

Abstract
Oxidative stress is closely associated with renal dysfunction following diabetes and hypertension. Angiotensin II (Ang II) can activate the NADPH- oxidase, increasing oxidative stress that is thought to blunt proximal tubular electrolyte transport and thereby oxygen consumption (QO2). We investigated the effect of Ang II on QO2 in immortalized mouse proximal tubular cells over-expressing the NADPH oxidase subunit p22phox; a model of increased oxidative stress. Cultured cells were exposed to either Ang II or H2O2 for 48 h. QO2 was determined during baseline (113 mmol/l NaCl; transport-dependent QO2) and during sodium-free conditions (transport-independent QO2). Ang II reduced transport-dependent QO2 in wild-types, but not in p22phox which also displayed increased QO2 at baseline. Transport-independent QO2 was increased in p22phox and Ang II had no additional effect, whereas it increased QO2 in wild-type. Addition of H2O2 reduced transport- dependent QO2 in wild-types, but not in p22phox. Transport-independent QO2 was unaffected by H2O2. The similar effects of Ang II and H2O2 to reduce transport- dependent QO2 suggest a direct regulatory role of oxidative stress. In accordance, the transport-dependent QO2 was reduced in p22phox already during baseline. The effects of Ang II on transport-independent QO2 was not replicated by H2O2, indicating direct regulation via Ang II-receptors independently of oxidative stress. However, the Ang II effect was absent in p22phox, suggesting that oxidative stress also modulates normal Ang II signaling. In conclusion, Ang II affects both transport-dependent and transport-independent QO2 in proximal tubular cells and may be an important pathway modulating renal QO2.
Keywords: Proximal tubule cell, Oxidative stress, Angiotensin-II, Oxygen consumption, Electrolyte transport
1 Introduction
Development of nephropathy due to diabetes and hypertension is strongly connected with concurrent development of renal hypoxia [1]. Renal hypoxia may develop with increased metabolic demand, renal anemia, or when renal peritubular capillary blood flow is decreased due to glomerular injury or vasoactive substances constricting the arterioles. Angiotensin II (Ang II), a vasoactive substance known to be increased in both hypertensive and diabetic kidneys [2, 3], induces oxidative stress and constriction of afferent as well as efferent arteriole [4]. Both these effects reduce renal blood flow and therefore reduce oxygen delivery, contributing to renal hypoxia. The metabolic demand by the kidney is largely determined by electrolyte reabsorption, accounting for 80 % of total kidney oxygen consumption (QO2) [5]. Interestingly, the energy-demanding Na+/K+-ATPase-activity is increased in diabetes [6], increasing QO2 and possibly limiting renal oxygenation. Diabetes-induced increased QO2 and development of renal hypoxia are closely linked to increased levels of oxidative stress, as demonstrated by the prevention of renal hypoxia using antioxidants [7]. Interestingly, Ang II activates the NADPH oxidase via the Ang II receptor subtype 1 (AT1-R), resulting in increased superoxide formation [8] and several studies have suggested a role for NADPH-oxidase in the development of both hypertension and nephropathy [9, 10].
In summary, renal QO2 is affected by mitochondrial activity, electrolyte transport and cellular QO2, all processes that are altered by oxidative stress. However, the detailed pathways of Ang II-mediated effects on proximal tubular QO2 and their relation to increased oxidative stress are presently unknown. Therefore, the present study separated the effects of Ang II on electrolyte transport-dependent QO2 from those on transport-independent QO2 in immortalized wild-type and p22phox overexpressing mouse proximal tubular cells. The latter is a model of increased oxidative stress and was utilized to separate the effects of AT1-R signaling per se from those of Ang II-induced oxidative stress.
2 Material and Methods
Immortalized proximal tubular cells with and without a stable over-expression of the NADPH oxidase subunit p22phox were maintained at 37°C and 5 % CO2 in Dulbecco’s Modified Eagle Medium/F12 medium containing 5 % fetal bovine serum. At 50 % confluency, cell splitting was routinely performed using 2.5 % trypsin. Both cell types were grown with or without Ang II (10−7 mol/l, Sigma-Aldrich, St Louis, MO, USA replaced every 12 h) and H2O2 (2.5 × 10−5 mol/l, Sigma-Aldrich, St Louis, MO, USA replaced every 12 h) for 48 h. Before measurements, cells were placed in suspension with 2.5 % trypsin followed by triple rinsing by slow centrifugation (100 × g, 4 min, 4°C) in ice-cold respiration buffer (in mmol/l: glucose 23.2; NaCl 113; KCl 4.0; NaHCO3 27.2; KH2PO4 1.0; MgCl2 1.2; CaCl2 1.0; HEPES 10.0; Ca2+-lactate 0.5; glutamine 2.0 and streptomycin 50 U/ml; osmolality 298 ± 2 mOsm checked with a freezing point osmometer, Fiske laboratories; pH 7.4) with or without Na+. In absence of NaCl and NaHCO3, osmolality was kept constant with 280.4 mOsm mannitol. The cell-suspension was kept on ice until QO2 was measured as described previously [11]. Briefly, a custom-made 1.1 ml gas-tight Plexiglas chamber thermostatically controlled at 37°C with continuous stirring from an air driven magnetic stirrer was used to determine QO2 on free-floating cells in respiration buffer with or without Na+ to evaluate transport-dependent QO2 and transport-independent QO2 respectively. QO2 was determined as rate of oxygen disappearance as measured by a modified Unisense-500 O2-sensor (Unisense A/S, Aarhus, Denmark) calibrated with the air-equilibrated buffer solution as 228 μmol/l O2 and Na2S2O5-saturated buffer as zero, and normalized to protein concentration. Statistical analyses were performed using GraphPad Prism software (GraphPad Software Inc., San Diego, CA, USA). Multiple comparisons between groups were performed using analysis of variance (ANOVA) followed by Sidak multiple comparisons test. All data are presented as mean ± standard error of the mean (SEM) and p < 0.05 was considered statistically significant.
3 Results
Cells overexpressing p22phox had reduced transport-dependent QO2 compared to wild-type. Ang II reduced transport-dependent QO2 in wild-type but not in p22phox. During baseline conditions, transport-independent QO2 was increased in p22phox compared to wild-type. Ang II displayed only a tendency to decrease transport- dependent oxygen consumption in p22phox whereas it increased transport- independent QO2 in wild-type (Fig. 21.1). Addition of H2O2 reduced transport-dependent QO2 in wild-type but only tended to reduce QO2 in p22phox. However, transport-independent QO2 was unaffected by H2O2 in both cell types (Fig. 21.2).
Fig. 21.1.
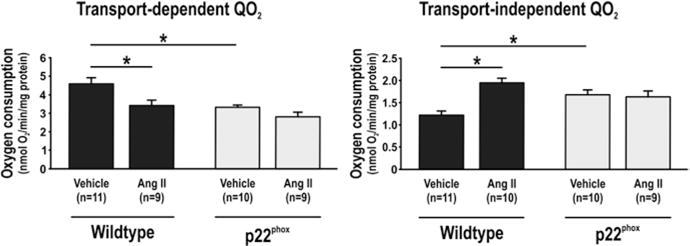
Transport-dependent (left) and transport-independent oxygen consumption (QO2, right) in immortalized wild-type mouse proximal tubular cells and corresponding cells overexpressing the NADPH oxidase subunit p22phox and the effect of 48 h exposure to angiotensin II. Asterisk denotes p < 0.05 and data are presented as mean ± SEM
Fig. 21.2.
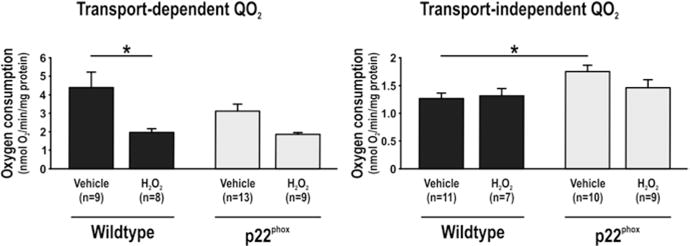
Transport-dependent (left) and transport-independent oxygen consumption (QO2, right) in immortalized wild-type mouse proximal tubular cells and corresponding cells overexpressing the NADPH oxidase subunit p22phox and the effect of 48 h exposure to H2O2. Asterisk denotes p < 0.05 and data are presented as mean ± SEM
4 Discussion
The present study demonstrates a role for Ang II in regulating transport-dependent as well as transport-independent QO2 in mouse proximal tubular cells. It is likely that Ang-II decreases transport-dependent QO2 by inducing oxidative stress, a hypothesis strengthened by the fact that the effect is mimicked by elevating oxidative stress via H2O2. Furthermore, the response to Ang II is reduced in cells with increased levels of oxidative stress during baseline, i.e. the p22phox, further highlighting oxidative stress as a crucial component in the mechanism of Ang II.
However, as the effects on transport-independent QO2 is not mimicked by H2O2 other mechanisms than oxidative stress may be involved.
It is known that Ang II increases superoxide formation by activating NADPH oxidase via AT-1R [8, 12]. Indeed, inhibition of AT-1R by olmesartan reduces oxidative stress independently of its blood pressure-lowering effect [13]. Several studies have suggested a role for NADPH oxidase in the development of hypertension and kidney disease [10, 14]. Oxidative stress is indeed increased in human hypertension [15] as well as in several hypertensive animal models [16]. AT1-Receptor blockade with candesartan has been reported equally effective as the superoxide dismutase mimetic tempol in normalizing renal pO2, implying that oxidative stress is directly involved [17]. Furthermore, inhibition of Ang II with angiotensin- converting enzyme (ACE) inhibitors and AT1-R blockers lowers renal QO2 [18]. Ang II acting on AT1-Receptors has been implied for the development in intrarenal hypoxia during hypertension, since 2 weeks of treatment with the AT1-R blocker candesartan to SHR rats normalize renal pO2 [19]. Finally, Ang II-dependent hypertension in rats increases transport-dependent QO2 in the thick ascending limb [20].
Cells with increased levels of oxidative stress displayed increased transport- independent QO2. This may be due to mitochondrial uncoupling by uncoupling protein (UCP)-2, a phenomenon known to be induced during conditions of increased oxidative stress, resulting in increased QO2 unrelated to ATP production [11, 21]. Importantly, diabetes-induced mitochondria uncoupling via UCP-2 is prevented by antioxidant treatment [22]. In the present study, addition of H2O2 to wildtype cells decreased transport-dependent oxygen consumption and did not affect transport- independent oxygen consumption, arguing against the presence of mitochondria uncoupling in these cells. However, it has been shown that mitochondria uncoupling is regulated by oxidative stress originating from the matrix side of the electron transport chain [23] and addition of H2O2 may therefore not be a strong enough signal to increase mitochondria uncoupling in cells with normal levels of oxidative stress. However, in cells with increased oxidative stress an increase in transport- independent oxygen consumption was evident, suggesting increased mitochondria uncoupling. Interestingly, the specific effect of Ang II on increasing transport- independent QO2 may indeed be due to increased mitochondrial uncoupling. In a study by Doughan et al., addition of Ang II to cultured bovine aortic endothelial cells increased mitochondrial superoxide production and mitochondrial uncoupling [24]. Recently, Ang II receptors have been localized to the mitochondrial inner membrane [25]. In kidneys from SHR rats, oxidative stress was increased and the mitochondria displayed increased H2O2 generation, decreased membrane potential and increased UCP-2 expression [26]. Importantly, the effect of Ang II on transport- independent QO2 was not replicated by increased oxidative stress per se, implying a crucial and specific role for AT1-R signaling. It is tempting to speculate that AT1-R located in the mitochondria are involved in these specific effects of Ang II. However, there was no effect of Ang II on transport-independent QO2 in cells with increased levels of oxidative stress, demonstrating that oxidative stress may still have a regulatory role in mediating the observed effect on transport-independent QO2 in wild-type cells. Reduced AT1-R expression or blunted receptor response in conditions with increased oxidative stress cannot be excluded.
In conclusion, the present study demonstrates that Ang II can affect both transport- dependent and transport-independent QO2 in mouse proximal tubular cells and may be an important pathway modulating renal QO2.
Contributor Information
Malou Friederich-Persson, Department of Medical Cell Biology, Uppsala University, Biomedical Center, Husargatan 3, Uppsala 751 23, Sweden.
William J. Welch, Department of Medicine, Georgetown University Medical Center, Washington DC, USA
Zaiming Luo, Department of Medicine, Georgetown University Medical Center, Washington DC, USA.
Fredrik Palm, Department of Medical Cell Biology, Uppsala University, Biomedical Center, Husargatan 3, Uppsala 751 23, Sweden; Department of Medical and Health Sciences, Linköping University, Linköping, Sweden; Center for Medical Image Science and Visualization, Linköping University, Linköping, Sweden.
Lina Nordquist, Department of Medical Cell Biology, Uppsala University, Uppsala, Sweden.
References
- 1.Nangaku M. Chronic hypoxia and tubulointerstitial injury: a final common pathway to end-stage renal failure. J Am Soc Nephrol. 2006;17:17–25. doi: 10.1681/ASN.2005070757. [DOI] [PubMed] [Google Scholar]
- 2.Kobori H, et al. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59:251–287. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 3.Nagai Y, et al. Temporary angiotensin II blockade at the prediabetic stage attenuates the development of renal injury in type 2 diabetic rats. J Am Soc Nephrol. 2005;16:703–711. doi: 10.1681/ASN.2004080649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arendshorst WJ, Brannstrom K, Ruan X. Actions of angiotensin II on the renal micro-vasculature. J Am Soc Nephrol. 1999;10:149–161. [PubMed] [Google Scholar]
- 5.Lassen NA, Munck O, Thaysen JH. Oxygen consumption and sodium reabsorption in the kidney. Acta Physiol Scand. 1961;51:371–384. doi: 10.1111/j.1748-1716.1961.tb02147.x. [DOI] [PubMed] [Google Scholar]
- 6.Korner A, et al. Increased renal metabolism in diabetes. Mechanism and functional implications. Diabetes. 1994;43:629–633. doi: 10.2337/diab.43.5.629. [DOI] [PubMed] [Google Scholar]
- 7.Palm F, et al. Reactive oxygen species cause diabetes-induced decrease in renal oxygen tension. Diabetologia. 2003;46:1153–1160. doi: 10.1007/s00125-003-1155-z. [DOI] [PubMed] [Google Scholar]
- 8.Chabrashvili T, et al. Effects of ANG II type 1 and 2 receptors on oxidative stress, renal NADPH oxidase, and SOD expression. Am J Physiol Regul Integr Comp Physiol. 2003;285:117–124. doi: 10.1152/ajpregu.00476.2002. [DOI] [PubMed] [Google Scholar]
- 9.Cifuentes ME, et al. Upregulation of p67(phox) and gp91(phox) in aortas from angiotensin II-infused mice. Am J Physiol Heart Circ Physiol. 2000;279:2234–2240. doi: 10.1152/ajpheart.2000.279.5.H2234. [DOI] [PubMed] [Google Scholar]
- 10.Rey FE, et al. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(−) and systolic blood pressure in mice. Circ Res. 2001;89:408–414. doi: 10.1161/hh1701.096037. [DOI] [PubMed] [Google Scholar]
- 11.Friederich M, et al. Diabetes-induced up-regulation of uncoupling protein-2 results in increased mitochondrial uncoupling in kidney proximal tubular cells. Biochim Biophys Acta. 2008;1777:935–940. doi: 10.1016/j.bbabio.2008.03.030. [DOI] [PubMed] [Google Scholar]
- 12.Chabrashvili T, et al. Expression and cellular localization of classic NADPH oxidase subunits in the spontaneously hypertensive rat kidney. Hypertension. 2002;39:269–274. doi: 10.1161/hy0202.103264. [DOI] [PubMed] [Google Scholar]
- 13.Fujimoto S, et al. Olmesartan ameliorates progressive glomerular injury in subtotal nephrectomized rats through suppression of superoxide production. Hypertens Res. 2008;31:305–313. doi: 10.1291/hypres.31.305. [DOI] [PubMed] [Google Scholar]
- 14.Landmesser U, et al. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension. 2002;40:511–515. doi: 10.1161/01.hyp.0000032100.23772.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar KV, Das UN. Are free radicals involved in the pathobiology of human essential hypertension? Free Radic Res Commun. 1993;19:59–66. doi: 10.3109/10715769309056499. [DOI] [PubMed] [Google Scholar]
- 16.Touyz RM. Oxidative stress and vascular damage in hypertension. Curr Hypertens Rep. 2000;2:98–105. doi: 10.1007/s11906-000-0066-3. [DOI] [PubMed] [Google Scholar]
- 17.Welch WJ, et al. Angiotensin-induced defects in renal oxygenation: role of oxidative stress. Am J Physiol Heart Circ Physiol. 2005;288:22–28. doi: 10.1152/ajpheart.00626.2004. [DOI] [PubMed] [Google Scholar]
- 18.Deng A, et al. Oxygen consumption in the kidney: effects of nitric oxide synthase isoforms and angiotensin II. Kidney Int. 2005;68:723–730. doi: 10.1111/j.1523-1755.2005.00450.x. [DOI] [PubMed] [Google Scholar]
- 19.Welch WJ, et al. Renal oxygenation defects in the spontaneously hypertensive rat: role of AT1 receptors. Kidney Int. 2003;63:202–208. doi: 10.1046/j.1523-1755.2003.00729.x. [DOI] [PubMed] [Google Scholar]
- 20.Silva GB, Garvin JL. Angiotensin II-dependent hypertension increases Na transport- related oxygen consumption by the thick ascending limb. Hypertension. 2008;52:1091–1098. doi: 10.1161/HYPERTENSIONAHA.108.120212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Echtay KS, et al. Superoxide activates mitochondrial uncoupling proteins. Nature. 2002;415:96–99. doi: 10.1038/415096a. [DOI] [PubMed] [Google Scholar]
- 22.Persson MF, et al. Coenzyme Q10 prevents GDP-sensitive mitochondrial uncoupling, glomerular hyperfiltration and proteinuria in kidneys from db/db mice as a model of type 2 diabetes. Diabetologia. 2012;55:1535–1543. doi: 10.1007/s00125-012-2469-5. [DOI] [PubMed] [Google Scholar]
- 23.Echtay KS, et al. Superoxide activates mitochondrial uncoupling protein 2 from the matrix side. Studies using targeted antioxidants. J Biol Chem. 2002;277:129–135. doi: 10.1074/jbc.M208262200. [DOI] [PubMed] [Google Scholar]
- 24.Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–496. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- 25.Abadir PM, et al. Identification and characterization of a functional mitochondrial angiotensin system. Proc Natl Acad Sci U S A. 2011;108:14849–14854. doi: 10.1073/pnas.1101507108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Cavanagh EM, et al. Renal mitochondrial dysfunction in spontaneously hypertensive rats is attenuated by losartan but not by amlodipine. Am J Physiol Regul Integr Comp Physiol. 2006;290:1616–1625. doi: 10.1152/ajpregu.00615.2005. [DOI] [PubMed] [Google Scholar]