

Abstract
Heterogeneous Stock (HS) populations allow for fine-resolution genetic mapping of a variety of complex traits. HS mice and rats were created from breeding together eight inbred strains, followed by maintaining the colony in a manner that minimizes inbreeding. After 50 or more generations of breeding, the resulting animals’ chromosomes represent a genetic mosaic of the founders’ haplotypes, with the average distance between recombination events in the centiMorgan range. This allows for genetic mapping to only a few Mb, a much smaller region than what can be identified using traditional F2 intercross or backcross mapping strategies. HS animals have been used to fine-map a variety of complex traits including anxiety and fear behaviors, diabetes, asthma, and heart disease, among others. Once a quantitative trait locus (QTL) has been identified, founder sequence and expression analysis can be used to identify underlying causal genes. In the following review, we provide an overview of how HS rats and mice have been used to identify genetic loci, and in some cases the causal genes, underlying complex traits. We discuss the creation and breeding strategies for both HS rats and mice. We then discuss the statistical analyses used to identify genetic loci, as well as strategies to identify causal genes underlying these loci. We end the chapter by discussing limitations faced when using HS populations, including several statistical challenges that have not been fully resolved.
Keywords: Resources for systems genetics, Genetic mapping, Outbred mice and rats, Expression analysis
1 Introduction
HS populations of mice and rats are created from breeding together eight inbred strains, followed by maintaining the colony in a manner that minimizes inbreeding (Fig. 1). After 50 or more generations of breeding, the resulting animals’ chromosomes are a genetic mosaic of the founders’ haplotypes, with the average distance between recombination events in the centiMorgan range allowing genetic mapping to only a few Mb [1, 2]. HS animals exhibit a high degree of both genetic and phenotypic diversity, allowing high-resolution genetic mapping for a wide variety of traits. While both HS rats and mice were originally created to be a resource population for experimental and selection studies [3, 4], Flint and colleagues demonstrated in 1999 that these populations can be used to narrow a previously identified quantitative trait locus (QTL) for anxiety [5] to only 0.8 cM [6], a large improvement over mapping studies using traditional F2 intercross or backcross approaches which generally map to 30 cM or more. Since that time, multiple studies have used HS rats and mice for genetic mapping of complex traits. Whilst this chapter focuses on mouse and rat HS, similar types of populations—in which each individual’s genome is a mosaic of the founders—have been made in these and other species [7–10]. Other outbred populations, such as advanced intercross lines [11, 12], the mouse Diversity Outcross (also created from eight inbred founder strains; [13]), and commercially available outbreds [14, 15], are also available and have been reviewed previously [16].
Fig. 1.
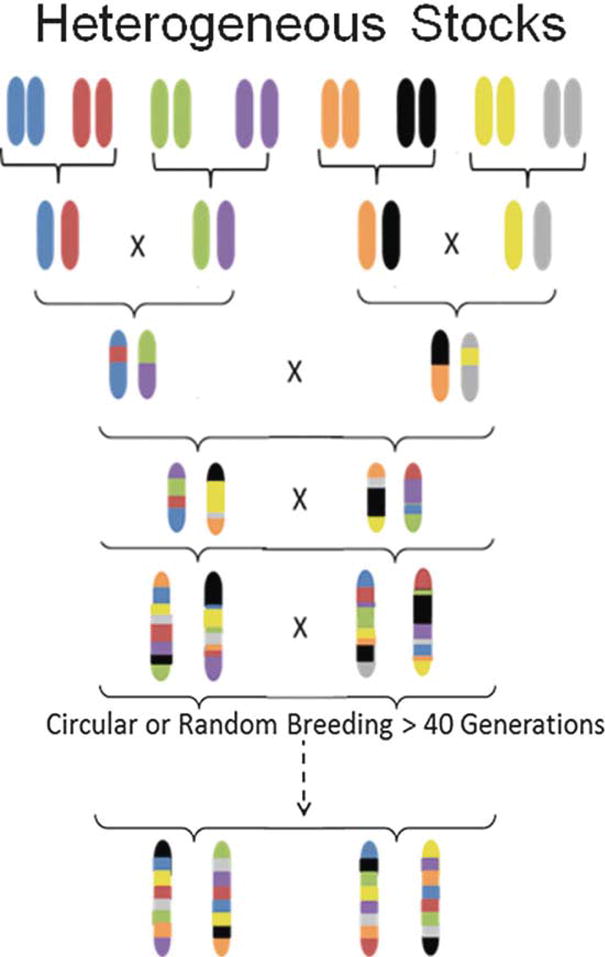
Breeding scheme for heterogeneous stock (HS) populations. HS are created by breeding together eight inbred strains. Once all eight genomes are combined, the animals are bred using either a circular strategy or random breeding. Existing HS mouse and rat colonies have been bred for over 50 generations. Figure adapted from Solberg Woods, 2014
Upon demonstrating success of the HS strategy for fine-mapping a single locus for a behavioral trait, the Flint lab went on to use HS mice to conduct a large multi-phenotype study, including traits involved in fear and anxiety behaviors, diabetes and asthma, and others. The study included the largest cohort of mice at that time (1904 mice) and resulted in the identification of 843 QTL at 25 % false discovery rate (FDR) with an average confidence interval of only 2.8 Mb 17]. In separate studies, HS mice have also been used to fine-map traits such as fear [18, 19], ethanol-induced locomotor activity [20], and arthritis [21]. Two HS mouse colonies have been created: the Boulder HS [4] and the Northport HS [20]. Six of the inbred founder strains are shared between these stocks, namely A/J, AKR/J, BALB/cJ, C3H/HeJ, C57BL/6J, DBA/2J. The additional founders of the Boulder HS are the strains Is/Bi and R111, while those of the Northport HS are CBA/2J and LP/J. Colonies were created and maintained with 24–40 breeder pairs using either a circular or random mating scheme.
With the success of HS mice, investigators began to also use HS rats for genetic fine-mapping. The HS rat colony (N:NIH-HS) was first established by the NIH in 1984 using the following inbred strains: ACI/N,BN/SsN, BUF/N, F344/N, M520/N, MR/N, WKY/N, WN/N [3]. The colony was maintained using 60 breeder pairs using a random mating strategy. Upon the retirement of the colony’s originator, Dr. Carl Hansen, in 2006 the colony was transferred to the laboratory of Dr. Eva Redei at Northwestern University where it was maintained for 2 years. In 2006, Dr. Redei transferred breeder pairs to the Medical College of Wisconsin in the United States and the Autonomous University of Barcelona in Spain. The Medical College of Wisconsin currently maintains 64 breeder pairs and is using a random breeding strategy, using kinship coefficent to ensure closely related pairs are not bred together. A smaller colony is also currently being maintained at Barcelona (Fernandez Teruel, personal communication). The first genetic mapping study in HS rats fine-mapped a single locus for glucose tolerance from 60 Mb to only 2.4 Mb [22]. Using expression QTL analysis and founder sequence data, Tpcn2 was identified as the likely causal gene within this region within only a few years [23]. Since that time, Flint and colleagues again conducted a large multi-phenotype study, including traits involved in anxiety, heart disease, and multiple sclerosis among others [24]. They were able to fine-map 355 QTL for 122 traits at 10 % FDR. An example of a scan for platelet aggregation is shown in Fig. 2. Using a merge analysis (described below) and protein modeling, they were able to identify 35 probable causative genes within these loci. These data are described in detail and publicly available at [25]. We note that different analysis methodologies were used in the mouse and rat HS experiments, reflecting methodological improvements (principally the development of mixed models, described below) in the interim. A recent study has also fine-mapped bone structure and strength in HS rats [26] and additional studies have demonstrated phenotypic variability, and thus suitability for future genetic studies, for additional phenotypic traits including kidney traits [27], drug abuse behaviors [28, 29], and behavioral and physiological responses to stress [30–32] and to ethanol [33–35]. Because of the rich history of the rat in behavioral studies, the HS rat will likely prove a useful model for genetic dissection of behaviors that are not easily modeled in the mouse (see ref. 36). The utility of the HS rat will also be enhanced by the recent availability of gene knockouts and other genetic manipulations now available in the rat [37, 38].
Fig. 2.
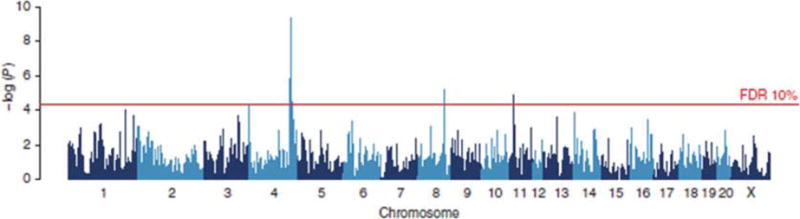
Genome scan for platelet aggregation as shown in Baud et al., 2013. The scan shows the results of a haplotype-based mixed model. The y-axis shows the negative log P values for association with variation in platelet aggregation. The association peak on chromosome 4 harbors the von Willebrand factor gene that was identified through sequence analysis as the causative gene
2 Methods: Statistical Analysis and Systems Genetics in HS Populations
In order to perform genetic mapping in HS populations, the underlying ancestral haplotype mosaics must first be determined. In this way, one can compute the probability that a particular locus in a given individual descended from which of the eight founder strains, thus providing increased information over simply analyzing genotype data (often based on biallelic single nucleotide polymorphisms or SNPs). HAPPY, a program developed by Mott and colleagues [2], uses a hidden Markov Model to determine the ancestral probabilities and has been shown to significantly improve genetic mapping results. Other programs, such as DOQTL which was more recently developed for use in the Diversity Outbred (DO) mouse population, a population of mice created from eight founder strains, very similar to the HS [39], can also be used to determine ancestral probabilities in the HS. Once probabilities are determined, regression modeling is conducted on the underlying mosaic structure to identify QTL.
2.1 Programs for QTL Analysis
Several programs are available for identifying QTL including Bagpipe [40], QTLRel [41], and DOQTL [39]. As with other highly recombinant populations, it is important to account for the complex family relationships within the HS population when conducting the analysis [24, 40, 42]. Most colonies are maintained using either a random or circular breeding strategy with anywhere from 40 to 60 breeding pairs, generally sufficient to minimize inbreeding and control genetic drift. Random mating strategies have the advantage of also avoiding reduced map expansion and therefore may be preferable to circular mating strategies [43]. As a result of the closed nature of the breeding strategy, animals within a colony are all related to each other but to differing degrees. If not accounted for, false positive QTL will be identified simply on the basis of relatedness, as opposed to actually pertaining to the phenotype. Moreover, in HS studies, in order to generate a large sample of animals for phenotyping, it is necessary to expand the colony size resulting in a large number of families. Thus the analysis of the phenotyped individuals acquires a mixture of linkage (due to family structure) and association (because all individuals are ultimately descended from eight founders). If the parents of the phenotyped generation are genotyped along with the phenotyped mice, then it is also possible to infer the maternal and paternal origin of the alleles, allowing the study of parent-of-origin effects [44].
There are several ways to account for unequal degrees of relatedness in outbred populations. These include mixed modeling approaches such as EMMA [45] or by resample model averaging [40]. When genome-wide genotype information is unavailable, and/or when the full pedigree is unknown, family can also be accounted for by including this as a random term in the model, as previously demonstrated for a single locus on rat chromosome 1 [22, 46]. When full pedigree information is known, QTLRel can be used to account for family relationships in highly recombinant animal populations such as the HS [41]. Resample model averaging approaches make use of genome-wide genotypes to determine genetic relatedness directly, and may prove advantageous under certain circumstances, particularly when pedigree information is unknown [40]. For mixed models, a kinship matrix is used to determine the genetic relatedness of each pair of individuals. This can be simply computed from SNP data (in the same way that it is computed in human studies) or from the ancestral haplotype mosaics [44]. Baud et al. [24] used a mixed model to control for differences in relatedness, whilst Valdar et al. 2006 [17] used resample model averaging (developed further in [40]). Each method has advantages and disadvantages. The mixed-model methodology is essentially equivalent to transforming both phenotypes and genotypes by multiplication by the square root of the inverse of the variance-covariance matrix, to create an uncorrelated dataset that can be analyzed by ordinary least squares. The method is well established and works well on phenotypes that are approximately normally distributed. Resampling methods, on the other hand, work well on phenotypes that are strongly skewed in distribution, but are slower than mixed models.
2.2 Identification of Causal Genes and Variants
Once QTLs are identified, there are several methods that can be used to identify the underlying causal gene(s) within the locus. These include a statistical merge analysis [47] (a form of genotype imputation), expression QTL mapping, and protein modeling. To date, complete genomes have been sequenced in the founder strains of the HS mice [48] and rats [24]. Relative to the respective reference genomes, more than four million SNPs per strain have been identified in the mouse [48] and more than two million SNPs per strain have been identified in the rat [24], in addition to structural variants and insertion/deletions. Because a repetitive portion of the genome (~15 % in mouse and ~12 % in rat) could not be reliably mapped to the reference strains, the number of variants identified is likely much larger than reported [24, 48]. The available sequence information can be used in several ways to identify candidate genes and/or variants within a fine-mapped QTL. By coupling founder sequences with relatively dense genotyping of the outbred population, it is possible to impute HS genotypes at all possible SNPs within a QTL. This can be followed by a merge analysis to narrow the potential causative variants within the QTL [47]. Briefly, a merge analysis uses probabilistically inferred descent to impute genotype dosages at unobserved loci, and then surveys those multiple imputed SNPs for their association with the phenotype. Using this method, two statistical models are compared: the haplotype model and the allelic model. In the haplotype model, the underlying ancestral probabilities at each SNP are used to model the QTL, with each founder haplotype permitted to carry a different phenotypic effect. This haplotype model is compared with one in which only the alleles for that SNP are used (allelic model). In the allelic model, the founder strain alleles are “merged” into two groups for each diallelic SNP: those containing allele “a” at a locus of interest and those containing allele “b” at this locus [47], with each group having a single phenotypic effect. Potentially causative variants are those in which the allelic model provides a better fit, that is, explaining the same amount of variation but with far fewer parameters, than the haplotype model (see ref. 21, 47). This method has proven useful in narrowing the number of causative variants within QTL mapped in HS mice [48] and rats [24]. A single causal variant, however, is rarely identified and follow-up studies are often needed to identify the causal variant. In addition, the method works less well when multiple causal variants underlie a single QTL [24], but can be used to show that a QTL cannot be explained by a single biallelic variant (as was the case for about half the QTLs detected by Baud et al.). Merge analysis is most useful for excluding genetic variants that cannot be causal.
Transcript abundance levels, based on RNAseq or microarray data, can also be used to identify causal genes underlying a QTL, as well as identification of gene networks that play a role in a given trait. Expression QTL (eQTL) analysis in HS populations allows investigators to map both cis and trans-eQTL to within only a few Mb of the transcript itself [49]. Overlap of cis-eQTL with physiological QTL can then be used to identify candidate genes within an interval, as demonstrated in HS rats by Tsaih et al. [23]. Our group identified Tpcn2 as a cis-eQTL within a physiological QTL for fasting glucose and insulin levels and demonstrated that glucose levels strongly correlated with Tpcn2 expression levels in the HS rats. We then demonstrated that fasting glucose and insulin in response to a glucose challenge were altered in Tpcn2 knock-out mice, and Tpcn2 was nominally correlated with fasting insulin levels in humans, providing evidence that Tpcn2 is the likely causal gene within this region. Transcript abundance levels can also be used to identify gene networks (groups of correlated transcripts) that may play a role in disease [50–53]. Although this strategy has not been applied specifically to HS populations to date, it offers a promising avenue of research for the future. Similarly, regions of the genome associated with open chromatin, such as DNAse-1 hypersensitive sites, identified in the mouse reference genome by the mouse ENCODE project [54] and across the HS founder strains [55] may be used to help identify causal variants and genes.
3 Further Considerations and Limitations
3.1 General Considerations
Using outbred HS populations offers several advantages over traditional F2 intercross or backcross strategies. The first is the ability to fine-map to only a few Mb, greatly decreasing the number of potential candidate genes within a given locus. Once loci are identified, full genome sequence is available for founder strains of the HS mouse [48] and HS rat [24] and this information has proven to be invaluable for identifying causative genes and variants within fine-mapped QTL. Despite these advantages, there are also several disadvantages that should be considered prior to embarking on a study with HS populations. Each animal is genetically and phenotypically distinct, so once a QTL, or even a gene, has been identified, there is no inbred model to go back to for functional testing, although the inbred founders might be used for this purpose. In addition, large numbers of animals are needed to have sufficient power for each mapping study and high density genotyping platforms (generally 10 thousand or greater) are required. Because each animal is unique, all animals need to be genotyped and phenotyped with each new study, as opposed to recombinant inbred lines where genotypes need be collected only once. As a result, it is beneficial to gather as much phenotype information as possible from the same group of animals, so that genotyping only needs to be done once and this information can be used to map multiple traits (e.g., [17, 24]). A further disadvantage is that these populations have been created through a single funnel (i.e., combining founder genomes only once), leading to loss of certain alleles and potentially unbalanced representation of the founder genomes. There are further considerations regarding confirmation of a potentially causal gene, and several statistical challenges that have not been resolved when using highly recombinant populations such as the HS.
3.2 Considerations in Determining Causality
Once a candidate gene is identified, follow-up studies are needed to confirm or disprove the role of that candidate in the trait. In addition to replication in a separate cohort, conducting a cross-species comparison can help provide support for the gene of interest. Of particular interest is human genome-wide association data which is often publicly available and can be mined to determine if a gene of interest falls just below the genome-wide significance threshold in humans. Once there is sufficient evidence to suspect a causal role for a specific gene, one of the most popular methods used to verify this gene is to study it in a knock-out model. Such methods have been available in the mouse since 1990 [56] and have recently become available in the rat [57]. Although popular because of their relative ease of constructing a knock-out, it is important to recognize that showing a change in phenotype in a knock-out model neither proves nor disproves a causative role of this gene at the QTL [58], particularly because there is no way to create a knock-out on the same background in which the QTL was identified. Methods such as quantitative complementation offer an alternative approach to test a causal role of the gene or variant [59, 60]. More importantly, new gene targeting approaches which allow for changes in single base pairs are now being used and offer a more realistic approach than a full gene knock-out (see ref. 61). The revolution in gene editing due to CRISPR/Cas9 technology (recently applied to rats in [38]) suggests that gene confirmation will become more straightforward in the future, at least for isolated coding variants. However, cases where multiple causal variants, carried on a single haplotype, are implicated will likely remain a challenge to prove causality, particularly if their effect is regulatory. It is therefore important for investigators to assess all available information, including expression, sequence, cross-species comparisons, results from a knock-out, allelic changes using CRISPR/Cas9 modifications, as well as possible in vitro studies to assess the potential causative role of a particular gene and/or variant.
3.3 Statistical Limitations
In addition to the above considerations, there are several statistical concerns that need to be taken into consideration when analyzing outbred HS populations. One of these is how best to determine significance thresholds. Cheng and Palmer [62] recently compared four different methods used in an advanced intercross population. They found that as long as an appropriate statistical model (i.e., one that takes into account the complex family relationships) is used, all methods worked relatively well, with gene-dropping (a simulation technique used in pedigree analysis) decreasing false QTL even when family is not taken into account. The best way to determine QTL confidence intervals is another challenge. Many studies use the 1.5 LOD drop method, (e.g., [46, 63, 64]), which was initially developed for use in F2 intercross populations. An alternative approach is to use nonparametric bootstrapping [65], in which the QTL is re-estimated under alternative datasets based on the original, with each alternative dataset created by resampling the individuals with replacement [66]. Although this method has been shown to be overly conservative [67], it does provide a complementary estimate of how sensitive the localization of the top QTL peak is to resampling, thus providing insight into whether more than one locus may underlie the QTL (see ref. 46).
Accurate determination of the joint effects of diplotypes (i.e., combinations of founder haplotypes—effectively any departure from the assumption of additivity in the haplotype effects) is also an on-going statistical challenge in outbred populations. Using the DO mouse population, investigators have looked at the effects of just the founder allele effects within the QTL [13, 63]. This has been useful in conducting haplotype analysis and narrowing the region of the QTL. However, within the HS or DO populations, there are in effect 36 possible diplotype combinations, and founder effects account for only eight of these. We [46] have recently published methods that accounts for all 36 possible diplotype effects and work in this area is on-going (see ref. 68).
A final statistical concern is that of statistical power. Although previous power calculations in multi-founder populations suggest that 1000–1500 animals provide sufficient power for mapping QTL explaining 5 % of the variance [2, 69, 70], these simulations do not account for the confounding effects of relatedness (e.g., [40, 42]), or marker ascertainment (e.g., [71]), and are therefore likely overstated. A previous study in the diversity outbred mouse population used as few as 150 mice; however this study provided sufficient power to map only 11 of 113 traits that were measured [13]. Studies in HS populations have used over 1000 animals, successfully mapping most traits analyzed [17, 24], demonstrating the increased power of these studies. In order to have more accurate power estimates for future studies in these populations, power calculations will need to account for both family structure and polygenes. That said, increasing sample size has many benefits: there is greater power to detect QTLs of small effect, QTL effect sizes are less likely to be overestimated due to Winner’s Curse, and confidence intervals are likely to be smaller and more accurate.
4 Conclusion
HS populations have proven useful for fine-mapping complex traits to only a few Mb, rapidly narrowing the number of potential candidate genes within the locus. Several strategies, including use of full founder sequence and expression QTL mapping, have been used to identify the underlying causal genes within these loci. It is important, however, to consider the cost and labor intensive challenges of working with HS populations, as well as the many unanswered statistical challenges that still remain. Despite these challenges, use of outbred models such as the HS has and will continue to enhance the knowledge of the genetic architecture of complex traits.
References
- 1.Mott R, Flint J. Dissecting quantitative traits in mice. Annu Rev Genomics Hum Genet. 2013;14:421–439. doi: 10.1146/annurev-genom-091212-153419. [DOI] [PubMed] [Google Scholar]
- 2.Mott R, Talbot CJ, Turri MG, Collins AC, Flint J. A method for fine mapping quantitative trait loci in outbred animal stocks. Proc Natl Acad Sci U S A. 2000;97(23):12649–12654. doi: 10.1073/pnas.230304397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hansen C, Spuhler K. Development of the National Institutes of Health genetically heterogeneous rat stock. Alcohol Clin Exp Res. 1984;8(5):477–479. doi: 10.1111/j.1530-0277.1984.tb05706.x. [DOI] [PubMed] [Google Scholar]
- 4.McClearn GE, Wilson JR, Meredith W. The use of isogenic and heterogenic mouse stocks in behavioral research. In: Lindzey G, Thiessen D, editors. Contributions to behavior-genetic analysis: the mouse as a prototype. Appleton Century Crofts; New York: 1970. pp. 3–22. [Google Scholar]
- 5.Flint J, Corley R, DeFries JC, Fulker DW, Gray JA, Miller S, Collins AC. A simple genetic basis for a complex psychological trait in laboratory mice. Science. 1995;269(5229):1432–1435. doi: 10.1126/science.7660127. [DOI] [PubMed] [Google Scholar]
- 6.Talbot CJ, Nicod A, Cherny SS, Fulker DW, Collins AC, Flint J. High-resolution mapping of quantitative trait loci in outbred mice. Nat Genet. 1999;21(3):305–308. doi: 10.1038/6825. [DOI] [PubMed] [Google Scholar]
- 7.Aylor DL, Valdar W, Foulds-Mathes W, Buus RJ, Verdugo RA, Baric RS, Ferris MT, Frelinger JA, Heise M, Frieman MB, Gralinski LE, Bell TA, Didion JD, Hua K, Nehrenberg DL, Powell CL, Steigerwalt J, Xie Y, Kelada SN, Collins FS, Yang IV, Schwartz DA, Branstetter LA, Chesler EJ, Miller DR, Spence J, Liu EY, McMillan L, Sarkar A, Wang J, Wang W, Zhang Q, Broman KW, Korstanje R, Durrant C, Mott R, Iraqi FA, Pomp D, Threadgill D, de Villena FP, Churchill GA. Genetic analysis of complex traits in the emerging Collaborative Cross. Genome Res. 2011;21(8):1213–1222. doi: 10.1101/gr.111310.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Durrant C, Tayem H, Yalcin B, Cleak J, Goodstadt L, de Villena FP, Mott R, Iraqi FA. Collaborative Cross mice and their power to map host susceptibility to Aspergillus fumigatus infection. Genome Res. 2011;21(8):1239–1248. doi: 10.1101/gr.118786.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kover PX, Valdar W, Trakalo J, Scarcelli N, Ehrenreich IM, Purugganan MD, Durrant C, Mott R. A multiparent advanced generation inter-cross to fine-map quantitative traits in Arabidopsis thaliana. PLoS Genet. 2009;5(7):e1000551. doi: 10.1371/journal.pgen.1000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Long AD, Macdonald SJ, King EG. Dissecting complex traits using the Drosophila Synthetic Population Resource. Trends Genet. 2014;30(11):488–495. doi: 10.1016/j.tig.2014.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Darvasi A, Soller M. Advanced inter-cross lines, an experimental population for fine genetic mapping. Genetics. 1995;141(3):1199–1207. doi: 10.1093/genetics/141.3.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parker CC, Cheng R, Sokoloff G, Palmer AA. Genome-wide association for methamphetamine sensitivity in an advanced intercross mouse line. Genes Brain Behav. 2012;11(1):52–61. doi: 10.1111/j.1601-183X.2011.00747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Svenson KL, Gatti DM, Valdar W, Welsh CE, Cheng R, Chesler EJ, Palmer AA, McMillan L, Churchill GA. High-resolution genetic mapping using the mouse diversity outbred population. Genetics. 2012;190(2):437–447. doi: 10.1534/genetics.111.132597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yalcin B, Flint J. Association studies in outbred mice in a new era of full-genome sequencing. Mamm Genome. 2012;23(9–10):719–726. doi: 10.1007/s00335-012-9409-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang W, Korstanje R, Thaisz J, Staedtler F, Harttman N, Xu L, Feng M, Yanas L, Yang H, Valdar W, Churchill GA, Dipetrillo K. Genome-wide association mapping of quantitative traits in outbred mice. G3 (Bethesda) 2012;2(2):167–174. doi: 10.1534/g3.111.001792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Solberg Woods LC. QTL mapping in outbred populations: successes and challenges. Physiol Genomics. 2014;46(3):81–90. doi: 10.1152/physiolgenomics.00127.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Valdar W, Solberg LC, Gauguier D, Burnett S, Klenerman P, Cookson WO, Taylor MS, Rawlins JN, Mott R, Flint J. Genome-wide genetic association of complex traits in heterogeneous stock mice. Nat Genet. 2006;38(8):879–887. doi: 10.1038/ng1840. [DOI] [PubMed] [Google Scholar]
- 18.Johannesson M, Lopez-Aumatell R, Stridh P, Diez M, Tuncel J, Blazquez G, Martinez-Membrives E, Canete T, Vicens-Costa E, Graham D, Copley RR, Hernandez-Pliego P, Beyeen AD, Ockinger J, Fernandez-Santamaria C, Gulko PS, Brenner M, Tobena A, Guitart-Masip M, Gimenez-Llort L, Dominiczak A, Holmdahl R, Gauguier D, Olsson T, Mott R, Valdar W, Redei EE, Fernandez-Teruel A, Flint J. A resource for the simultaneous high-resolution mapping of multiple quantitative trait loci in rats: the NIH heterogeneous stock. Genome Res. 2009;19(1):150–158. doi: 10.1101/gr.081497.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Talbot CJ, Radcliffe RA, Fullerton J, Hitzemann R, Wehner JM, Flint J. Fine scale mapping of a genetic locus for conditioned fear. Mamm Genome. 2003;14(4):223–230. doi: 10.1007/s00335-002-3059-5. [DOI] [PubMed] [Google Scholar]
- 20.Demarest K, Koyner J, McCaughran J, Jr, Cipp L, Hitzemann R. Further characterization and high-resolution mapping of quantitative trait loci for ethanol-induced locomotor activity. Behav Genet. 2001;31(1):79–91. doi: 10.1023/a:1010261909853. [DOI] [PubMed] [Google Scholar]
- 21.Johnsen AK, Valdar W, Golden L, Ortiz-Lopez A, Hitzemann R, Flint J, Mathis D, Benoist C. Genome-wide and species-wide dissection of the genetics of arthritis severity in heterogeneous stock mice. Arthritis Rheum. 2011;63(9):2630–2640. doi: 10.1002/art.30425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Solberg Woods LC, Holl K, Tschannen M, Valdar W. Fine-mapping a locus for glucose tolerance using heterogeneous stock rats. Physiol Genomics. 2010;41(1):102–108. doi: 10.1152/physiolgenomics.00178.2009. doi:10.1152/physiolgenomics.00178.2009, 00178.2009 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsaih SW, Holl K, Jia S, Kaldunski M, Tschannen M, He H, Andrae JW, Li SH, Stoddard A, Wiederhold A, Parrington J, Ruas da Silva M, Galione A, Meigs J, Hoffmann RG, Simpson P, Jacob H, Hessner M, Solberg Woods LC. Identification of a novel gene for diabetic traits in rats, mice, and humans. Genetics. 2014;198(1):17–29. doi: 10.1534/genetics.114.162982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baud A, Hermsen R, Guryev V, Stridh P, Graham D, McBride MW, Foroud T, Calderari S, Diez M, Ockinger J, Beyeen AD, Gillett A, Abdelmagid N, Guerreiro-Cacais AO, Jagodic M, Tuncel J, Norin U, Beattie E, Huynh N, Miller WH, Koller DL, Alam I, Falak S, Osborne-Pellegrin M, Martinez-Membrives E, Canete T, Blazquez G, Vicens-Costa E, Mont-Cardona C, Diaz-Moran S, Tobena A, Hummel O, Zelenika D, Saar K, Patone G, Bauerfeind A, Bihoreau MT, Heinig M, Lee YA, Rintisch C, Schulz H, Wheeler DA, Worley KC, Muzny DM, Gibbs RA, Lathrop M, Lansu N, Toonen P, Ruzius FP, de Bruijn E, Hauser H, Adams DJ, Keane T, Atanur SS, Aitman TJ, Flicek P, Malinauskas T, Jones EY, Ekman D, Lopez-Aumatell R, Dominiczak AF, Johannesson M, Holmdahl R, Olsson T, Gauguier D, Hubner N, Fernandez-Teruel A, Cuppen E, Mott R, Flint J. Combined sequence-based and genetic mapping analysis of complex traits in outbred rats. Nat Genet. 2013;45(7):767–775. doi: 10.1038/ng.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baud A, Guryev V, Hummel O, Johannesson M, Flint J. Genomes and phenomes of a population of outbred rats and its progenitors. Sci Data. 2014;1:140011. doi: 10.1038/sdata.2014.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alam I, Koller DL, Canete T, Blazquez G, Mont-Cardona C, Lopez-Aumatell R, Martinez-Membrives E, Diaz-Moran S, Tobena A, Fernandez-Teruel A, Stridh P, Diez M, Olsson T, Johannesson M, Baud A, Econs MJ, Foroud T. Fine mapping of bone structure and strength QTLs in heterogeneous stock rat. Bone. 2015;81:417–426. doi: 10.1016/j.bone.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Solberg Woods LC, Stelloh C, Regner KR, Schwabe T, Eisenhauer J, Garrett MR. Heterogeneous stock rats: a new model to study the genetics of renal phenotypes. Am J Physiol Renal Physiol. 2010;298(6):F1484–F1491. doi: 10.1152/ajprenal.00002.2010. doi: 10.1152/ajprenal.00002.2010, 00002.2010 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richards JB, Lloyd DR, Kuehlewind B, Militello L, Paredez M, Solberg Woods L, Palmer AA. Strong genetic influences on measures of behavioral-regulation among inbred rat strains. Genes Brain Behav. 2013;12(5):490–502. doi: 10.1111/gbb.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang T, Han W, Wang B, Jiang Q, Solberg-Woods LC, Palmer AA, Chen H. Propensity for social interaction predicts nicotine-reinforced behaviors in outbred rats. Genes Brain Behav. 2014;13(2):202–212. doi: 10.1111/gbb.12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diaz-Moran S, Palencia M, Mont-Cardona C, Canete T, Blazquez G, Martinez-Membrives E, Lopez-Aumatell R, Tobena A, Fernandez-Teruel A. Coping style and stress hormone responses in genetically heterogeneous rats: comparison with the Roman rat strains. Behav Brain Res. 2012;228(1):203–210. doi: 10.1016/j.bbr.2011.12.002. doi: 10.1016/j.bbr.2011.12.002, S0166-4328(11)00844-8 [pii] [DOI] [PubMed] [Google Scholar]
- 31.Lopez-Aumatell R, Guitart-Masip M, Vicens-Costa E, Gimenez-Llort L, Valdar W, Johannesson M, Flint J, Tobena A, Fernandez-Teruel A. Fearfulness in a large N/Nih genetically heterogeneous rat stock: differential profiles of timidity and defensive flight in males and females. Behav Brain Res. 2008;188(1):41–55. doi: 10.1016/j.bbr.2007.10.015. doi: 10.1016/j.bbr.2007.10.015, S0166-4328(07)00559-1 [pii] [DOI] [PubMed] [Google Scholar]
- 32.Lopez-Aumatell R, Vicens-Costa E, Guitart-Masip M, Martinez-Membrives E, Valdar W, Johannesson M, Canete T, Blazquez G, Driscoll P, Flint J, Tobena A, Fernandez-Teruel A. Unlearned anxiety predicts learned fear: a comparison among heterogeneous rats and the Roman rat strains. Behav Brain Res. 2009;202(1):92–101. doi: 10.1016/j.bbr.2009.03.024. doi: 10.1016/j.bbr.2009.03.024, S0166-4328(09)00185-5 [pii] [DOI] [PubMed] [Google Scholar]
- 33.Bice PJ, Liang T, Zhang L, Graves TJ, Carr LG, Lai D, Kimpel MW, Foroud T. Fine mapping and expression of candidate genes within the chromosome 10 QTL region of the high and low alcohol-drinking rats. Alcohol. 2010;44(6):477–485. doi: 10.1016/j.alcohol.2010.06.004. doi: 10.1016/j.alcohol.2010.06.004, S0741-8329(10)00079-0 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foroud T, Bice P, Castelluccio P, Bo R, Miller L, Ritchotte A, Lumeng L, Li TK, Carr LG. Identification of quantitative trait loci influencing alcohol consumption in the high alcohol drinking and low alcohol drinking rat lines. Behav Genet. 2000;30(2):131–140. doi: 10.1023/a:1001955205117. [DOI] [PubMed] [Google Scholar]
- 35.Spuhler K, Deitrich RA. Correlative analysis of ethanol-related phenotypes in rat inbred strains. Alcohol Clin Exp Res. 1984;8(5):480–484. doi: 10.1111/j.1530-0277.1984.tb05707.x. [DOI] [PubMed] [Google Scholar]
- 36.Parker CC, Chen H, Flagel SB, Geurts AM, Richards JB, Robinson TE, Solberg Woods LC, Palmer AA. Rats are the smart choice: rationale for a renewed focus on rats in behavioral genetics. Neuropharmacology. 2014;76Pt B:250–258. doi: 10.1016/j.neuropharm.2013.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katter K, Geurts AM, Hoffmann O, Mates L, Landa V, Hiripi L, Moreno C, Lazar J, Bashir S, Zidek V, Popova E, Jerchow B, Becker K, Devaraj A, Walter I, Grzybowksi M, Corbett M, Filho AR, Hodges MR, Bader M, Ivics Z, Jacob HJ, Pravenec M, Bosze Z, Rulicke T, Izsvak Z. Transposon-mediated transgenesis, transgenic rescue, and tissue-specific gene expression in rodents and rabbits. FASEB J. 2013;27(3):930–941. doi: 10.1096/fj.12-205526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao L, Oliver E, Maratou K, Atanur SS, Dubois OD, Cotroneo E, Chen CN, Wang L, Arce C, Chabosseau PL, Ponsa-Cobas J, Frid MG, Moyon B, Webster Z, Aldashev A, Ferrer J, Rutter GA, Stenmark KR, Aitman TJ, Wilkins MR. The zinc transporter ZIP12 regulates the pulmonary vascular response to chronic hypoxia. Nature. 2015;524(7565):356–360. doi: 10.1038/nature14620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gatti DM, Svenson KL, Shabalin A, Wu LY, Valdar W, Simecek P, Goodwin N, Cheng R, Pomp D, Palmer A, Chesler EJ, Broman KW, Churchill GA. Quantitative trait locus mapping methods for diversity outbred mice. G3 (Bethesda) 2014;4(9):1623–1633. doi: 10.1534/g3.114.013748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valdar W, Holmes CC, Mott R, Flint J. Mapping in structured populations by resample model averaging. Genetics. 2009;182(4):1263–1277. doi: 10.1534/genetics.109.100727. doi: 10.1534/genetics.109.100727, genetics.109.100727 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng R, Abney M, Palmer AA, Skol AD. QTLRel: an R package for genome-wide association studies in which relatedness is a concern. BMC Genet. 2011;12:66. doi: 10.1186/1471-2156-12-66. doi: 10.1186/1471-2156-12-66 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng R, Lim JE, Samocha KE, Sokoloff G, Abney M, Skol AD, Palmer AA. Genome-wide association studies and the problem of relatedness among advanced inter-cross lines and other highly recombinant populations. Genetics. 2010;185(3):1033–1044. doi: 10.1534/genetics.110.116863. doi: 10.1534/genetics.110.116863, genetics.110.116863 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rockman MV, Kruglyak L. Breeding designs for recombinant inbred advanced inter-cross lines. Genetics. 2008;179(2):1069–1078. doi: 10.1534/genetics.107.083873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mott R, Yuan W, Kaisaki P, Gan X, Cleak J, Edwards A, Baud A, Flint J. The architecture of parent-of-origin effects in mice. Cell. 2014;156(1–2):332–342. doi: 10.1016/j.cell.2013.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, Eskin E. Efficient control of population structure in model organism association mapping. Genetics. 2008;178(3):1709–1723. doi: 10.1534/genetics.107.080101. doi: 10.1534/genetics.107.080101, doi:178/3/1709 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Solberg Woods LC, Holl KL, Oreper D, Xie Y, Tsaih SW, Valdar W. Fine-mapping diabetes-related traits, including insulin resistance, in heterogeneous stock rats. Physiol Genomics. 2012;44(21):1013–1026. doi: 10.1152/physiolgenomics.00040.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yalcin B, Flint J, Mott R. Using progenitor strain information to identify quantitative trait nucleotides in outbred mice. Genetics. 2005;171(2):673–681. doi: 10.1534/genetics.104.028902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, Furlotte NA, Eskin E, Nellaker C, Whitley H, Cleak J, Janowitz D, Hernandez-Pliego P, Edwards A, Belgard TG, Oliver PL, McIntyre RE, Bhomra A, Nicod J, Gan X, Yuan W, van der Weyden L, Steward CA, Bala S, Stalker J, Mott R, Durbin R, Jackson IJ, Czechanski A, Guerra-Assuncao JA, Donahue LR, Reinholdt LG, Payseur BA, Ponting CP, Birney E, Flint J, Adams DJ. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477(7364):289–294. doi: 10.1038/nature10413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang GJ, Shifman S, Valdar W, Johannesson M, Yalcin B, Taylor MS, Taylor JM, Mott R, Flint J. High resolution mapping of expression QTLs in heterogeneous stock mice in multiple tissues. Genome Res. 2009;19(6):1133–1140. doi: 10.1101/gr.088120.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen Y, Zhu J, Lum PY, Yang X, Pinto S, MacNeil DJ, Zhang C, Lamb J, Edwards S, Sieberts SK, Leonardson A, Castellini LW, Wang S, Champy MF, Zhang B, Emilsson V, Doss S, Ghazalpour A, Horvath S, Drake TA, Lusis AJ, Schadt EE. Variations in DNA elucidate molecular networks that cause disease. Nature. 2008;452(7186):429–435. doi: 10.1038/nature06757. doi: 10.1038/nature06757, nature06757 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ghazalpour A, Doss S, Zhang B, Wang S, Plaisier C, Castellanos R, Brozell A, Schadt EE, Drake TA, Lusis AJ, Horvath S. Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet. 2006;2(8):e130. doi: 10.1371/journal.pgen.0020130. doi: 10.1371/journal.pgen.0020130, 06-PLGE-RA-0128R2 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keller MP, Choi Y, Wang P, Belt Davis D, Rabaglia ME, Oler AT, Stapleton DS, Argmann C, Schueler KL, Edwards S, Steinberg HA, Chaibub Neto E, Kleinhanz R, Turner S, Hellerstein MK, Schadt EE, Yandell BS, Kendziorski C, Attie AD. A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res. 2008;18(5):706–716. doi: 10.1101/gr.074914.107. doi: 10.1101/gr.074914.107, gr.074914.107 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schadt EE, Lamb J, Yang X, Zhu J, Edwards S, Guhathakurta D, Sieberts SK, Monks S, Reitman M, Zhang C, Lum PY, Leonardson A, Thieringer R, Metzger JM, Yang L, Castle J, Zhu H, Kash SF, Drake TA, Sachs A, Lusis AJ. An integrative genomics approach to infer causal associations between gene expression and disease. Nat Genet. 2005;37(7):710–717. doi: 10.1038/ng1589. doi: 10.1038/ng1589, ng1589 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stamatoyannopoulos JA, Snyder M, Hardison R, Ren B, Gingeras T, Gilbert DM, Groudine M, Bender M, Kaul R, Canfield T, Giste E, Johnson A, Zhang M, Balasundaram G, Byron R, Roach V, Sabo PJ, Sandstrom R, Stehling AS, Thurman RE, Weissman SM, Cayting P, Hariharan M, Lian J, Cheng Y, Landt SG, Ma Z, Wold BJ, Dekker J, Crawford GE, Keller CA, Wu W, Morrissey C, Kumar SA, Mishra T, Jain D, Byrska-Bishop M, Blankenberg D, Lajoie BR, Jain G, Sanyal A, Chen KB, Denas O, Taylor J, Blobel GA, Weiss MJ, Pimkin M, Deng W, Marinov GK, Williams BA, Fisher-Aylor KI, Desalvo G, Kiralusha A, Trout D, Amrhein H, Mortazavi A, Edsall L, McCleary D, Kuan S, Shen Y, Yue F, Ye Z, Davis CA, Zaleski C, Jha S, Xue C, Dobin A, Lin W, Fastuca M, Wang H, Guigo R, Djebali S, Lagarde J, Ryba T, Sasaki T, Malladi VS, Cline MS, Kirkup VM, Learned K, Rosenbloom KR, Kent WJ, Feingold EA, Good PJ, Pazin M, Lowdon RF, Adams LB. An encyclopedia of mouse DNA elements (Mouse ENCODE) Genome Biol. 2012;13(8):418. doi: 10.1186/gb-2012-13-8-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hosseini M, Goodstadt L, Hughes JR, Kowalczyk MS, de Gobbi M, Otto GW, Copley RR, Mott R, Higgs DR, Flint J. Causes and consequences of chromatin variation between inbred mice. PLoS Genet. 2013;9(6):e1003570. doi: 10.1371/journal.pgen.1003570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thomas KR, Capecchi MR. Targeted disruption of the murine int-1 proto-oncogene resulting in severe abnormalities in midbrain and cerebellar development. Nature. 1990;346(6287):847–850. doi: 10.1038/346847a0. [DOI] [PubMed] [Google Scholar]
- 57.Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM, Jenkins SS, Wood A, Cui X, Meng X, Vincent A, Lam S, Michalkiewicz M, Schilling R, Foeckler J, Kalloway S, Weiler H, Menoret S, Anegon I, Davis GD, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Jacob HJ, Buelow R. Knockout rats via embryo microinjection of zinc-finger nucleases. Science. 2009;325(5939):433. doi: 10.1126/science.1172447. doi: 10.1126/science.1172447, doi:325/5939/433 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Flint J, Eskin E. Genome-wide association studies in mice. Nat Rev Genet. 2012;13(11):807–817. doi: 10.1038/nrg3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Long AD, Mullaney SL, Mackay TF, Langley CH. Genetic interactions between naturally occurring alleles at quantitative trait loci and mutant alleles at candidate loci affecting bristle number in Drosophila melanogaster. Genetics. 1996;144(4):1497–1510. doi: 10.1093/genetics/144.4.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yalcin B, Willis-Owen SA, Fullerton J, Meesaq A, Deacon RM, Rawlins JN, Copley RR, Morris AP, Flint J, Mott R. Genetic dissection of a behavioral quantitative trait locus shows that Rgs2 modulates anxiety in mice. Nat Genet. 2004;36(11):1197–1202. doi: 10.1038/ng1450. [DOI] [PubMed] [Google Scholar]
- 61.Adams DJ, van der Weyden L. Contemporary approaches for modifying the mouse genome. Physiol Genomics. 2008;34(3):225–238. doi: 10.1152/physiolgenomics.90242.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cheng R, Palmer AA. A simulation study of permutation, bootstrap, and gene dropping for assessing statistical significance in the case of unequal relatedness. Genetics. 2013;193(3):1015–1018. doi: 10.1534/genetics.112.146332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Logan RW, Robledo RF, Recla JM, Philip VM, Bubier JA, Jay JJ, Harwood C, Wilcox T, Gatti DM, Bult CJ, Churchill GA, Chesler EJ. High-precision genetic mapping of behavioral traits in the diversity outbred mouse population. Genes Brain Behav. 2013;12(4):424–437. doi: 10.1111/gbb.12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Samocha KE, Lim JE, Cheng R, Sokoloff G, Palmer AA. Fine mapping of QTL for prepulse inhibition in LG/J and SM/J mice using F(2) and advanced intercross lines. Genes Brain Behav. 2010;9(7):759–767. doi: 10.1111/j.1601-183X.2010.00613.x. doi: 10.1111/j.1601-183X.2010.00613.x, GBB613 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hastie T, Tibshirani R, Friedman J. The elements of statistical learning: data mining, inference, and prediction. 2nd. Springer; New York: 2009. [Google Scholar]
- 66.Visscher PM, Thompson R, Haley CS. Confidence intervals in QTL mapping by bootstrapping. Genetics. 1996;143(2):1013–1020. doi: 10.1093/genetics/143.2.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Manichaikul A, Dupuis J, Sen S, Broman KW. Poor performance of bootstrap confidence intervals for the location of a quantitative trait locus. Genetics. 2006;174(1):481–489. doi: 10.1534/genetics.106.061549. doi: 10.1534/genetics.106.061549, genetics.106.061549 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Durrant C, Mott R. Bayesian quantitative trait locus mapping using inferred haplotypes. Genetics. 2010;184(3):839–852. doi: 10.1534/genetics.109.113183. doi: 10.1534/genetics.109.113183, genetics.109.113183 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mott R, Flint J. Simultaneous detection and fine mapping of quantitative trait loci in mice using heterogeneous stocks. Genetics. 2002;160(4):1609–1618. doi: 10.1093/genetics/160.4.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Valdar WS, Flint J, Mott R. QTL fine-mapping with recombinant-inbred heterogeneous stocks and in vitro heterogeneous stocks. Mamm Genome. 2003;14(12):830–838. doi: 10.1007/s00335-003-3021-1. [DOI] [PubMed] [Google Scholar]
- 71.Valdar W, Flint J, Mott R. Simulating the collaborative cross: power of quantitative trait loci detection and mapping resolution in large sets of recombinant inbred strains of mice. Genetics. 2006;172(3):1783–1797. doi: 10.1534/genetics.104.039313. doi: 10.1534/genetics.104.039313, doi:genetics.104.039313 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]