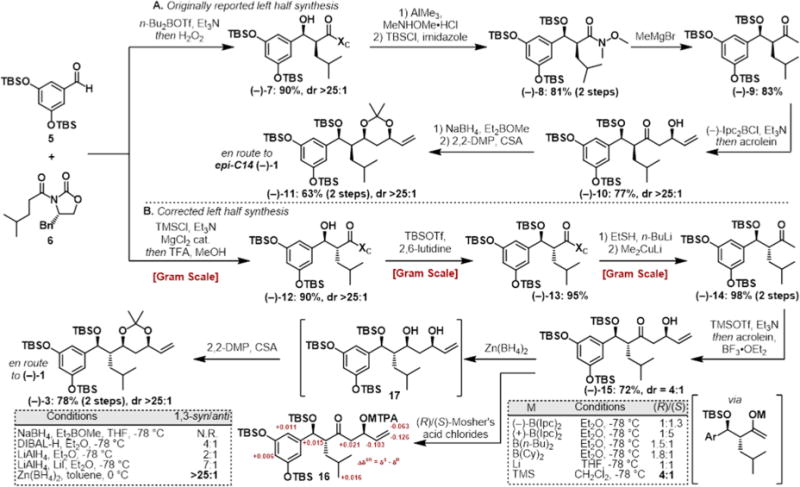
(a) Synthetic Approach to the Fragment Corresponding to the Originally Reported Baulamycin Structure (Figure 1B, epi-C14 (−)-1). (b) Revised Route To Access the Left Half Corresponding to the Corrected Baulamycin Structurea
aN.R. = no reaction.
At this point, the publication by Goswami and co-workers14 reported that the structure assigned to baulamycin A did not match the natural compound. A conspicuously aberrant coupling constant (J1′-14 = 3.7 Hz) in their synthesized molecule did not match the reported value (J1′-14 = 7.0 Hz). With this discrepancy in mind, we referred to the literature in regards to Evans syn and anti-aldol adducts and found that larger coupling constants were associated with an anti-configuration.15 This hypothesis was further supported by Aggarwal et al., who published the correct structure during the course of our synthesis,13 and a more recent study by Sim and co-workers.16
Our revised route to the corrected structure of (−)-1 began with an Evans anti aldol reaction between building blocks 5 and 6 to provide the desired aldol adduct (−)-12 as a single diastereomer (Scheme 1B). Masking the resulting alcohol as the silyl ether yielded (−)-13 followed by displacement by lithium ethyl thiolate yielded the corresponding thioester. Treatment of this intermediate with lithium dimethyl cuprate afforded methyl ketone (−)-14 in nearly quantitative yield over two steps. Next, we turned to the key aldol reaction between methyl ketone (−)-14 and acrolein. The previously selective conditions ((−)-Ipc2BCI, Et3N) yielded a mixture of diastereomeric products (dr = 1:1.3). Undeterred, we separated both resultant diastereomers by column chromatography and determined the stereochemical outcome by converting the secondary alcohol to the corresponding Mosher’s esters 16. This analysis showed that the previously optimized conditions were now in favor of the undesired stereoisomer. Surprisingly, when achiral boron reagents were employed, the product ratio was overturned in favor of the desired secondary alcohol, with a modest 2:1 selectivity. Gratifyingly, Mukaiyama aldol reaction between the corresponding silyl enol ether and acrolein improved the product ratio to 4:1 in favor of the desired (R)-alcohol (−)-15. The 1,3-syn reduction of the β-hydroxyketone (−)-15 was then investigated. After much optimization, it was found that zinc borohydride in toluene provided the desired 1,3-diol with high diastereoselectivity at C13 (>25:1). The resultant unstable diol 17 was immediately converted to acetonide (−)-3. The scalable route afforded the key building block (−)-3 in six steps and 31% overall yield from known material.
Based on the initial ambiguity surrounding the deoxypropionate triad, we sought to use Myers alkylation chemistry to iteratively investigate the structure in a flexible manner. Beginning with known iodide (−)-18, a two-step Myers alkylation/reduction sequence,17 employing pseudoephedrine amide (+)-19, led to alcohol (+)-20 in 88% yield over two steps as a single diastereomer. A second Myers alkylation/reduction sequence after iodination provided alcohol (+)-21 in 64% overall yield. Sequential Swern oxidation and Wittig reaction then furnished olefin (−)-4 with no erosion of the α-methyl stereochemistry (Scheme 2). Benzyl ether (−)-4 was then converted to the corresponding ethyl ketone (+)-22 to afford a more convergent route toward baulamycin A and the DTS of analogues (see Scheme S1). Deoxypropionate precursors are now readily available via this route in 11 steps and 23% overall yield from known material.
Scheme 2.
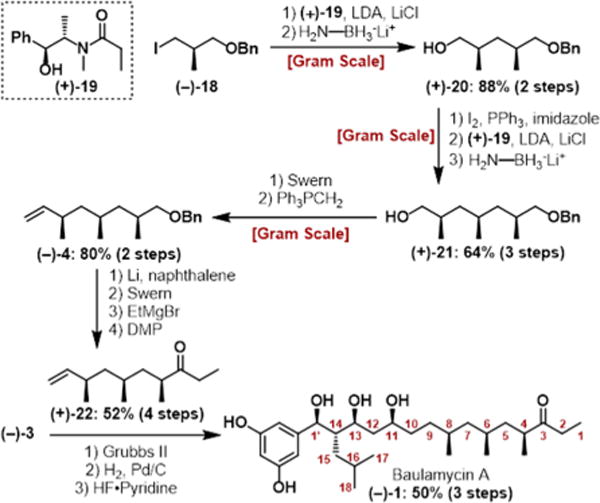
Route to Deoxypropionate Fragment (−)-4 and Final Steps Leading to (−)-1
Having constructed both halves, the cross-metathesis step was achieved using Grubbs’ second-generation catalyst. The report by Goswami et al. suggested this disconnection was not feasible; however, another report by Chandrasekhar et al. claimed the opposite.14,18 In our hands, multiple additions of catalyst over the course of the reaction were required to obtain acceptable yields. From there, hydrogenation and global deprotection in the presence of HF–pyridine afforded (−)-baulamycin A in a longest linear sequence of 14 steps, matching the natural product in all respects ([α]20D − 15.1 (c 0.44, MeOH)). Once the correct structure was verified spectroscopically, we sought to leverage DTS to investigate the SAR of the baulamycins and investigate the mechanism of action. From the outset, we were intrigued by the amphipathic nature of the natural product with its polar headgroup (two phenols, three hydroxyl groups) and lipophilic tail. Thus, we postulated that the baulamycins’ broad-spectrum inhibitory activity in iron-rich media could be due to nonselective lysis or damage to cell membranes, akin to a detergent.
To test this hypothesis, we systematically altered several of the natural products’ key structural features. In an effort to alter the hydrophilic portion, conversion of the phenols to methyl ethers ((−)-23) provided an aromatic system devoid of hydrogen bond donors (Figure 2). Ketone analogue (−)-24 allowed us to test the importance of the hydroxyl located at C13. We also considered the stereochemistry at C14 ((−)-25) and hypothesized that if this epimer retained activity this would lend credence to our membrane hypothesis, as this drastic structural alteration is unlikely to be tolerated by an enzyme active site. In a similar vein, the hydrophobic fragment was converted from the ethyl to the propyl ketone ((−)-26). As baulamycin A is more active than B (ethyl vs methyl), we were curious if other extensions would be tolerated. We also removed all of the stereogenic methyl groups while either retaining ((−)-27) or removing ((−)-28) the ketone in an effort to develop a simplified analogue with improved potency. Finally, we sought to further probe this hypothesis by either removing the entire hydrophobic portion ((−)-29) or adding a polar component ((−)-30).
Figure 2.
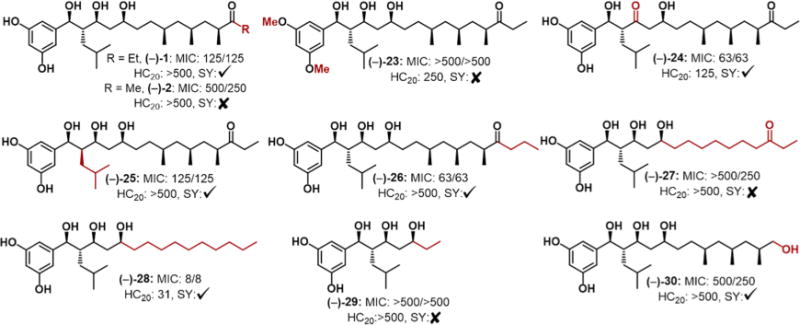
Analogue structures and selected biological results. The two MIC values (in μM) refer to IRM and IDM, respectively (S. aureus SH1000). HC20 concentrations given in μM. Checkmark and X symbols refer to positive and negative uptake results, respectively.
After completion of our synthetic analogue campaign, we tested all compounds in three different strains of S. aureus using both iron-rich and iron-depleted conditions (Figure 2: S. aureus SH1000 MIC data, for USA300 and ATCC33591, see Table S2). We used minimum inhibitory concentration (MIC) measurements in contrast to Sherman and co-workers’ IC50 growth data for reproducibility reasons.6 All analogues were tested alongside baulamycin A and B and with vancomycin as a positive control. In all cases, we saw minimal differences in growth inhibition between IRM and IDM in accordance with Sherman. This again hinted at an alternative mechanism that did not involve siderophore biosynthesis or iron acquisition. Compounds with similar mechanisms in vitro have been shown to display high (100-fold) differences between IRM and IDM.19 Therefore, although the baulamycins inhibit SbnE in vitro we do not think that it is biologically relevant in vivo and instead sought to test our membrane-targeting hypothesis.
Toward this end, we employed each of our synthetic compounds in a SYTOX uptake assay. SYTOX exhibits a large increase in fluorescence when bacterial membranes become permeabilized, whereby the dye crosses the cell membrane and complexes with DNA.20 As a positive control we used one of the most potent quaternary ammonium compounds (12,3,2,3,12) from our laboratory as a positive control, as this compound is a well-known inducer of bacterial cell lysis.21a,b We found that baulamycin A as well as all active analogues tested positive for membrane disruption via this assay (see the Supporting Information for details). Baulamycin B was one exception; however, this discrepancy may be a result of its modest MIC against S. aureus (500 μM). These results support our hypothesis that the compounds are potentially inducing membrane damage and explains their broad-spectrum activity in IRM. Current studies are underway to investigate the specific mechanism by which these compounds are able to disrupt membrane composition and will be reported in due course. Membrane lytic antibacterial compounds are also known to lyse mammalian red blood cells; therefore, we tested each compound for hemolysis activity. Baulamycins A and B did not induce any hemolysis up to 500 μM, along with the propyl analogue (−)-26. However, ketone (−)-24 and our most potent compound, (−)-28, displayed significant hemolytic activity (Figure 2). Future studies will focus on the optimization of (−)-26, the most potent compound that did not induce hemolysis.
Herein, we report the diverted total synthesis of the baulamycins in a longest linear sequence of 14 steps and 11% yield. The high overall efficiency allowed for the expedited synthesis of eight rationally designed analogues. Whole cell biological assays revealed a common chemotype that hinted at membrane lysis as the likely mode of action, which was further supported by uptake experiments. Future work will require optimization of the baulamycin scaffold to mitigate nonselective membrane lysis. The duality of mechanisms that the baulamycins display demonstrates the importance of supplementing in vitro data with in vivo assays whenever possible.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of General Medical Sciences (GM119426) and the National Science Foundation (CHE1755698). The NMR instruments used in this work were supported by the National Science Foundation (CHE1531620).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:10.1021/acs.orglett.8b00054.
Detailed synthetic procedures and compound characterization (PDF)
ORCID
William M. Wuest: 0000-0002-5198-7744
Notes
The authors declare no competing financial interest.
References
- 1.Alanis AJ. Arch Med Res. 2005;36:697. doi: 10.1016/j.arcmed.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 2.Ventola CL. Pharm Ther. 2015;40:277. [Google Scholar]
- 3.Rossiter SE, Fletcher MH, Wuest WM. Chem Rev. 2017;117:12415. doi: 10.1021/acs.chemrev.7b00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miethke M, Marahiel MA. Microbiol Mol Biol Rev. 2007;71:413. doi: 10.1128/MMBR.00012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Litwin CM, Calderwood SB. Clin Microbiol Rev. 1993;6:137. doi: 10.1128/cmr.6.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tripathi A, Schofield MM, Chlipala GE, Schultz PJ, Yim I, Newmister SA, Nusca TD, Scaglione JB, Hanna PC, Tamayo-Castillo G, Sherman DH. J Am Chem Soc. 2014;136:1579. doi: 10.1021/ja4115924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tripathi A, Schofield MM, Chlipala GE, Schultz PJ, Yim I, Newmister SA, Nusca TD, Scaglione JB, Hanna PC, Tamayo-Castillo G, Sherman DH. J Am Chem Soc. 2014;136:10541. doi: 10.1021/ja4115924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Njardarson JnT, Gaul C, Shan D, Huang X-Y, Danishefsky SJ. J Am Chem Soc. 2004;126:1038. doi: 10.1021/ja039714a. [DOI] [PubMed] [Google Scholar]
- 9.Wilson RM, Danishefsky SJ. J Org Chem. 2006;71:8329. doi: 10.1021/jo0610053. [DOI] [PubMed] [Google Scholar]
- 10.Solinski AE, Koval AB, Brzozowski RS, Morrison KR, Fraboni AJ, Carson CE, Eshraghi AR, Zhou G, Quivey RGJr, Voelz VA, Buttaro BA, Wuest WM. J Am Chem Soc. 2017;139:7188. doi: 10.1021/jacs.7b03879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steele AD, Keohane CE, Knouse KW, Rossiter SE, Williams SJ, Wuest WM. J Am Chem Soc. 2016;138:5833. doi: 10.1021/jacs.6b03373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steele AD, Knouse KW, Keohane CE, Wuest WM. J Am Chem Soc. 2015;137:7314. doi: 10.1021/jacs.5b04767. [DOI] [PubMed] [Google Scholar]
- 13.(a) Wu J, Lorenzo P, Zhong S, Ali M, Butts CP, Myers EL, Aggarwal VK. Nature. 2017;547 doi: 10.1038/nature23265. [DOI] [PubMed] [Google Scholar]; (b) Lorenzo P, Butts CP, Myers EL, Aggarwal VK. Biochemistry. 2017;56:6177. doi: 10.1021/acs.biochem.7b00994. [DOI] [PubMed] [Google Scholar]
- 14.Guchhait S, Chatterjee S, Ampapathi RS, Goswami RK. J Org Chem. 2017;82:2414. doi: 10.1021/acs.joc.6b02838. [DOI] [PubMed] [Google Scholar]
- 15.Harada K, Kubo M, Horiuchi H, Ishii A, Esumi T, Hioki H, Fukuyama Y. J Org Chem. 2015;80:7076. doi: 10.1021/acs.joc.5b00945. [DOI] [PubMed] [Google Scholar]
- 16.Sengupta S, Bae M, Oh DC, Dash U, Kim HJ, Song WY, Shin I, Sim T. J Org Chem. 2017;82:12947. doi: 10.1021/acs.joc.7b01719. [DOI] [PubMed] [Google Scholar]
- 17.Myers AG, Yang BH, Chen H, McKinstry L, Kopecky DJ, Gleason JL. J Am Chem Soc. 1997;119:6496. [Google Scholar]
- 18.Paladugu S, Mainkar PS, Chandrasekhar S. Tetrahedron Lett. 2017;58:2784. [Google Scholar]
- 19.Neres J, Labello NP, Somu RV, Boshoff HI, Wilson DJ, Vannada J, Chen L, Barry CE, 3rd, Bennett EM, Aldrich CC. J Med Chem. 2008;51:5349. doi: 10.1021/jm800567v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roth BL, Poot M, Yue ST, Millard PJ. Appl Environ Microbiol. 1997;63:2421. doi: 10.1128/aem.63.6.2421-2431.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Jennings MC, Ator LE, Paniak TJ, Minbiole KP, Wuest WM. ChemBioChem. 2014;15:2211. doi: 10.1002/cbic.201402254. [DOI] [PubMed] [Google Scholar]; (b) Wuest WM, Minbiole KP. Biscationic and Triscationic Amphiphiles as Antimicrobial Agents. 2016 Sep 29; US20160278375 A1. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.