

Abstract
Objective
A decline in mitochondrial function and biogenesis as well as increased reactive oxygen species (ROS) are important determinants of aging. With advancing age, there is a concomitant reduction in circulating levels of insulin-like growth factor-1 (IGF-1) that is closely associated with neuronal aging and neurodegeneration. In this study, we investigated the effect of the decline in IGF-1 signaling with age on astrocyte mitochondrial metabolism and astrocyte function and its association with learning and memory.
Methods
Learning and memory was assessed using the radial arm water maze in young and old mice as well as tamoxifen-inducible astrocyte-specific knockout of IGFR (GFAP-CreTAM/igfrf/f). The impact of IGF-1 signaling on mitochondrial function was evaluated using primary astrocyte cultures from igfrf/f mice using AAV-Cre mediated knockdown using Oroboros respirometry and Seahorse assays.
Results
Our results indicate that a reduction in IGF-1 receptor (IGFR) expression with age is associated with decline in hippocampal-dependent learning and increased gliosis. Astrocyte-specific knockout of IGFR also induced impairments in working memory. Using primary astrocyte cultures, we show that reducing IGF-1 signaling via a 30–50% reduction IGFR expression, comparable to the physiological changes in IGF-1 that occur with age, significantly impaired ATP synthesis. IGFR deficient astrocytes also displayed altered mitochondrial structure and function and increased mitochondrial ROS production associated with the induction of an antioxidant response. However, IGFR deficient astrocytes were more sensitive to H2O2-induced cytotoxicity. Moreover, IGFR deficient astrocytes also showed significantly impaired glucose and Aβ uptake, both critical functions of astrocytes in the brain.
Conclusions
Regulation of astrocytic mitochondrial function and redox status by IGF-1 is essential to maintain astrocytic function and coordinate hippocampal-dependent spatial learning. Age-related astrocytic dysfunction caused by diminished IGF-1 signaling may contribute to the pathogenesis of Alzheimer's disease and other age-associated cognitive pathologies.
Keywords: Primary astrocytes, IGF-1, Amyloid, Mitochondria, ROS, Alzheimer's disease
Graphical abstract

Highlights
-
•
Altered mitochondrial structure and function with IGFR deficiency in astrocytes is proposed.
-
•
Increased reactive oxygen species production and susceptibility to peroxide induced cytotoxicity.
-
•
Decreased Aβ uptake and impairment in spatial working memory.
1. Introduction
Aging is associated with global reductions in the brain's adaptive response to injury, global increases in oxidative load [reviewed by [1], [2]], reductions in mitochondrially-derived energy production [3], and a related decline in neuronal and astroglial function [4], [5], [6]. Mitochondria consume approximately 90% of cellular oxygen during cellular respiration generating a constant flux of free radicals which, when dysregulated, leads to sustained oxidative stress/damage [7]. Diminished ATP levels, excessive radical production, and dysregulated apoptosis are part of the etiology of many age-related human diseases, including neurodegenerative disorders such as Alzheimer's disease [7], [8].
Insulin-like growth factor-1 (IGF-1) is a potent trophic factor, the levels of which substantially decline with age [9]. Circulating IGF-1 is reduced by ∼60% in advanced age while levels of IGF-1 in the cerebrospinal fluid decline by ∼30%. IGF-1 signals through its cognate receptor, IGFR, and is produced and secreted into the bloodstream by the liver in response to growth hormone (GH) signaling. IGF-1 ligand binding induces receptor dimerization and trans-autophosphorylation of the intracellular kinase domains thereby activating downstream signaling cascades including the prototypical PI3K/AKT pathway, which drives cell survival and growth. IGF-1 has been reported to have pleiotropic effects on various tissues and cell types across the organism in a tissue- and gender–specific manner. Deficiency of GH/IGF-1 throughout adult life has been shown to extend lifespan in many animal models including nematodes, flies, and mice [10], [11]. Conversely, reduced IGF-1 has been associated with, among others, oxidative stress [12], [13] and increased risk of cardiovascular disease [14], [15], stroke [16], [17], and cognitive decline [18], [19], [20], [21]. As a result, it is necessary to study the actions of IGF-1 on specific cell types to understand the complex role of this peptide in aging and age-related disease.
In addition to circulating IGF-1 produced by the liver, this peptide is also produced locally in tissues and has autocrine/paracrine actions on cells. Astrocytes play an integral role in the maintenance and modulation of neuronal health and function by supplying important trophic factors, such as IGF-1, and energy substrates necessary for neuronal function [22], [23], [24]. IGF-1 signaling regulates key aspects of astrocyte function, including glucose uptake [25], [26], [27], regulation of glutamate transport [28], and protection from oxidative stress [29] in the brain. Reduced serum IGF-1 and its signaling in astrocytes is also associated with Alzheimer's disease progression and pathology [30], [31]. In support of the protective effect of IGF-1, decreased IGF-1 availability in the brain by ectopic expression of IGF-1 binding protein (IGFBP-1) has been shown to reduce astrocyte response to injury [32]. IGF-1 has also been shown to protect astrocytes and thereby neurons against oxidative stress [29], [33]. Moreover, astrocyte-specific overexpression of IGF-1 protects hippocampal neurons and ameliorates cognitive impairments resulting from oxidative stress due to traumatic brain injury [34]. Furthermore, a decline in astrocytic mitochondrial function is associated with memory impairments [6], [35], [36] and stroke [37], [38]. These observations demonstrate that IGF-1 regulation of astrocyte redox homeostasis is a key aspect of brain function that may be dysregulated with age. To date, however, there is little information on IGF-1 regulation of astrocyte mitochondrial energetics and redox homeostasis in astrocytes during brain aging.
In this study, we investigated the role of IGF-1 signaling in astrocytic function. We show that spatial learning deficits in aged mice is associated with decreased IGFR expression in the hippocampus and increased gliosis. More importantly, astrocyte-specific knockout of IGFR in mice show impairments in hippocampal-dependent working memory. To elucidate the effects of IGF-1 signaling on astrocyte mitochondrial function, dysregulation of which is proposed to underlie tissue dysfunction during aging, we utilized primary cultured astrocytes and astrocyte-specific IGFR knockout mice. We show that astrocytes with IGFR knockout exhibit decreased mitochondrial energy charge and respiration, rounded perinuclear localization of mitochondrial networks, and increased mitochondrial ROS production accompanied by increased antioxidant response. Furthermore, IGFR deficiency in astrocytes lead to increased susceptibility to oxidant stress and impaired Aβ1−42 uptake. Finally, astrocytic knockout of IGFR in mice show impairment in hippocampal-dependent spatial learning, suggesting that targeted intervention of mitochondrial function in astrocytes may lead to improved therapeutics for age-related mitochondrial dysfunction and Alzheimer's disease.
2. Materials and methods
2.1. Animals
All procedures were approved by and followed the guidelines of the Institutional Animal Care and Use Committee of OUHSC. Young (4–6 months) and old (22–24 months) C57Bl/6 mice were obtained from the NIA colony at the National Institutes of Health and Igfrf/f (B6;129-Igf1rtm2Arge/J; loxP sites flanking exon 3) and GFAP-Cre (B6.Cg-TgGFAP-cre/ERT2/505Fmv/J) mice were obtained from Jackson laboratories. Mice were housed (3–4 per cage) in Allentown XJ cages with Anderson's Enrich-o-cob bedding (Maumee, OH). Igfrf/f mice were bred in house to generate experimental cohorts. These mice were housed in the Rodent Barrier Facility (RBF) at OUHSC, which is a specific pathogen free (including helicobacter and parvovirus) facility. Mice were bred on a 14-h light/10-h dark cycle and weaned mice were maintained in a 12-h light/12-h dark cycle at 21 °C and were given access to standard irradiated bacteria-free rodent chow (5053 Pico Lab, Purina Mills, Richmond, IN) and reverse osmosis filtered water ad libitum. GFAP-CreERT2 (males) mice were bred with Igfrf/f (females) for to generate GFAP-CreERT2/Igfr+/− males, which were bred with Igfrf/f female mice to obtain the founder colony of Cre+/Igfr homozygous floxed mice. These mice were allowed to breed with Igfrf/f mice to generate experimental cohorts of Cre+ and Cre-/Igfrf/f mice. Mice were injected intraperitoneally with tamoxifen (75 mg tamoxifen/kg body weight) dissolved in corn oil or sham (corn oil) only for 5 days at 3 months. Mice were allowed to recover for 2 months before behavioral assessments.
2.2. Radial arm water maze (RAWM)
Mice (young and old C57Bl/6; n = 10/group) were tested for spatial learning using an 8-arm radial arm water maze. Mice were given 8 × 60-second trials a day for 3 days to find a hidden platform in one arm of an 8-arm radial arm water maze that is 67 cm in diameter filled about 2/3 rds with an opaque liquid (water with food coloring). Animals were randomly placed in an arm other than the target arm. Mice were guided to the platform if they failed to find the target at the end of each 60-sec trial. Animal movements in the maze were recorded by a camera and tracked using an automated tracking system (Noldus Ethovision XT 11, Wageningen, Netherlands). The number of errors (number of entries into incorrect arms) and pathlength (cm; total distance traveled) to target were recorded. An error was counted when the animal had traversed 2/3 the length of the arm that did not have the platform.
Similarly, GFAP-CreERT2/Igfrf/f (TAM-Cre; n = 11) and controls (TAM-Igfr: n = 10 and Sham-Cre: n = 6) were tested for spatial learning in the RAWM using a working memory task. Mice were randomly placed in one arm of the RAWM and were given 4 × 60-second trials a day (beginning at 09:00 h) to learn the location of the hidden platform that is submerged in one arm. As before, mice in the maze were tracked and their behavior assessed. The number of incorrect entries to platform were recorded as before. After the short initial learning phase of the study on day 1, the submerged platform was moved to a different arm on day 2, thereby simulating a short term/working memory paradigm [39] and mice were evaluated in the number entries into the incorrect arms recorded. Data were analyzed using SigmaPlot 10.0 (Systat Software Inc., Germany) and Graphpad Prism 6.0.
2.3. Astrocyte primary cultures
Primary astrocytes and neurons were isolated from Igfrf/f mice on postnatal day 1–3 in accordance with the approved Institutional Animal Care and Use Committees guidelines at the University of Oklahoma HSC. Astrocytes and neurons were isolated as described in [40]. In brief, following papain enzymatic digestion and trituration, cortical cells were resuspended in growth media [DMEM containing 2% NuSerum, 10% fetal bovine serum, penicillin (10 units/ml), streptomycin (10 μg/ml), and l-glutamine (29.2 μg/ml)] and seeded on 50 μg/ml poly-d-lysine-coated at a density of 1.5 × 106/10 cm2 plate. Cells were fed every 3–4 days, with half of the media being replaced with fresh growth media. For astrocyte cultures, after 7 days in vitro (DIV7), cells were trypsinized and plated on to poly-D lysine coated plates at a density of 1.5 × 106/10 cm2 plate. Cultures were treated with AAV9-CMV-GFP (GFP) or AAV-CMV-GFP-CRE (CRE) at 8 × 103 GC/cell (Penn Vector Core) on day 3 for neurons and day 2 for astrocytes after splitting. Transduction efficiency was determined to be between 50 and 60% of the total cells for astrocytes and 70–80% for neurons. Increasing viral particles for better efficiency was not feasible since preliminary studies demonstrated increased toxicity from viral particle overload. Astrocytes were harvested for further analysis on day 5 post viral treatment for all experiments unless otherwise stated.
2.4. Energy charge measurements by HPLC
Control (GFP) and Igfr knockout (CRE) astrocytes (n = 5/group) were rinsed with ice cold 10 mM PBS, scraped into 1 ml of PBS and flash frozen in liquid nitrogen and stored at −80 °C. Samples were thawed slowly on ice, vortexed vigorously for 10 s, then 300 μl of the sample was extracted with an equal volume of 300 mM potassium hydroxide. The resultant extract was centrifuged at 20,000 × g for 15 min, then filtered and resolved by High-performance liquid chromatography and a UV/VIS diode array spectrometer used to detect AMP, ADP, and ATP [41]. For more information on HPLC procedures please see Supplementary Methods.
2.5. Mitotracker and MitoSOX staining
Astrocytes were treated with 100 nM of Mitotracker (Molecular Probes) for 30 min at 37 °C, following which the cells were fixed and immuno-labeled for the astrocytic marker, GFAP. Mitochondrial distribution in GFAP labeled cells was identified using confocal imaging as described in Supplementary Methods. Control and IGFR-KO astrocytes were treated with 1 μM of MitoSOX Red (Molecular Probes) for 30 min at 37 °C following which astrocytes were analyzed by flow cytometry as described below.
2.6. Seahorse methods
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured as previously described [42] using a Seahorse XF24 Flux Analyzer (Agilent Technologies). 50K cells/well were seeded in an XF24 culture plate, grown to confluency then treated with AAV-GFP, -CRE. After 5 days, the culture media was changed to DMEM supplemented with glucose, pyruvate, and glutamax, and the plate was incubated at 37 °C for 1 h in a non-CO2 dry incubator. OCR and ECAR measurements of mitochondrial function were obtained using sequential injections of 1 μM oligocmycin, 1 μM FCCP, and 1 μM antimycin A. After three basal measurements of OCR and ECAR were recorded, oligomycin was injected to inhibit ATP synthase and two more measurements were recorded to assess proton leak. Next, FCCP was injected to uncouple respiration and measure maximal respiration. Finally, antimycin A was injected to measure non-mitochondrial respiration. All OCR measurements were normalized to non-mitochondrial respiration and the final values normalized to total cell numbers plated in each well as live cell numbers were not different between the groups. Reserve capacity is the difference between maximal respiration and basal respiration, while ATP-linked OCR is the difference between basal and proton leak. Data represented are n = 7 repeated on 3 separate Seahorse plates.
2.7. Aβ uptake
Control (GFP) or Igfr knockout (CRE) astrocytes in culture were treated with 0.1 μM of human HiLyte™ Fluor 555 – labeled Aβ (1–42) (Anaspec, Fremont, CA) for 4 h in serum free media as previously described [43]. Following treatment, astrocytes were rinsed twice with 1X HBSS without calcium and magnesium, harvested with 0.25% trypsin, and spun at 1000 × g for 10 min. The pelleted cells were re-suspended in 1X HBSS with 0.1% BSA. For imaging, cells on coverslips were fixed and processed as described above in “Immunocytochemistry” methods.
2.8. Flow cytometry
Cells were analyzed by Influx Cell Sorter (BD Biosciences) for dual fluorescence with 488nm/530nm/30 and 561nm/593nm/40 (excitation/Emission/bandpass) and visualized using Summit 4.3 software (Dako Colorado, Inc, Fort Collins, CO). Fluorescence intensity was calculated as the geometric mean of the Aβ-555 or MitoSOX Red (568) intensity in GFP + cells. Unstained cells were used to gate for forward and side-scatter measurements.
2.9. Statistical analyses
All experiments were performed in multiple independent replicates per group stated for each experiment. Statistical differences between experimental groups were analyzed using a multivariate ANOVA followed by post hoc pairwise comparisons using Graphpad Prism 6.0, and Student's T-test. Heteroscedastic data were either log or square root transformed. Data are represented as the mean ± SEM. Significance is indicated by P value measurements with a P < 0.05 considered significant; *P < 0.05; **P < 0.01; ***P < 0.001. Additional methods are provided in Supplementary Methods section.
3. Results
3.1. Age-related decline in learning is associated with reduced hippocampal IGFR expression and gliosis
To assess the age-associated decline in cognitive function, we evaluated spatial learning in young (4–6 m) and old (22–24 m) C57Bl/6 mice in the radial arm water maze. Old mice made significantly more errors (Figure 1A) and took a longer pathlength (Figure 1B) to find the hidden target compared to young mice, indicating a slower rate of learning. We then harvested the brains from these mice, dissected the hippocampus, and evaluated IGFR mRNA expression. Hippocampi from old mice showed a 40% reduction in IGFR expression (Figure 1C) which was associated with an increase in GFAP expression, a marker for gliosis. Levels of GFAP mRNA (Figure 1D) were significantly increased with a 30% increase in protein expression by western blotting (Figure 1E) in old hippocampi compared to young. Immunohistochemical localization of GFAP (Figure 1F) in the hippocampi showed an increase in GFAP expression in old compared to young. Aldehyde dehydrogenase (ALDH1L1), another marker for astrocytes, showed no significant change in mRNA expression. We therefore assessed the role of IGF-1R signaling in astroglial function and learning and memory.
Figure 1.

Age-dependent impairment in learning is associated with decreased hippocampal IGFR expression and gliosis. C57Bl/6 young (4–6 m) and old (22–24 m) mice were tested on the radial arm water maze for spatial learning (n = 10/group). Old mice showed significantly more errors (A) and pathlength (B) to target and display a shallow learning curve compared to young mice. Hippocampal IGFR (C) expression was significantly decreased, while gliosis marker, GFAP, was significantly increased (D) in old mice compared to young. Increase in GFAP protein expression is validated via western blotting (E; n = 6/group) as well as by immunohistochemistry (n = 3/group) in young and old mice. Scale bar = 100 μm. Data are represented as the Mean ± SEM. *P < 0.05.
3.2. Astrocyte-specific IGFR knockout mice show impairments in working memory
To determine the role of astrocyte IGFR signaling in learning and memory, we generated the tamoxifen-inducible astrocyte-specific knockout mice (GFAP-CreTAM/Igfrf/f; TAM-Cre). We induced recombination of the floxed allele in mice expressing the Cre transgene by administering tamoxifen intraperitoneally. Sham-injected GFAP-CreTAM/Igfrf/f (Sham-Cre) and tamoxifen-injected Igfrf/f (TAM-Igfr) littermates were used as controls. We induced Cre expression at 3–4 months of age to avoid any compensatory developmental effects from IGFR deficiency. Astrocyte-specific IGFR knockout mice (TAM-Cre) showed a reduction (∼10–15%) in hippocampal Igfr expression (Figure 2A) compared to controls (TAM-Igfr and Sham-Cre). Hippocampal-dependent learning was assessed at 6 months of chronological age in the radial arm maze using a modified working memory task due to difficulty in parsing out learning deficiency in young mice based on our previous observations. When tested in the radial arm water maze, no differences were noted on the first day (4 trials) in number of errors (Figure 2B) between the controls (Sham-Cre and TAM-Igfr) and the knockout (TAM-Cre). However, using this working memory task, the TAM-Cre mice made significantly more errors compared to controls (Figure 2C), suggesting the inability to extinguish a previous memory and relearn the new target location. These results, for the first time, show the importance of IGFR signaling in astrocytes and model the loss in expression seen during brain aging.
Figure 2.

Astrocyte-specific knockout of IGFR impairs working memory. (A) IGFR-KO (TAM-Cre) mice showed 10–15% less expression of total hippocampal levels of IGFR compared to controls (Sham-Cre and TAM-Igfr). (B) Mice tested in the RAWM (4 trials/60 s each) showed no difference in spatial learning between the groups on day 1 for the number of errors. (C) Tamoxifen-induced astrocyte-specific IGFR knockout (TAM-Cre) mice showed significantly increased number of errors to find the platform compared to controls (Sham-Cre and TAM-Igfr littermates). Data are represented as the mean ± SEM; *P < 0.05; TAM-Cre (n = 11); TAM-Igfr (n = 10); Sham-Cre (n = 6).
3.3. IGFR-KO in astrocytes alters mitochondrial structure and function
To understand the molecular basis of IGFR deficiency in astrocyte function, we used primary astrocyte cultures from Igfrf/f mice to induce cell-specific knockout. This allowed us to enrich for astrocytes within our cultures with minimal numbers of other brain cells. Purity of astrocytes and neurons in culture is illustrated by western blotting and photomicrographs of cultured cells expressing GFP (Figure S1 A–B). Differentiated astrocytes were passaged once, grown to confluency, then transduced with AAV9-CMV-GFP (GFP; Control) or AAV9-CMV-GFP-CRE (CRE; IGFR-KO) viral particles, and expression of igfr was quantified using qPCR (Figure 3A) 5 days after infection. This methodology allowed us to investigate the effects of IGFR knockout without affecting development or ability to fully mature into an astrocyte and hence models the loss of age-related loss of IGF-1 signaling. Astrocytes transduced with CRE showed a 30–50% decline in igfr expression compared to GFP controls, mirroring the level of decline in IGF-1 signaling seen with aging. We then analyzed control and knockout astrocytes for levels of ATP, AMP, and ADP using high performance liquid chromatography (HPLC), which showed that astrocytes with igfr knockout had a ∼20% decline in energy charge ([ATP] + 0.5[ADP])/([ATP] + [ADP] + [AMP]; Figure 3B). To verify whether the changes in energy levels within astrocytes were due to changes in mitochondrial content, we quantified the mitochondrial DNA copy number using digital PCR and normalized to nuclear DNA content. Interestingly, there were no differences in mitochondrial copy numbers between the two groups (Figure 3C). IGFR knockout was confirmed by western blotting that showed reduction in ERK phosphorylation (Figure 3D–E), albeit AKT (s573) phosphorylation was not changed suggesting MAPK-dependent modulation of mitochondrial function. Interestingly, we did not see differences in energy charge or mitochondrial copy numbers in neurons with a 70% reduction in igfr expression (Figures S1C–E), suggesting disparate role of IGFR in astrocytes and neurons. These results suggest that reduction in energy levels with IGFR deficiency in astrocytes may be due to either reduced availability of substrates for energy function or deficiencies in mitochondrial function.
Figure 3.

IGFR signaling deficiency modulates mitochondrial energy charge in astrocytes in complete growth media. Knockout of IGF-1R in astrocytes (A–C) showed 30% reduction in mRNA levels (A), ∼20% reduction in energy charge (B) with no significant change in mitochondrial DNA/nuclear DNA ratio (C) in the overall population. (D) Representative western blots for IGFRβ and ERK showing reduction IGFR in the knockout and reduced p44 ERK levels quantified in (E), while no differences were found in AKT phosphorylation. Data were analyzed by two-tailed student T-test; Mean ± SEM; n = 5 per group. (F) Representative confocal images of untransduced (UT; top panel), GFP-transduced (GFP; middle panel) and IGF-1R knockout (CRE; bottom panel) astrocytes labeled for GFAP (blue) and Mitotracker (Red) showing a vesicular perinuclear mitochondrial localization in the knockout. Scale bar = 20 μm; n = 3/group.
Fused mitochondria have been reported to be a compensatory mechanism to maximize energy production [44] during stressed conditions and to dilute mitochondrial damage [45], [46]. However, fused mitochondria transiently buffer ETC dysfunction [44] and cellular stress, such as during periods of nutrient starvation and protect against autophagic degradation or mitophagy [47], [48], [49]. Prolonged stress or starvation can result in a sustained hyperfused state that ultimately results in mitochondrial dysfunction [50]. We therefore examined the morphology of mitochondrial structure using Mitotracker (Figure 3F). Untreated (UT; top panel) and GFP-transduced (GFP; middle panel) controls showed comparable spread of mitochondria throughout the cell. Astrocytes with IGFR knockout (CRE; bottom panel) appeared clustered and displayed a perinuclear localization. Thus, IGF-1 signaling deficiency via IGFR knockout in astrocytes or that which occurs with age from reduced circulating IGF-1 levels may confer a state of distress leading to altered mitochondrial structure and thereby function.
3.4. IGFR-KO in astrocytes decreases OCR and increases mitochondrial ROS
To further characterize mitochondrial function, we evaluated the effect of IGFR knockout on mitochondrial electron transport chain (ETC) function and oxidative phosphorylation [51]. Mitochondria were isolated from control and IGFR-KO astrocytes and mitochondrial respiration, ROS generation, and OXPHOS coupling efficiency were evaluated using O2K respirometry and fluorometry. Isolated mitochondria from IGFR-KO astrocytes showed a trend towards a decrease in oxygen consumption rate (OCR; Figure 4A), as well as OXPHOS coupling efficiency (Figure 4B), which is a measure of the efficiency of the ETC to generate ATP, compared to control (GFP) astrocytes. The trends in data and lack of significance may arise from cells that were not transduced in the population of astrocytes. In the absence of exogenous substrates, mitochondria from IGFR-KO astrocytes showed significantly increased ROS, measured as the production of H2O2 by Amplex UltraRed reactivity, in State 1 respiration (Figure 4C). With the addition of glutamate/malate (S2; complex I (CI)-linked leak respiration), ROS production was still elevated; however, the difference was not statistically significant. The transfer of electrons to oxygen from the activity of the ETC generates ROS in mitochondria. There are multiple sources of ROS within the ETC [52], generating superoxide. Because CI, CII, and CI&II-linked respiration are reduced in the CRE, we further hypothesize that defects in IGFR-KO may be occurring later in the chain (CIII or CIV). To corroborate the increase in mitochondrial ROS in intact cells, we used MitoSOX, the live-cell permeant indicator of mitochondrial superoxide. IGFR-KO astrocytes showed increased MitoSOX-Red fluorescence intensity compared to controls (Figure 4D). These results show that mitochondrial function is skewed towards increased ROS production with IGFR knockout.
Figure 4.

IGFR knockout in astrocytes impairs mitochondrial respiration and increases ROS production. Knockout of IGFR (CRE) in cultured astrocytes show a trend towards (A) decreased oxygen consumption rate and (B) mitochondrial OXPHOS coupling efficiency in isolated mitochondria as measured by Oroboros respirometry. Leak respiration was measured with substrates in the absence of ADP, while OXPHOS capacity was measured with substrates for complex I & II, ADP, and cytochrome C. IGFR knockout astrocytes also exhibit: (C) a significant increase in mitochondrial State 1 (S1) ROS without exogenous substrates or ADP, while ROS in State 2 (S2), or Leak, respiration is increased but not statistically significant. (D) In a live cell assay, IGFR knockout astrocytes show increased MitoSOX fluorescent intensity measured by flow cytometry. Data are the Mean ± SEM; n = 6/group; *P < 0.05. A–C are expressed as levels per μg of mitochondrial protein (from isolated mitochondria).
To investigate whether the steady state levels of mitochondrial proteins were altered with IGFR-KO, we examined the abundance of enzymes involved in mitochondrial metabolism by mass spectrometry. Relative abundance of Kreb's cycle enzymes (Idh1, Idh2), ATP synthase (Atp5a1, Atp5b) and others measured were not altered between groups (Figure S2; Table S1).
To further assess mitochondria and metabolic function in a live cell assay, we measured oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) in astrocytes by Seahorse XF-24 (Figure 5A). Data from the Seahorse were normalized to total cell numbers plated per well since IGF-1 has been reported to affect protein synthesis [53], [54]. Total cell numbers per well 5 days post transduction were comparable between groups as measured by the Neutral Red uptake assay for cell viability (Figure S3). Basal, ATP-linked, proton leak, and non-mitochondrial respiration were significantly reduced in IGFR-KO astrocytes compared to controls (GFP). Maximal respiration (following the addition of FCCP) showed a trending decline (P < 0.06), while reserve capacity was not different between groups (Figure 5B). The reduction in ATP-linked respiration was in concurrence with reduced ATP (energy charge) in IGFR-KO astrocytes reported above. Oligomycin induced ECAR, indicative of glycolytic activity, was decreased significantly in IGFR-KO astrocytes (Figure 5C), while basal ECAR was not different between groups. OCR to ECAR ratio of basal to maximal respiration showed decreased induction of respiratory capacity in IGFR-KO (CRE) compared to the induction in control (GFP) astrocytes (Figure 5D). These data suggest that IGFR-KO in astrocytes would impair ability to respond to stress stimuli and/or metabolic demand.
Figure 5.

IGFR knockout in astrocytes increases maximal respiration by Seahorse. (A) Knockout of IGFR (CRE) in astrocytes significantly decreased oxygen consumption. (B) OCR was reduced significantly in several stages of respiration in IGFR knockout (CRE) astrocytes including, basal, ATP-linked, proton-leak and non-mitochondrial respiration. Maximal respiration was reduced but not significantly. (C) Oligomycin-induced extracellular acidification (ECAR) was significantly decreased in IGFR-KO, while basal ECAR was not different between groups. (D) Basal and maximal OCR to ECAR ratio depicts lower induction of response with FCCP in CRE astrocytes compared to control. Data are represented as the Mean ± SEM (n = 7/group), *P < 0.05.
3.5. Increased antioxidant protein response in IGFR-KO astrocytes
The decrease in energy charge ratio reflects a relative increase in AMP levels. AMP stimulates AMP-activated protein kinase (AMPK) and its downstream signaling cascade promoting sirtuin activity. Sirtuins (SIRTs) are NAD + -dependent protein deacetylases and function as cellular sensors of the redox status of the cell. Mitochondrial SIRT3 activity has been shown to increase under energy deficient conditions while the reverse is true during energy replete conditions. We measured sirtuin activity in the mitochondrial fractions from IGFR-KO and control astrocytes. Mitochondrial sirtuin activity was significantly higher in the IGFR-KO compared to controls (Figure 6A), while there was no significant difference in sirtuin activity in the nuclear fraction (Figure 6B). The increase in sirtuin activity is further supported by increase in NADH oxidase activity in IGFR-KO astrocytes generating NAD + substrates from the oxidation of NADH (Figure 6C). These results suggest that IGF-1 signaling modulates sirtuin activity possibly through AMPK-dependent regulation of cellular stress pathways.
Figure 6.

IGFR knockout in astrocytes increases antioxidant response but increases susceptibility to oxidant stress. (A) Sirtuin activity was significantly increased in the mitochondrial fraction while it trended lower in the nuclear fraction (B) in IGFR-KO astrocytes. (C) NADH oxidase activity was increased in IGFR-KO astrocytes as well as an increase in anti-oxidant protein expression: (D) SOD abundance (by Mass Spectrometry), and (E) SOD1 activity were also significantly increased in IGFR-KO lysates compared to controls. (F) Peroxide-induced cytotoxicity was significantly greater at 150 μM and 250 μM H2O2 in IGFR-KO astrocytes compared to controls with a trending decline at 200 μM. Data are represented as the mean ± SEM; *P < 0.05; **P < 0.01; n = 6/group; ns = not significant.
One of the major functions of SIRT3 is to mount a mitochondrial antioxidant response. Superoxide dismutases (SOD) scavenge and reduce superoxide to H2O2, and SOD activities are promoted by SIRT-mediated deacetylation. Manganese SOD (MnSOD; SOD2) is the primary mitochondrial superoxide dismutase, while copper-zinc SOD (CuZnSOD; SOD1) is also found in the mitochondrial intermembrane space and cytosol. We investigated whether increased ROS production and sirtuin activity resulted in an increase in antioxidant response in IGFR-KO astrocytes. We measured the abundance of antioxidant proteins using mass spectrometry (Figure 6D). SOD2 protein abundance was elevated in astrocytes with IGFR-KO compared to controls. SOD1 levels, which are also involved in quenching of mitochondrial and cytosolic superoxide, were increased in IGFR-KO astrocytes compared to controls. The increase in SOD1 protein levels was mirrored by the increase in SOD1 activity in IGFR-KO astrocytes compared to controls (Figure 6E).
We further evaluated the impact of increased mitochondrial ROS levels on glutathione levels, both reduced (GSH) and oxidized (GSSG) forms, using HPLC in controls and IGFR-KO astrocytes. GSH production by the enzyme glutathione reductase is induced upon oxidative stress. IGFR-KO astrocytes showed no significant change in GSH levels (Figure S4 A), albeit GSSG levels declined (Figure S4 B), indicating the induction of the glutathione reductase activity to reduce GSSG. These data concur with increased mitochondrial antioxidant response from underlying oxidative stress due to IGFR-KO and suggest a protective phenotype.
To determine whether knockout of IGFR in astrocytes alters their susceptibility to oxidant stress, we assessed cytotoxicity to H2O2 induced oxidant stress using the neutral red staining protocol as previously described [55]. No differences were found in cell viability between control and knockout groups (Figure S3) under low concentrations of H2O2 (0–100 μM). Cell viability was significantly reduced at 150 μM and 250 μM of extracellular H2O2 and trended lower at 200 μM (P = 0.06) in IGFR knockout astrocytes (Figure 6F). While cell numbers continued to decline with increasing concentrations of peroxide, the differences were not significant between groups beyond 250 μM. These data suggest a narrow threshold of susceptibility to oxidant stress, between 150 μM and 250 μM of extracellular H2O2, in IGFR-KO astrocytes under these experimental conditions. The gradient between extracellular-to-intracellular H2O2 has been reported to be 650-fold [56], while the non-pathogenic intracellular H2O2 concentration is reported to be in the range of 1–700 nM [57]. Based on the 650-fold concentration differential between extracellular/intracellular H2O2, the treatment range of 100–250 μM equates to intracellular H2O2 concentrations of 154–385 nM. Thus, the induced toxicity from 150 to 250 μM of exogenous H2O2 indicates increased sensitivity to non-pathological concentration of H2O2 in IGFR-KO astrocytes.
3.6. IGFR-KO astrocytes are deficient in glucose and Aβ uptake
Decreased IGF-1 signaling has been shown to confer insulin resistance thus hindering the ability of a cell to internalize and metabolize glucose [58], [59], [60]. Astrocytic uptake of glucose is essential to supply the energy needs of neurons by releasing lactate that promotes hippocampal learning and memory [61]. We tested whether astrocytes deficient in IGF-1/IGFR signaling were able to internalize a modified glucose, 2-deoxy-d-glucose (2DG). IGFR-KO astrocytes had significantly reduced 2DG6P (Figure 7A) levels compared to controls (untransduced and GFP transduced), which is in agreement with decreased ECAR with oligomycin in the Seahorse assessment. In addition to reduced glucose uptake, glucose transporter (Glut1) expression was also significantly reduced with IGFR-KO (Figure S5). These results suggest an important role for IGF-1 signaling in regulating glucose metabolism by astrocytes, a major source of energy in the brain.
Figure 7.

IGFR knockout in astrocytes decreases glucose and Aβ uptake. (A) Astroyctes were treated with 1 mM of 2D-glucose (2DG) and accumulation of 2DG6P was measured using luminescence. IGFR-KO astrocytes were significantly impaired in glucose uptake compared to GFP and un-transduced (UT) controls. (B) Flow cytometry for Aβ1−42-555 uptake showed reduced total fluorescence intensity in IGFR-KO (CRE) culture compared to GFP control culture. Unstained astrocytes were used for forward and side scatter gating. (C) Geometric mean of the fluorescence intensity of 555 in GFP + cells was also significantly reduced in IGFR-KO astrocytes compared to controls. (D) Representative confocal images of astrocytes (n = 4/group) treated with Aβ1−42-555 show reduced uptake of Aβ1−42 in IGFR-KO astrocytes (Lower panel) compared to GFP controls (Upper panel). GFP fluorescence represents endogenous expression of viral transduction while staining for GFAP was performed by immunocytochemistry. Scale bar = 20 μm; n = 5/group Data (A–C) are represented as the mean ± SEM; *P < 0.05; n = 5/group.
One of the major functions of astrocytes is the uptake and clearance of Aβ from the synapse to maintain normal brain physiology. We tested whether knockout of IGFR in astrocytes affects Aβ uptake, a fundamental aspect of astrocyte function that is aberrant in Alzheimer's disease. Astrocytes with IGFR-KO and controls were treated with 100 nM of HiLyte™ Fluor 555-labeled oligomeric human Aβ1−42 overnight, following which cells were washed and trypsinized and internalized Aβ1−42 was analyzed by flow cytometry. GFP+/Aβ+ cells were separated by flow and the fluorescence intensity was quantified. Total fluorescence for Aβ1−42 was reduced in IGFR-KO compared to GFP controls (Figure 7B). Fluorescence intensity for Aβ1−42 (calculated as the geometric mean of Aβ1−42 -555 intensity in GFP + cells) was significantly reduced in astrocytes with IGFR-KO (Figure 7C) compared to controls (GFP). Immunocytochemistry revealed reduced Aβ1−42 puncta in IGFR-KO astrocytes (Figure 7C). These results show that Aβ1−42 uptake is impaired in astrocytes with IGFR signaling deficiency.
4. Discussion
Alterations in the metabolic state of astrocytes are a key underlying factor in brain aging and neurodegenerative disorders. Aging is associated with decreased glucose and oxygen metabolic rates in brain cells that is further exacerbated in neurodegenerative conditions including AD and Parkinson's disease. Importantly, reduced IGF-1 levels with age is linked to reduced cognitive function and altered glucose metabolism in a tissue-specific context. However, the specific actions of IGF-1 in astrocyte function are poorly understood. In this study, we demonstrate that learning impairments with aging is associated with a concomitant reduction in IGFR expression and increased gliosis. Furthermore, we show that astrocyte-specific knockdown of IGFR in mice show impairment in a working memory task at a younger age, deficits that have been reported with aging.
Energy levels (ATP:AMP ratio) have been reported to decrease during periods of cellular stress, exercise, caloric restriction, and aging [62], [63]. Astrocyte integrity is integral to regulation of neuronal function since these cells provide energy substrates, e.g. lactate, that are coupled to neuronal activity and have been shown to improve learning and memory correlates [24], [64]. Thus, it is well accepted that glucose metabolism and mitochondrial energy production in astrocytes are critical for the preservation of cognitive function with age. We show that IGF-1 signaling through its cognate receptor IGFR regulates astrocyte mitochondrial energy production and mitochondrial architecture. IGFR knockout in astrocytes resulted in reduced OCR and decreased energy charge through a reduction in MAPK signaling. These results are the first to directly demonstrate the regulation of astrocytic mitochondrial function is mediated by IGF-1 signaling. Our findings are supported by previous reports of regulation of ATP and glucose utilization in the brain with systemic IGF-1 replacement [65]. Furthermore, we show that IGFR knockout alters mitochondrial structure, inducing a more vesicular and perinuclear localization of the mitochondria. Interestingly, altered mitochondrial bioenergetics [8] and, specifically, altered mitochondrial structure and impaired mitochondrial fission-fusion dynamics have been linked to the progression of Alzheimer's disease (AD) [66]. Importantly, the flux between the two states is an important determinant of healthy mitochondrial function. Thus, the vesicular localization of mitochondria and increased antioxidant response in IGFR knockout astrocytes may be a compensatory mechanism in response to reduced energy. Our data suggest that reduction in IGFR signaling alters fundamental aspects of mitochondrial structure and function such that effects are detectable in isolated mitochondria and intact cells without alterations in mitochondrial genome/DNA copy numbers. Future studies exploring fission and fusion dynamics in response to IGFR knockout would likely reveal whether modulating these pathways could provide an avenue for therapeutic intervention for reduced energy with age.
In addition to functional alterations in energy metabolism, mitochondria are the major source of reactive oxygen species (ROS) that impair cellular function by inducing oxidative damage to macromolecules. The contribution of insulin and IGF-1 signaling to the regulation of ROS remains controversial. Mammalian models of circulating IGF-1 deficiency have been reported to lead to reduced ROS, increased antioxidant defense systems, and resistance to stress induced mortality, yet they sustained more diquat-induced oxidative damage and tissue-specific loss of function [67], [68], [69], [70], [71], [72], [73], [74], [75]. Conversely, IGF-1 replacement has been shown to improve spatial memory [76] and reduce oxidation of glutathione in aged mice [77]. These studies highlight some of the controversies in the field and the importance of unraveling the cell-specific role of IGF-1 signaling and its regulation of oxidative stress in cognitive function. ROS production generally increases with age and specifically in aged astrocytes [78] contributing to dysregulation of stress response pathways that can have adverse effects on cell and tissue function. In addition to showing that IGFR knockout in astrocytes reduced energy, we show that that it increased mitochondrial ROS production and MitoSOX staining in intact cells, providing evidence for increased superoxide production. Furthermore, astrocytes with a knockout of IGFR show increases in the abundance of SOD1 and SOD2 antioxidant proteins, both of which have direct effects on mitochondrial function and influence lifespan [79], [80], [81]. This observation suggests that underlying oxidative stress is present in response to reduced IGF signaling and supports the finding of increased superoxide staining in IGFR knockout astrocytes.
The dysregulated antioxidant response can result in sustained oxidative stress and damage ultimately leading to impairments in cellular function. Here, we show that astrocytes with a knockout of IGFR are more susceptible to peroxide-induced cytotoxicity, thereby indicating that the existing antioxidant response is insufficient to counter the effects of oxidant stress in the absence of IGFR signaling. ROS have been shown to activate Jun-N-terminal Kinase (JNK)/Forkhead box (FoxO) signaling as well as nuclear factor kB (NF-kB)-mediated activation of anti-oxidant genes (SOD1, SOD2) [82], [83]. Chronic activation of JNK/FoxO [84], [85] or NF-kB reduce cellular function and longevity and aberrant astrogliosis; the latter mediating the glial pathology reported in neurodegenerative diseases [86]. Thus, the activation of these signaling pathways with IGFR signaling deficiency would shed light on possible targets for intervention to alleviate age-related glial pathology.
Astrocytes form a complex network with astrocytic end-feet innervating both neuronal synapses and endothelial cells in the blood brain barrier facilitating the transport of glucose and other molecules to support brain function. Astrocytes have a fundamental role in glucose uptake into the brain and provide energy substrates, such as lactate, to support neuronal activity. These cells are the site of glycogen storage that is mobilized upon hypoglycemic stimulation or neuronal activity. Studies by Hernandez-Garzon and colleagues [26] report that, in response to whisker stimulation, a chronic reduction in IGFR in astrocytes results in increased glucose uptake. Although it is difficult to reconcile this study with our own, the presence of hybrid insulin and IGF receptors in the brain [6], [7], [8] and or the duration of the IGFR receptor deficiency may contribute to these differences. Importantly, the studies by Hernandez-Garzon did not investigate compensatory changes in IR or their signaling pathways. In our studies, glucose uptake was reduced in astrocytes in response to IGFR deficiency in agreement with the difference in lactate production measured by the extracellular acidification rate (ECAR) with the addition of oligomycin to inhibit ATP synthase. These findings are further supported by reports of regulation of ATP and glucose utilization in the brain with systemic IGF-1 replacement [9]. Further studies will be required to resolve the apparent differences in response to IGFR deficiency on glucose uptake and their mechanisms of action. Previous studies indicate that lactate production from glycogenolysis has an important role in memory-processing [87]. Glycogen synthesis is stimulated by IGF-1 [88] and serves as an energy reserve, which, when mobilized, supports short-term metabolic needs. However, sustained glycogenolysis and depletion of glycogen stores, as with sleep deprivation [89], leads to impaired cognition [61] and synaptic loss [90] observed with aging and age-related brain disorders. Reduced glycogen stores are also associated with neuronal hyperactivity and increased Aβ production in AD leading to cognitive dysfunction [64]. Our data suggest that with reduced glucose uptake in IGFR deficiency may result in decreased glycogen stores that may negatively impact cognitive function.
Uptake and clearance of Aβ from the synapse are key functions mediated by astrocytes [91]. Accumulation of extracellular Aβ forming neuritic plaques is a key determinant in the pathogenesis of AD [92]. We show that astrocytes with a knockout of IGFR are deficient in Aβ1−42 uptake indicated by flow cytometry and further confirmed by immunocytochemistry. This key finding is supported by numerous studies with IGF-1 replacement that have neuroprotective and pro-cognitive effects in aged animal models [9], [93], [94], [95], [96], [97], [98], [99] and in AD [30], [100], [101]. ATP-binding cassette (ABC) transporters, fueled by ATP, facilitate the transport of Aβ across the plasma membrane through modulating cholesterol efflux [102]. Increased ABCA1 function decreases astrocytic Aβ deposition, thereby reducing Alzheimer's pathology [103], [104]. Thus, the reduction in ATP levels with IGFR-KO could reduce the activity of these transporters, thereby adversely affecting Aβ uptake into astrocytes and clearance across the blood brain barrier, increasing susceptibility for neuronal toxicity. Some studies report reduced Aβ deposition in neuronal-specific IGFR knockout [105], [106]. Since Aβ release from neurons is coupled to neuronal stimulation, dampening neuronal activity with IGFR knockdown may prove beneficial in such cases. Our data also support the disparate effects of IGFR knockout in neurons and astrocytes on ATP synthesis.
While the effect of reduction in IGF-1 signaling on mitochondrial function in vivo remain to be elucidated, it is evident that IGFR signaling declines with age and is critical for hippocampal learning and memory. In conclusion, our data suggest that astrocytes may serve as key sensors of peripheral hormonal fluctuations bridging the cerebral vasculature with neuronal activity in response to endocrine status. Therefore, positive regulation of IGFR signaling and its interacting partners affecting energy metabolism and transport (Figure 8) may provide therapeutic targets to improve astrocytic function and alleviate age-associated pathologies, including AD.
Figure 8.

Model summarizing effects of IGFR signaling deficiency in astrocytes. IGFR knockout in astrocytes alters mitochondrial structure, decreases oxygen consumption, induces antioxidant response, increases ROS production and susceptibility to oxidant stress. Decreased ATP with IGFR signaling deficiency can reduce ABC transporter (ABCA) function thereby impairing Aβ uptake and clearance by astrocytes. Neuronal activity-dependent Aβ release can thereby increase synaptic toxicity and loss of synapses with age and in neurodegenerative diseases such as Alzheimer's disease.
Acknowledgements
The authors declare no potential conflicts of interest. The authors would like to thank Dr. Nicole M. Ashpole for conception of the GFAP-Cre/Igfrf/f animal model. The authors would like to thank the Laboratory for Molecular Biology and Cytometry Research at OUHSC for the use of the Flow Cytometry and Imaging facility, which provided the use of instruments for Imaging and flow cytometry services and analyses. The authors would also like to acknowledge T32 training grant (T32AG052363) awarded to Drs. Logan and Masser, and Gavin Pharaoh and Alexander Yeganeh. We would also like to thank the Nathan Shock Center Core of Oklahoma P30 AG050911. This work was supported by the funding sources: NIH R01AG038747, R01NS056218 to WES.
Footnotes
Supplementary data related to this article can be found at https://doi.org/10.1016/j.molmet.2018.01.013.
Conflict of interest
The authors declare no potential conflicts of interest.
Appendix A. Supplementary data
The following are the supplementary data related to this article:
Figure S1.

(A) Western blotting for Synaptophysin (SYP) and GFAP markers showing purity of primary neuronal (<10% astrocytes) and astrocyte (no visible neuronal contamination) cultures, respectively. (B) Photomicrograph of neurons (left) and astrocytes (right) transduced with AAV9-CMV-GFP. (C) Neurons with IGFR knockout (∼70% reduction in mRNA expression) showed no significant changes in mitochondrial energy charge (D) or mtDNA copy numbers (E). Data were analyzed by two-tailed student T-test; mean ± SEM; n = 5 per group.
Figure S2.

Abundance of enzymes in mitochondrial metabolism. Levels of some enzymes were increased in abundance in IGFR-KO compared to controls. However, there was no statistical significance between groups for the enzymes measured in this panel by Mass spectrometry. Data (mean ± SEM) are presented as abundance (%) relative to GFP (controls); n = 6/group. Abbreviations for proteins in this panel are identified in Supplementary Table 1.
Figure S3.
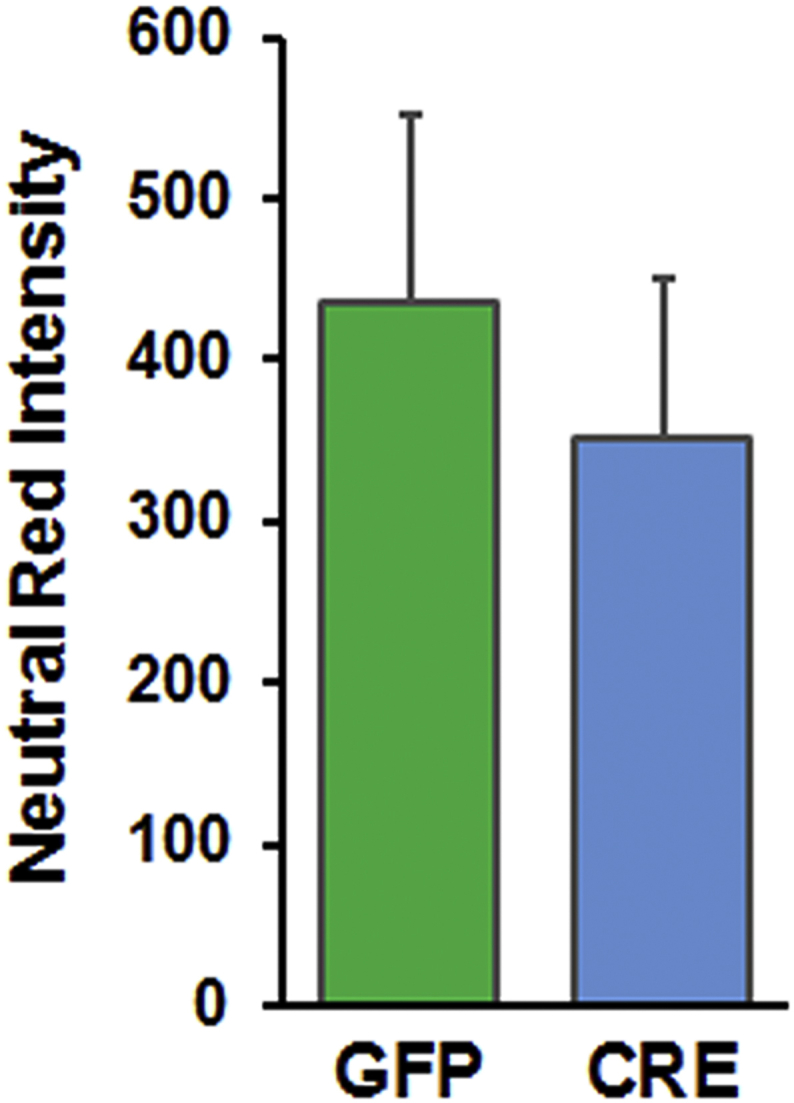
Neutral red staining of astrocytes 5 days post viral transduction showing comparable cell numbers in both groups. Neutral red staining was performed for 2 h and the intensity of dye incorporated within live cells measured spectrophotometrically using ex 530 nm and em 645 nm.
Figure S4.

Oxidized glutathione levels are reduced with IGFR-KO in astrocytes. (A) Levels of reduced (GSH) were not different between groups but (B) oxidized (GSSG) glutathione levels were significantly reduced in IGFR-KO (CRE) astrocytes. Data are represented as the mean ± SEM; (n = 5/group); *P < 0.05; **P < 0.01.
Figure S5.

Glucose transporter expression is reduced with IGFR-KO in astrocytes. Message levels of Glut1 glucose transporter was significantly reduced by ∼13% with IGFR-KO (∼40% knockout). Glut3 levels were also reduced but not statistically significant. Data are represented as the mean ± SEM; (n = 5/group); **P < 0.01, ***P < 0.001.
Figure S6.

Genotyping for GFAP-Cre/Igfrf/f. PCR and agarose separation of 200 bp product for the GFAP-Cre (lanes 1–6) transgene identifying mice that are positive (lanes 1–3 and 6) or mice that lack the transgene (lanes 4–5). PCR identification of Igfrf/f mice (lanes 7–12) show a 220 bp homozygous-floxed band and a 124 bp product for the wild-type band for Igfrf/f mice. Mice homozygous for the floxed gene (lanes 10–11) were used for further breeding with GFAP-Cre/Igfrf/f to generate experimental colonies. Lanes 1–12 indicate individual mice and a 100 bp DNA ladder was used to identify band sizes.
References
- 1.Poon H.F., Calabrese V., Scapagnini G., Butterfield D.A. Free radicals and brain aging. Clinics in Geriatric Medicine. 2004;20(2):329–359. doi: 10.1016/j.cger.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Poon H.F., Calabrese V., Scapagnini G., Butterfield D.A. Free radicals: key to brain aging and heme oxygenase as a cellular response to oxidative stress. The Journal of Gerontology. Series A, Biological Sciences and Medical Sciences. 2004;59(5):478–493. doi: 10.1093/gerona/59.5.m478. [DOI] [PubMed] [Google Scholar]
- 3.Vancova O., Baciak L., Kasparova S., Kucharska J., Palacios H.H., Horecky J. In vivo and in vitro assessment of brain bioenergetics in aging rats. Journal of Cellular and Molecular Medicine. 2010;14(11):2667–2674. doi: 10.1111/j.1582-4934.2009.00879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parihar M.S., Brewer G.J. Simultaneous age-related depolarization of mitochondrial membrane potential and increased mitochondrial reactive oxygen species production correlate with age-related glutamate excitotoxicity in rat hippocampal neurons. Journal of Neuroscience Research. 2007;85(5):1018–1032. doi: 10.1002/jnr.21218. [DOI] [PubMed] [Google Scholar]
- 5.Parihar M.S., Kunz E.A., Brewer G.J. Age-related decreases in NAD(P)H and glutathione cause redox declines before ATP loss during glutamate treatment of hippocampal neurons. Journal of Neuroscience Research. 2008;86(10):2339–2352. doi: 10.1002/jnr.21679. [DOI] [PubMed] [Google Scholar]
- 6.Kubik L.L., Philbert M.A. The role of astrocyte mitochondria in differential regional susceptibility to environmental neurotoxicants: tools for understanding neurodegeneration. Toxicological Sciences. 2015;144(1):7–16. doi: 10.1093/toxsci/kfu254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo C., Sun L., Chen X., Zhang D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regeneration Research. 2013;8(21):2003–2014. doi: 10.3969/j.issn.1673-5374.2013.21.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parihar M.S., Brewer G.J. Mitoenergetic failure in Alzheimer disease. American Journal of Physiology – Cell Physiology. 2007;292(1):C8–C23. doi: 10.1152/ajpcell.00232.2006. [DOI] [PubMed] [Google Scholar]
- 9.Sonntag W.E., Ramsey M., Carter C.S. Growth hormone and insulin-like growth factor-1 (IGF-1) and their influence on cognitive aging. Ageing Research Reviews. 2005;4(2):195–212. doi: 10.1016/j.arr.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 10.Rojanathammanee L., Rakoczy S., Kopchick J., Brown-Borg H.M. Effects of insulin-like growth factor 1 on glutathione S-transferases and thioredoxin in growth hormone receptor knockout mice. Age (Dordrecht, Netherlands) 2014;36(4):9687. doi: 10.1007/s11357-014-9687-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sell C. Minireview: the complexities of IGF/Insulin signaling in aging: why flies and worms are not humans. Molecular Endocrinology. 2015;29(8):1107–1113. doi: 10.1210/me.2015-1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Higashi Y., Sukhanov S., Anwar A., Shai S.Y., Delafontaine P. IGF-1, oxidative stress and atheroprotection. Trends in Endocrinology and Metabolism. 2010;21(4):245–254. doi: 10.1016/j.tem.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gong Z., Kennedy O., Sun H., Wu Y., Williams G.A., Klein L. Reductions in serum IGF-1 during aging impair health span. Aging Cell. 2014;13(3):408–418. doi: 10.1111/acel.12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vasan R.S., Sullivan L.M., D'Agostino R.B., Roubenoff R., Harris T., Sawyer D.B. Serum insulin-like growth factor I and risk for heart failure in elderly individuals without a previous myocardial infarction: the Framingham Heart Study. Annals of Internal Medicine. 2003;139(8):642–648. doi: 10.7326/0003-4819-139-8-200310210-00007. [DOI] [PubMed] [Google Scholar]
- 15.Toth P., Tarantini S., Ashpole N.M., Tucsek Z., Milne G.L., Valcarcel-Ares N.M. IGF-1 deficiency impairs neurovascular coupling in mice: implications for cerebromicrovascular aging. Aging Cell. 2015;14(6):1034–1044. doi: 10.1111/acel.12372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parker D.R., Lapane K.L., Lasater T.M., Carleton R.A. Short stature and cardiovascular disease among men and women from two southeastern New England communities. International Journal of Epidemiology. 1998;27(6):970–975. doi: 10.1093/ije/27.6.970. [DOI] [PubMed] [Google Scholar]
- 17.Aberg N.D., Olsson S., Aberg D., Jood K., Stanne T.M., Nilsson M. Genetic variation at the IGF1 locus shows association with post-stroke outcome and to circulating IGF1. European Journal of Endocrinology. 2013;169(6):759–765. doi: 10.1530/EJE-13-0486. [DOI] [PubMed] [Google Scholar]
- 18.Tumati S., Burger H., Martens S., van der Schouw Y.T., Aleman A. Association between cognition and serum insulin-like growth factor-1 in middle-aged & older men: an 8 year follow-up study. PLoS One. 2016;11(4):e0154450. doi: 10.1371/journal.pone.0154450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitschelen M., Yan H., Farley J.A., Warrington J.P., Han S., Herenu C.B. Long-term deficiency of circulating and hippocampal insulin-like growth factor I induces depressive behavior in adult mice: a potential model of geriatric depression. Neuroscience. 2011;185:50–60. doi: 10.1016/j.neuroscience.2011.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ashpole N.M., Logan S., Yabluchanskiy A., Mitschelen M.C., Yan H., Farley J.A. IGF-1 has sexually dimorphic, pleiotropic, and time-dependent effects on healthspan, pathology, and lifespan. Geroscience. 2017;39(2):129–145. doi: 10.1007/s11357-017-9971-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ashpole N.M., Sanders J.E., Hodges E.L., Yan H., Sonntag W.E. Growth hormone, insulin-like growth factor-1 and the aging brain. Experimental Gerontology. 2015;68:76–81. doi: 10.1016/j.exger.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pitt J., Wilcox K.C., Tortelli V., Diniz L.P., Oliveira M.S., Dobbins C. Neuroprotective astrocyte-derived insulin/insulin-like growth factor 1 stimulates endocytic processing and extracellular release of neuron-bound Abeta oligomers. Molecular Biology of the Cell. 2017;28(20):2623–2636. doi: 10.1091/mbc.E17-06-0416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barros L.F., Weber B. CrossTalk proposal: an important astrocyte-to-neuron lactate shuttle couples neuronal activity to glucose utilisation in the brain. The Journal of Physiology. 2018 doi: 10.1113/JP274944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Belanger M., Allaman I., Magistretti P.J. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metabolism. 2011;14(6):724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 25.Fernandez A.M., Hernandez-Garzon E., Perez-Domper P., Perez-Alvarez A., Mederos S., Matsui T. Insulin regulates astrocytic glucose handling through cooperation with IGF-I. Diabetes. 2017;66(1):64–74. doi: 10.2337/db16-0861. [DOI] [PubMed] [Google Scholar]
- 26.Hernandez-Garzon E., Fernandez A.M., Perez-Alvarez A., Genis L., Bascunana P., Fernandez de la Rosa R. The insulin-like growth factor I receptor regulates glucose transport by astrocytes. Glia. 2016;64(11):1962–1971. doi: 10.1002/glia.23035. [DOI] [PubMed] [Google Scholar]
- 27.Fernandez A.M., Hernandez E., Guerrero-Gomez D., Miranda-Vizuete A., Torres Aleman I. A network of insulin peptides regulate glucose uptake by astrocytes: potential new druggable targets for brain hypometabolism. Neuropharmacology. 2017 doi: 10.1016/j.neuropharm.2017.08.034. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki K., Ikegaya Y., Matsuura S., Kanai Y., Endou H., Matsuki N. Transient upregulation of the glial glutamate transporter GLAST in response to fibroblast growth factor, insulin-like growth factor and epidermal growth factor in cultured astrocytes. Journal of Cell Science. 2001;114(Pt 20):3717–3725. doi: 10.1242/jcs.114.20.3717. [DOI] [PubMed] [Google Scholar]
- 29.Genis L., Davila D., Fernandez S., Pozo-Rodrigalvarez A., Martinez-Murillo R., Torres-Aleman I. Astrocytes require insulin-like growth factor I to protect neurons against oxidative injury. F1000Research. 2014;3:28. doi: 10.12688/f1000research.3-28.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vidal J.S., Hanon O., Funalot B., Brunel N., Viollet C., Rigaud A.S. Low serum insulin-like growth factor-I predicts cognitive decline in Alzheimer's disease. Journal of Alzheimers Disease. 2016;52(2):641–649. doi: 10.3233/JAD-151162. [DOI] [PubMed] [Google Scholar]
- 31.Moloney A.M., Griffin R.J., Timmons S., O'Connor R., Ravid R., O'Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer's disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiology of Aging. 2010;31(2):224–243. doi: 10.1016/j.neurobiolaging.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 32.Ni W., Rajkumar K., Nagy J.I., Murphy L.J. Impaired brain development and reduced astrocyte response to injury in transgenic mice expressing IGF binding protein-1. Brain Research. 1997;769(1):97–107. doi: 10.1016/s0006-8993(97)00676-8. [DOI] [PubMed] [Google Scholar]
- 33.Davila D., Fernandez S., Torres-Aleman I. Astrocyte resilience to oxidative stress induced by insulin-like growth factor I (IGF-I) involves preserved AKT (protein kinase B) activity. Journal of Biological Chemistry. 2016;291(5):2510–2523. doi: 10.1074/jbc.M115.695478. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34.Madathil S.K., Carlson S.W., Brelsfoard J.M., Ye P., D'Ercole A.J., Saatman K.E. Astrocyte-specific overexpression of insulin-like growth factor-1 protects hippocampal neurons and reduces behavioral deficits following traumatic brain injury in mice. PLoS One. 2013;8(6):e67204. doi: 10.1371/journal.pone.0067204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gibbs M.E., Bowser D.N. Astrocytes and interneurons in memory processing in the chick hippocampus: roles for G-coupled protein receptors, GABA(B) and mGluR1. Neurochemical Research. 2009;34(10):1712–1720. doi: 10.1007/s11064-009-9980-1. [DOI] [PubMed] [Google Scholar]
- 36.Gibbs M.E., Gibbs Z., Hertz L. Rescue of Abeta(1-42)-induced memory impairment in day-old chick by facilitation of astrocytic oxidative metabolism: implications for Alzheimer's disease. Journal of Neurochemistry. 2009;109(Suppl 1):230–236. doi: 10.1111/j.1471-4159.2009.05800.x. [DOI] [PubMed] [Google Scholar]
- 37.Diekman C.O., Fall C.P., Lechleiter J.D., Terman D. Modeling the neuroprotective role of enhanced astrocyte mitochondrial metabolism during stroke. Biophysical Journal. 2013;104(8):1752–1763. doi: 10.1016/j.bpj.2013.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng W., Watts L.T., Holstein D.M., Prajapati S.I., Keller C., Grass E.H. Purinergic receptor stimulation reduces cytotoxic edema and brain infarcts in mouse induced by photothrombosis by energizing glial mitochondria. PLoS One. 2010;5(12):e14401. doi: 10.1371/journal.pone.0014401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Light K.R., Kolata S., Wass C., Denman-Brice A., Zagalsky R., Matzel L.D. Working memory training promotes general cognitive abilities in genetically heterogeneous mice. Current Biology. 2010;20(8):777–782. doi: 10.1016/j.cub.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ashpole N.M., Song W., Brustovetsky T., Engleman E.A., Brustovetsky N., Cummins T.R. Calcium/calmodulin-dependent protein kinase II (CaMKII) inhibition induces neurotoxicity via dysregulation of glutamate/calcium signaling and hyperexcitability. Journal of Biological Chemistry. 2012;287(11):8495–8506. doi: 10.1074/jbc.M111.323915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lane R.S., Fu Y., Matsuzaki S., Kinter M., Humphries K.M., Griffin T.M. Mitochondrial respiration and redox coupling in articular chondrocytes. Arthritis Research and Therapy. 2015;17:54. doi: 10.1186/s13075-015-0566-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pharaoh G., Pulliam D., Hill S., Sataranatarajan K., Van Remmen H. Ablation of the mitochondrial complex IV assembly protein Surf1 leads to increased expression of the UPR(MT) and increased resistance to oxidative stress in primary cultures of fibroblasts. Redox Biology. 2016;8:430–438. doi: 10.1016/j.redox.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iram T., Trudler D., Kain D., Kanner S., Galron R., Vassar R. Astrocytes from old Alzheimer's disease mice are impaired in Abeta uptake and in neuroprotection. Neurobiology of Disease. 2016;96:84–94. doi: 10.1016/j.nbd.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 44.Rolland S.G., Motori E., Memar N., Hench J., Frank S., Winklhofer K.F. Impaired complex IV activity in response to loss of LRPPRC function can be compensated by mitochondrial hyperfusion. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(32):E2967–E2976. doi: 10.1073/pnas.1303872110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tondera D., Grandemange S., Jourdain A., Karbowski M., Mattenberger Y., Herzig S. SLP-2 is required for stress-induced mitochondrial hyperfusion. The EMBO Journal. 2009;28(11):1589–1600. doi: 10.1038/emboj.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Westermann B. Bioenergetic role of mitochondrial fusion and fission. Biochimica et Biophysica Acta. 2012;1817(10):1833–1838. doi: 10.1016/j.bbabio.2012.02.033. [DOI] [PubMed] [Google Scholar]
- 47.Rambold A.S., Kostelecky B., Elia N., Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(25):10190–10195. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gomes L.C., Di Benedetto G., Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nature Cell Biology. 2011;13(5):589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gomes L.C., Scorrano L. Mitochondrial elongation during autophagy: a stereotypical response to survive in difficult times. Autophagy. 2011;7(10):1251–1253. doi: 10.4161/auto.7.10.16771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Friedman J.R., Nunnari J. Mitochondrial form and function. Nature. 2014;505(7483):335–343. doi: 10.1038/nature12985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chance B., Williams G.R. Respiratory enzymes in oxidative phosphorylation. III. The steady state. Journal of Biological Chemistry. 1955;217(1):409–427. [PubMed] [Google Scholar]
- 52.Brand M.D. The sites and topology of mitochondrial superoxide production. Experimental Gerontology. 2010;45(7–8):466–472. doi: 10.1016/j.exger.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fang C.H., Li B.G., Wang J.J., Fischer J.E., Hasselgren P.O. Insulin-like growth factor 1 stimulates protein synthesis and inhibits protein breakdown in muscle from burned rats. JPEN – Journal of Parenteral and Enteral Nutrition. 1997;21(5):245–251. doi: 10.1177/0148607197021005245. [DOI] [PubMed] [Google Scholar]
- 54.Burgos S.A., Cant J.P. IGF-1 stimulates protein synthesis by enhanced signaling through mTORC1 in bovine mammary epithelial cells. Domestic Animal Endocrinology. 2010;38(4):211–221. doi: 10.1016/j.domaniend.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 55.Repetto G., del Peso A., Zurita J.L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nature Protocols. 2008;3(7):1125–1131. doi: 10.1038/nprot.2008.75. [DOI] [PubMed] [Google Scholar]
- 56.Huang B.K., Sikes H.D. Quantifying intracellular hydrogen peroxide perturbations in terms of concentration. Redox Biology. 2014;2:955–962. doi: 10.1016/j.redox.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stone J.R., Yang S. Hydrogen peroxide: a signaling messenger. Antioxidants and Redox Signaling. 2006;8(3–4):243–270. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- 58.Talbot K., Wang H.Y., Kazi H., Han L.Y., Bakshi K.P., Stucky A. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. Journal of Clinical Investigation. 2012;122(4):1316–1338. doi: 10.1172/JCI59903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huffman D.M., Farias Quipildor G., Mao K., Zhang X., Wan J., Apontes P. Central insulin-like growth factor-1 (IGF-1) restores whole-body insulin action in a model of age-related insulin resistance and IGF-1 decline. Aging Cell. 2016;15(1):181–186. doi: 10.1111/acel.12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aguirre G.A., De Ita J.R., de la Garza R.G., Castilla-Cortazar I. Insulin-like growth factor-1 deficiency and metabolic syndrome. Journal of Translational Medicine. 2016;14:3. doi: 10.1186/s12967-015-0762-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stobart J.L., Anderson C.M. Multifunctional role of astrocytes as gatekeepers of neuronal energy supply. Frontiers in Cellular Neuroscience. 2013;7:38. doi: 10.3389/fncel.2013.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hardie D.G., Ross F.A., Hawley S.A. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nature Reviews Molecular Cell Biology. 2012;13(4):251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Canto C., Auwerx J. Calorie restriction: is AMPK a key sensor and effector? Physiology (Bethesda) 2011;26(4):214–224. doi: 10.1152/physiol.00010.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bass B., Upson S., Roy K., Montgomery E.L., Jalonen T.O., Murray I.V. Glycogen and amyloid-beta: key players in the shift from neuronal hyperactivity to hypoactivity observed in Alzheimer's disease? Neural Regenerative Research. 2015;10(7):1023–1025. doi: 10.4103/1673-5374.160059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sonntag W.E., Bennett C., Ingram R., Donahue A., Ingraham J., Chen H. Growth hormone and IGF-I modulate local cerebral glucose utilization and ATP levels in a model of adult-onset growth hormone deficiency. American Journal of Physiology. Endocrinology and Metabolism. 2006;291(3):E604–E610. doi: 10.1152/ajpendo.00012.2006. [DOI] [PubMed] [Google Scholar]
- 66.Zhang L., Trushin S., Christensen T.A., Bachmeier B.V., Gateno B., Schroeder A. Altered brain energetics induces mitochondrial fission arrest in Alzheimer's Disease. Scientific Reports. 2016;6:18725. doi: 10.1038/srep18725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brown-Borg H., Johnson W.T., Rakoczy S., Romanick M. Mitochondrial oxidant generation and oxidative damage in Ames dwarf and GH transgenic mice. Journal of American Aging Association. 2001;24(3):85–96. doi: 10.1007/s11357-001-0012-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brown-Borg H.M., Johnson W.T., Rakoczy S.G. Expression of oxidative phosphorylation components in mitochondria of long-living Ames dwarf mice. Age (Dordrecht, Netherlands) 2012;34(1):43–57. doi: 10.1007/s11357-011-9212-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kennedy M.A., Rakoczy S.G., Brown-Borg H.M. Long-living Ames dwarf mouse hepatocytes readily undergo apoptosis. Experimental Gerontology. 2003;38(9):997–1008. doi: 10.1016/s0531-5565(03)00164-5. [DOI] [PubMed] [Google Scholar]
- 70.Ren J., Brown-Borg H.M. Impaired cardiac excitation-contraction coupling in ventricular myocytes from Ames dwarf mice with IGF-I deficiency. Growth Hormone & IGF Research. 2002;12(2):99–105. doi: 10.1054/ghir.2002.0267. [DOI] [PubMed] [Google Scholar]
- 71.Rojanathammanee L., Rakoczy S., Brown-Borg H.M. Growth hormone alters the glutathione S-transferase and mitochondrial thioredoxin systems in long-living Ames dwarf mice. The Journal of Gerontology. Series A, Biological Sciences and Medical Sciences. 2014;69(10):1199–1211. doi: 10.1093/gerona/glt178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bokov A.F., Lindsey M.L., Khodr C., Sabia M.R., Richardson A. Long-lived ames dwarf mice are resistant to chemical stressors. The Journal of Gerontology. Series A, Biological Sciences and Medical Sciences. 2009;64(8):819–827. doi: 10.1093/gerona/glp052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Csiszar A., Labinskyy N., Perez V., Recchia F.A., Podlutsky A., Mukhopadhyay P. Endothelial function and vascular oxidative stress in long-lived GH/IGF-deficient Ames dwarf mice. American Journal of Physiology – Heart and Circulatory Physiology. 2008;295(5):H1882–H1894. doi: 10.1152/ajpheart.412.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hauck S.J., Aaron J.M., Wright C., Kopchick J.J., Bartke A. Antioxidant enzymes, free-radical damage, and response to paraquat in liver and kidney of long-living growth hormone receptor/binding protein gene-disrupted mice. Hormone and Metabolic Research. 2002;34(9):481–486. doi: 10.1055/s-2002-34787. [DOI] [PubMed] [Google Scholar]
- 75.Romanick M.A., Rakoczy S.G., Brown-Borg H.M. Long-lived Ames dwarf mouse exhibits increased antioxidant defense in skeletal muscle. Mechanism of Ageing and Development. 2004;125(4):269–281. doi: 10.1016/j.mad.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 76.Nieves-Martinez E., Sonntag W.E., Wilson A., Donahue A., Molina D.P., Brunso-Bechtold J. Early-onset GH deficiency results in spatial memory impairment in mid-life and is prevented by GH supplementation. Journal of Endocrinology. 2010;204(1):31–36. doi: 10.1677/JOE-09-0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Donahue A.N., Aschner M., Lash L.H., Syversen T., Sonntag W.E. Growth hormone administration to aged animals reduces disulfide glutathione levels in hippocampus. Mechanism of Ageing and Development. 2006;127(1):57–63. doi: 10.1016/j.mad.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 78.Jiang T., Cadenas E. Astrocytic metabolic and inflammatory changes as a function of age. Aging Cell. 2014;13(6):1059–1067. doi: 10.1111/acel.12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang Y., Liu Y., Walsh M., Bokov A., Ikeno Y., Jang Y.C. Liver specific expression of Cu/ZnSOD extends the lifespan of Sod1 null mice. Mechanism of Ageing and Development. 2016;154:1–8. doi: 10.1016/j.mad.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu Y., Qi W., Richardson A., Van Remmen H., Ikeno Y., Salmon A.B. Oxidative damage associated with obesity is prevented by overexpression of CuZn- or Mn-superoxide dismutase. Biochemical and Biophysical Research Communications. 2013;438(1):78–83. doi: 10.1016/j.bbrc.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Styskal J., Van Remmen H., Richardson A., Salmon A.B. Oxidative stress and diabetes: what can we learn about insulin resistance from antioxidant mutant mouse models? Free Radical Biology and Medicine. 2012;52(1):46–58. doi: 10.1016/j.freeradbiomed.2011.10.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bubici C., Papa S., Pham C.G., Zazzeroni F., Franzoso G. The NF-kappaB-mediated control of ROS and JNK signaling. Histology & Histopathology. 2006;21(1):69–80. doi: 10.14670/HH-21.69. [DOI] [PubMed] [Google Scholar]
- 83.Das K.C., Lewis-Molock Y., White C.W. Activation of NF-kappa B and elevation of MnSOD gene expression by thiol reducing agents in lung adenocarcinoma (A549) cells. American Journal of Physiology. 1995;269(5 Pt 1):L588–L602. doi: 10.1152/ajplung.1995.269.5.L588. [DOI] [PubMed] [Google Scholar]
- 84.Waeber G., Delplanque J., Bonny C., Mooser V., Steinmann M., Widmann C. The gene MAPK8IP1, encoding islet-brain-1, is a candidate for type 2 diabetes. Nature Genetics. 2000;24(3):291–295. doi: 10.1038/73523. [DOI] [PubMed] [Google Scholar]
- 85.Hirosumi J., Tuncman G., Chang L., Gorgun C.Z., Uysal K.T., Maeda K. A central role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 86.Hsiao H.Y., Chen Y.C., Chen H.M., Tu P.H., Chern Y. A critical role of astrocyte-mediated nuclear factor-kappaB-dependent inflammation in Huntington's disease. Human Molecular Genetics. 2013;22(9):1826–1842. doi: 10.1093/hmg/ddt036. [DOI] [PubMed] [Google Scholar]
- 87.Newman L.A., Korol D.L., Gold P.E. Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS One. 2011;6(12):e28427. doi: 10.1371/journal.pone.0028427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Muhic M., Vardjan N., Chowdhury H.H., Zorec R., Kreft M. Insulin and insulin-like growth factor 1 (IGF-1) modulate cytoplasmic glucose and glycogen levels but not glucose transport across the membrane in astrocytes. Journal of Biological Chemistry. 2015;290(17):11167–11176. doi: 10.1074/jbc.M114.629063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Franken P., Gip P., Hagiwara G., Ruby N.F., Heller H.C. Changes in brain glycogen after sleep deprivation vary with genotype. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2003;285(2):R413–R419. doi: 10.1152/ajpregu.00668.2002. [DOI] [PubMed] [Google Scholar]
- 90.Sheng M., Sabatini B.L., Sudhof T.C. Synapses and Alzheimer's disease. Cold Spring Harbour Perspectives in Biology. 2012;4(5) doi: 10.1101/cshperspect.a005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Thal D.R., Schultz C., Dehghani F., Yamaguchi H., Braak H., Braak E. Amyloid beta-protein (Abeta)-containing astrocytes are located preferentially near N-terminal-truncated Abeta deposits in the human entorhinal cortex. Acta Neuropathologica. 2000;100(6):608–617. doi: 10.1007/s004010000242. [DOI] [PubMed] [Google Scholar]
- 92.Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 93.Lichtenwalner R.J., Forbes M.E., Bennett S.A., Lynch C.D., Sonntag W.E., Riddle D.R. Intracerebroventricular infusion of insulin-like growth factor-I ameliorates the age-related decline in hippocampal neurogenesis. Neuroscience. 2001;107(4):603–613. doi: 10.1016/s0306-4522(01)00378-5. [DOI] [PubMed] [Google Scholar]
- 94.Lynch C.D., Lyons D., Khan A., Bennett S.A., Sonntag W.E. Insulin-like growth factor-1 selectively increases glucose utilization in brains of aged animals. Endocrinology. 2001;142(1):506–509. doi: 10.1210/endo.142.1.8053. [DOI] [PubMed] [Google Scholar]
- 95.Markowska A.L., Mooney M., Sonntag W.E. Insulin-like growth factor-1 ameliorates age-related behavioral deficits. Neuroscience. 1998;87(3):559–569. doi: 10.1016/s0306-4522(98)00143-2. [DOI] [PubMed] [Google Scholar]
- 96.Deak F., Sonntag W.E. Aging, synaptic dysfunction, and insulin-like growth factor (IGF)-1. The Journal of Gerontology. Series A, Biological Sciences and Medical Sciences. 2012;67(6):611–625. doi: 10.1093/gerona/gls118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ungvari Z., Sonntag W.E. Brain and cerebrovascular aging–new mechanisms and insights. The Journal of Gerontology. Series A, Biological Sciences and Medical Sciences. 2014;69(11):1307–1310. doi: 10.1093/gerona/glu187. [DOI] [PubMed] [Google Scholar]
- 98.Sonntag W.E., Deak F., Ashpole N., Toth P., Csiszar A., Freeman W. Insulin-like growth factor-1 in CNS and cerebrovascular aging. Frontiers in Aging Neuroscience. 2013;5:27. doi: 10.3389/fnagi.2013.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sonntag W.E., Lynch C., Thornton P., Khan A., Bennett S., Ingram R. The effects of growth hormone and IGF-1 deficiency on cerebrovascular and brain ageing. Journal of Anatomy. 2000;197(Pt 4):575–585. doi: 10.1046/j.1469-7580.2000.19740575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.McGinley L.M., Sims E., Lunn J.S., Kashlan O.N., Chen K.S., Bruno E.S. Human cortical neural stem cells expressing insulin-like growth Factor-I: a novel cellular therapy for Alzheimer's disease. Stem Cells Translational Medicine. 2016;5(3):379–391. doi: 10.5966/sctm.2015-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pitt J., Wilcox K.C., Tortelli V., Diniz L.P., Oliveira M.S., Dobbins C. Neuroprotective astrocyte-derived insulin/IGF-1 stimulate endocytic processing and extracellular release of neuron-bound Abeta oligomers. Molecular Biology of the Cell. 2017 doi: 10.1091/mbc.E17-06-0416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hartz A.M., Bauer B. ABC transporters in the CNS - an inventory. Current Pharmaceutical Biotechnology. 2011;12(4):656–673. doi: 10.2174/138920111795164020. [DOI] [PubMed] [Google Scholar]
- 103.Fan J., Donkin J., Wellington C. Greasing the wheels of Abeta clearance in Alzheimer's disease: the role of lipids and apolipoprotein E. BioFactors. 2009;35(3):239–248. doi: 10.1002/biof.37. [DOI] [PubMed] [Google Scholar]
- 104.Repa J.J., Li H., Frank-Cannon T.C., Valasek M.A., Turley S.D., Tansey M.G. Liver X receptor activation enhances cholesterol loss from the brain, decreases neuroinflammation, and increases survival of the NPC1 mouse. Journal of Neuroscience. 2007;27(52):14470–14480. doi: 10.1523/JNEUROSCI.4823-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Freude S., Hettich M.M., Schumann C., Stohr O., Koch L., Kohler C. Neuronal IGF-1 resistance reduces Abeta accumulation and protects against premature death in a model of Alzheimer's disease. The FASEB Journal. 2009;23(10):3315–3324. doi: 10.1096/fj.09-132043. [DOI] [PubMed] [Google Scholar]
- 106.Gontier G., George C., Chaker Z., Holzenberger M., Aid S. Blocking IGF signaling in adult neurons alleviates Alzheimer's disease pathology through amyloid-beta clearance. Journal of Neuroscience. 2015;35(33):11500–11513. doi: 10.1523/JNEUROSCI.0343-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.