

Abstract
Objective
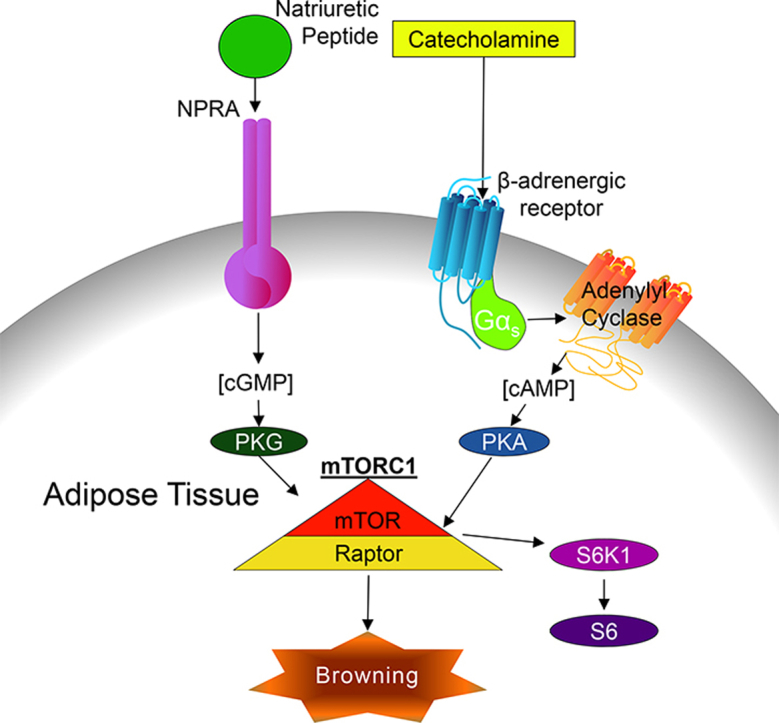
Activation of thermogenesis in brown adipose tissue (BAT) and the ability to increase uncoupling protein 1 (UCP1) levels and mitochondrial biogenesis in white fat (termed ‘browning’), has great therapeutic potential to treat obesity and its comorbidities because of the net increase in energy expenditure. β-adrenergic-cAMP-PKA signaling has long been known to regulate these processes. Recently PKA-dependent activation of mammalian target of rapamycin complex 1 (mTORC1) was shown to be necessary for adipose ‘browning’ as well as proper development of the interscapular BAT. In addition to cAMP-PKA signaling pathways, cGMP-PKG signaling also promotes this browning process; however, it is unclear whether or not mTORC1 is also necessary for cGMP-PKG induced browning.
Method
Activation of mTORC1 by natriuretic peptides (NP), which bind to and activate the membrane-bound guanylyl cyclase, NP receptor A (NPRA), was assessed in mouse and human adipocytes in vitro and mouse adipose tissue in vivo.
Results
Activation of mTORC1 by NP-cGMP signaling was observed in both mouse and human adipocytes. We show that NP-NPRA-PKG signaling activate mTORC1 by direct PKG phosphorylation of Raptor at Serine 791. Administration of B-type natriuretic peptide (BNP) to mice induced Ucp1 expression in inguinal adipose tissue in vivo, which was completely blocked by the mTORC1 inhibitor rapamycin.
Conclusion
Our results demonstrate that NP-cGMP signaling activates mTORC1 via PKG, which is a component in the mechanism of adipose browning.
Keywords: Thermogenesis, UCP1, Kinases
Abbreviations: βAR, β-adrenergic receptor; 8-Br-cAMP, 8-Bromoadenosine 3′,5′-cyclic monophosphate; 8-Br-cGMP, 8-Bromoguanosine 3′,5′-cyclic monophosphate; Akt, protein kinase B; ANP, Atrial natriuretic peptide; BNP, B-type natriuretic peptide; cAMP, cyclic adenosine monophosphate; cGMP, cyclic guanosine monophosphate; gWAT, gonadal white adipose tissue; hMADS, human mesenchymal adipose derived stem cells; iBAT, interscapular brown adipose tissue; iWAT, inguinal white adipose tissue; ISO, isoproterenol; mTORC1, mammalian target of rapamycin complex 1; NPRA, natriuretic peptide receptor-A; PKA, protein kinase A; PKG, protein kinase G; RAPA, rapamycin; S6K1, ribosomal protein S6 kinase 1; sGC, soluble guanylyl cyclase; SNS, sympathetic nervous system; UCP1, uncoupling protein 1; VASP, vasodilator-stimulated protein; WAT, white adipose tissue
Graphical abstract
Highlights
-
•
Natriuretic peptide-cGMP signaling activates mTORC1 in adipocytes in vitro.
-
•
Natriuretic peptide-cGMP signaling activates mTORC1 in adipose tissue in vivo.
-
•
Natriuretic peptides signaling stimulates Raptor phosphorylation.
-
•
mTORC1 is necessary for BNP-evoked browning of inguinal white adipose tissue in vivo.
1. Introduction
Atrial natriuretic peptide (ANP) and B-type natriuretic peptide (BNP) are endocrine hormones that are released from the heart in response to increases in cardiac wall stress and other local factors [1], [2], [3]. NPs were first discovered when it was shown that extracts of rat myocardial homogenates were able to lower blood pressure and greatly promote salt and water excretions and urine production [4]. In addition to the vasodepressor, diuretic, and natriuretic effects of NPs, they also play an important role in energy balance [5]. Of particular interest is the role of NPs in adipocyte metabolism. For example, NPs are potent lipolytic agents in adipocytes, facilitating the liberation of free fatty acids from triglycerides thereby promoting lipid mobilization [6], [7], [8]. NPs bind to a membrane-bound receptor that possesses guanylyl cyclase activity (Natriuretic Peptide Receptor A, NPRA), generating cyclic GMP (cGMP) that activates protein kinase G (PKG) [1], [3]. This pathway functions in parallel with the more well-known ability of norepinephrine (NE) to activate β-adrenergic receptors (βAR) to increase cAMP and PKA activity. It is now appreciated that the NPs are capable of increasing the thermogenic activity of adipocytes, via increasing intracellular cGMP through NPRA, to function in a parallel manner in adipose tissue [9]. In addition, we have demonstrated that NPs, similar to NE, promote brown adipocyte respiration, thermogenesis, and the ‘browning’ of white adipose depots through a common p38 MAPK signaling node [9], [10]. Together these pathways drive transcriptional events to increase the density of mitochondria, UCP1 and the metabolic enzymes that are necessary for this energy-intensive process of non-shivering thermogenesis in brown adipocytes [1], [11].
Recently we showed that mTORC1, which is generally recognized as a key hub in the insulin signaling pathway, is activated by βAR increases in PKA activity [12]. PKA directly phosphorylates mTOR and its partner protein Raptor, leading to the subsequent activation of p70 ribosomal S6 kinase (S6K1) [12]. This PKA-mTORC1 signaling pathway is necessary for cold-induced ‘browning’ of white adipose tissue (WAT). Similar to cold exposure, the ability of the β3AR agonist, CL316,243, to induce browning is also blocked by the mTORC1 inhibitor, rapamycin [12], [13]. Given the similarities that exist between β-adrenergic and NP activation of p38 MAPK, we sought to determine whether this pattern extends to the activation of mTORC1. In this study, we find that NPs increase the phosphorylation of the mTORC1 downstream target S6K1 in a rapamycin-sensitive manner that, as in the case of catecholamines, does not involve the canonical Akt pathway. Instead, PKG phosphorylates Raptor on Ser791, this site also being phosphorylated by PKA. Given these findings and the ubiquitous expression of mTOR, PKA, and PKG in many cell types, we speculate that our observations in adipose tissue may have broader significance by regulating a variety of cell signaling events in other cell types and organs.
2. Materials and methods
2.1. Reagents and antibodies
ANP (1–28) was obtained from AnaSpec Inc. (Fremont, CA, #AS-20648) and BNP was from ProSpec (East Brunswick, NJ, #CYT-369-B). 8-Br-cAMP, 8-Br-cGMP, ATP, cGMP, Isoproterenol (ISO), KT5720, and anti-Myc antibody-conjugated beads were from Sigma Aldrich (St. Louis, MO). Rapamycin was from LC Laboratories (Woburn, MA). Purified PKG was from Promega (Madison, WI, cat#V5171). Polyethylenimine was from Polysciences, Inc. (Warrington, PA). The following antisera were obtained from Cell Signaling Biotechnology (Danvers, MA): p-VASP (S239, #3114), VASP (#3132), p-S6K1 (T389, t#9205), S6K1 (#9202), p-S6 (T240/244, #2215), S6 (#2217), p-Akt (T308, #9275; S473, #4060), Akt (#9272), β-actin (#4967), p-RRXS/T (#9624), myc-tag antibody (#2278). Anti-Rabbit IgG-alkaline phosphatase (Sigma Aldrich, #A3687) and Clean Blot IP Detection Reagent (AP) (ThermoFisher, Grand Island, NY, #21233) were used as secondary antibodies. Immortalized mouse brown adipocytes (Bat8) were a gift from Dr. Bruce Spiegelman [14], human multipotent adipose-derived stem cells (hMADS) were a gift from Dr. Ez-Zoubir Amri [15], and HEK 293 cells stably expressing NPRA-GFP (NPRA-HEK) were a gift from Dr. John Burnett [16]. The myc-Raptor plasmid [17] was obtained from Addgene (plasmid #1859, Cambridge, MA), and construction of the myc-Raptor (S791A) plasmid was described previously [12].
2.2. Cell culture and transfection
Bat8 cells were cultured and differentiated as described [14]. Briefly, cells were grown in DMEM/F12 GlutaMAX™ (ThermoFisher, #10565018) containing 15% FBS, 2 mM Hepes, and 50 units/ml penicillin and streptomycin. To induce differentiation, the media was replaced with DMEM/F12 GlutaMAX™ containing 10% FBS, 5 μM dexamethasone, 0.5 μg/ml insulin, 0.5 mM isobutylmethylxanthine, 1 μM rosiglitazone, and 1 nM T3. On day 4 of differentiation, the cells were switched to a maintenance media of DMEM/F12 GlutaMAX™ containing 10% FBS with 0.5 μg/ml insulin and 1 nM T3. On day 7, the differentiated Bat8 cells were serum-starved overnight in DMEM/F12 GlutaMAX™ for treatments on day 8. Cells were treated with 100 nM rapamycin 30 min before the additional 60-minute treatment with 10 nM insulin, 1 μM ISO, or 200 μM 8-Br-cAMP.
hMADS cells were maintained in low glucose DMEM (Lonza, #12–707F), 10% fetal bovine serum, 2 mM l-glutamine, 10 mM HEPES buffer, 50 units/ml penicillin, and 50 μg/ml streptomycin, supplemented with 2.5 ng/ml human fibroblast growth factor-2. Cell growth and differentiation were performed as described previously [9]. 8-Br-cAMP (200 μM), 8-Br-cGMP (200 μM), and ANP (200 nM) were added to the differentiated cells for 30 min. Where indicated, KT5720 (2.5 μM) and rapamycin (100 nM) was added 30 min in advance of the respective treatments.
NPRA-HEK and HEK 293T cells were cultured in high glucose DMEM (ThermoFisher, #11995065), 2 mM l-glutamine, 10% FBS, supplemented with 50 units/ml penicillin and 50 μg/ml streptomycin. The HEK cells were grown in six-well plates and the appropriate myc-Raptor was transiently overexpressed by transfecting 2 μg plasmid using 6 μl of 25 μg/ml polyethylenimine per well. Twenty-four hours later, the cells were stimulated with 200 nM ANP or BNP for 30 min.
2.3. Western blotting
Cells or tissues were lysed and sonicated in buffer containing 25 mM HEPES (pH 7.4), 150 mM NaCL, 5 mM EDTA, 5 mM EGTA, 5 mM glycerophosphate, 0.9% Triton X-100, 0.1% IGEPAL, 5 mM sodium pyrophosphate, 10% glycerol, plus 1 tablet each of complete protease inhibitor cocktail (Roche) and PhoSTOP phosphatase inhibitors (Roche) per 10 ml of lysis buffer. For immunoprecipitations, the cell lysate was incubated with myc antibody conjugated beads in 800 μg of cell lysate overnight. The beads were washed three times with the lysis buffer for 5 min each. For Western blot analysis, the immunoprecipitate or 50 μg total protein was resolved in 10% Tris–glycine gels, transferred to nitrocellulose membranes, and probed overnight at 4 °C with specific primary antibodies, followed by secondary antibody incubation for 1 h at room temperature. Image acquisition was performed on a Typhoon FLA9000 variable mode imager and analyzed using ImageQuant TL software.
2.4. In vitro kinase assay
Myc-Raptor protein was produced by transfecting myc-Raptor and myc-Raptor (S791A) plasmids into HEK 293T cells and purified with myc antibody conjugated beads as during immunoprecipitation. Following a wash with the Enzyme Reaction Buffer of 40 mM Tris–HCl and 20 mM magnesium acetate, pH 7.5, the myc-Raptor bound beads were incubated in Enzyme Reaction Buffer with 200 units of PKG, 10 μM cGMP, and 200 μM ATP. The reaction was incubated at 30 °C for 30 min while rotating and western blotting was then performed.
2.5. Animal experiments
Male and female C57BL/6J mice were obtained from the Jackson Laboratory. All experiments were conducted in mice at 8–10 weeks of age. To activate PKG through NPRA, 5 mg/kg/day BNP was administered by once daily intraperitoneal injection for one week in the absence or presence of rapamycin 4 mg/kg/day. Rapamycin was prepared as described previously [12]. Mice were euthanized by CO2 asphyxiation at the end of the study, and tissues were collected. All mouse experiments were conducted in accordance with federal guidelines and were approved by the Institutional Animal Care and Use Committee of the SBP Medical Discovery Institute at Lake Nona.
2.6. RNA isolation and analysis
Total RNA was extracted from mouse adipose tissues using TRIzol Reagent (ThermoFisher) and purified with the RNeasy Mini Kit (Qiagen, Hilden, Germany). Reverse transcription of 2 μg RNA was performed with the High-Capacity cDNA Reverse Transcription Kit (ThermoFisher). All qPCR assays were run on a Roche LightCycler® 480 II. Gene expression data were collected from 2 replicates of each sample. Data were normalized to mRplp0 (36B4) and quantitative measurement was obtained using the ΔCT method. The primer sequences are as follow: Ucp1 forward 5′-GGCCTCTACGACTCAGTCCA-3′, reverse 5′-TAAGCCGGCTGAGATCTTGT-3′; Ppargc1a (PGC-1α) forward 5′-CGGAAATCATATCCAACCAG-3′, reverse 5′-TGAGAACCGCTAGCAAGTTTG-3′, and Rplp0 (36B4) forward 5′-GATGCCCAGGGAAGACAG-3′, reverse 5′-ACAATGAAGCATTTTGGATAATCA-3′.
2.7. Histology
The inguinal white adipose tissue (iWAT) was collected from male mice at the end of the experiment and fixed with 4% paraformaldehyde in PBS overnight, dehydrated, embedded in paraffin, and sectioned at 5 μm thickness. UCP1 immunostaining was performed as previously described by the Sanford Burnham Prebys Medical Discovery Institute at Lake Nona Histology Core [9], [18]. Slides were imaged at 20× with a ScanScope XT (Aperio, Vista, CA) at the histology core.
2.8. Statistics
Results are presented as mean ± SEM. Data were analyzed using 2-tailed Student's t test. Differences were considered significant at P < 0.05. Prism 7.0 software was used for statistical analyses; P values for significance are indicated for each data set.
3. Results
3.1. Natriuretic peptides activate mTORC1 in cultured adipocytes
The immortalized mouse brown adipocyte cell line, Bat8, was treated with insulin, ISO, or the membrane-permeable cAMP analogue, 8-Br-cAMP. As shown in Figure 1A and B, we observed increased phosphorylation of the mTORC1 downstream targets, S6K1 and S6. Rapamycin completely ablated the phosphorylation, reaffirming the role of mTORC1 in this process. We then asked whether cGMP and natriuretic peptides could elicit the same response in human adipocytes. Phosphorylation of the mTORC1 downstream targets S6K1 and S6 were increased by ANP, and the PKA inhibitor KT5720 had no effect, illustrating that natriuretic peptide-PKG activation of these targets occurs independent of PKA (Figure 1C). Figure 1D shows that the phosphorylation of S6K1 and S6 by ANP and by 8-Br-cGMP were completely blocked by rapamycin, the mTORC1 inhibitor. Also shown here is that ANP and 8-Br-cGMP increased phosphorylation of vasodilator-stimulated protein (VASP) at Ser239. VASP is a substrate for both PKA and PKG, with Ser239 being highly selective for PKG [19]. Since PKG is upstream of mTORC1, phosphorylation of VASP is not affected by rapamycin. (Figure 1D). Finally, Figure 1D also shows that AKT was not activated in response to either ANP or 8-Br-cGMP, as indicated by the unchanged phosphorylation state of Akt at Thr308 and Ser473 (Figure 1D). (Phosphorylation of Akt on Ser473 is known to be higher in the presence of rapamycin [20]). Thus, similar to what we have observed for PKA [12], PKG is able to activate mTORC1 in adipocytes in vitro.
Figure 1.

cAMP- and cGMP-dependent kinases activate mTORC1 signaling in adipocytes. (A) mTORC1 activity, measured by phosphorylation of S6K1 and S6, in Bat8 adipocytes, an immortalized mouse brown adipocyte cell line, in response to insulin (Ins) or isoproterenol (ISO) in the absence or presence of rapamycin (RAPA). (B) Bat8 adipocytes treated with ISO or 8-Br-cAMP with or without RAPA. (C) mTORC1 activity in hMADS, a human immortalized adipocyte cell line, in response to ANP, 8-Br-cAMP, or insulin, in the absence or presence of the PKA inhibitor, KT5720. (D) hMADS cells treated with ANP or 8-Br-cGMP in the absence or presence of RAPA. ANP treatment increases phosphorylation of VASP at Ser239, an established PKG phosphorylation site.
3.2. Protein kinase G directly phosphorylates Raptor at Ser791
We previously showed that PKA directly phosphorylates Raptor at Ser791, and this phosphorylation is responsible for mTORC1 activation by βARs and PKA, without any involvement of insulin-Akt signaling [12]. In order to determine whether the ANP- and BNP-evoked activation of mTORC1 is due to direct phosphorylation of Raptor at Ser791 by PKG, we employed an in vitro kinase assay. As shown in Figure 2A, for this assay, we produced myc-Raptor proteins by transfecting into HEK-293T cells, plasmids containing either the wild-type sequence or a point mutation in which Ser791 was converted to Ala. As illustrated by the cartoon in Figure 2A, the myc-tagged proteins were immunoprecipitated with anti-myc antibody-conjugated agarose beads and incubated with purified PKG together with cGMP to activate the kinase, with ATP serving as the phosphate donor. The reaction was stopped and western blotting was performed using an antibody directed against the phosphorylated RRXS/T motif, which is the canonical substrate of arginine-directed kinases such as PKA and PKG. There was robust phosphorylation of wild type Raptor, but when Ser791 was mutated, phosphorylation was no longer detected (Figure 2A). Therefore, PKG can directly phosphorylate Raptor at Ser791. Following these in vitro kinase studies we employed HEK 293 cells that stably express NPRA [16], in which the myc-Raptor (wild type) or myc-Raptor S791A were expressed. Twenty-four hours after transfection, the cells were treated with 200 nM ANP or BNP for 30 min. As shown in Figure 2B, both NPs resulted in phosphorylation of VASP at Ser239, indicating that NP-PKG signaling was increased as expected. The myc-Raptor proteins were immunoprecipitated from the cell lysates as in Figure 2A, and western blotting revealed that there was a dramatic increase in phosphorylation of the wild type myc-Raptor, which was absent from myc-Raptor S791A (Figure 2B). As we observed for PKA [12], the residual faint p-RRXS/T band seen in the ANP and BNP treated Raptor S791A immunoprecipitate is a non-specific protein that is recovered with the myc-antibody bead (Figure 2B right panel). Thus, ANP- and BNP-evoked PKG activity leads to the phosphorylation of Raptor at Ser791 in an analogous fashion to PKA.
Figure 2.

Protein kinase G phosphorylates Raptor at Serine 791. (A) myc-Raptor (wild type) or myc-Raptor in which Ser791 of Raptor is mutated to Ala (S791A) were purified and incubated with PKG in an in vitro kinase assay. Phosphorylation of Raptor was performed as described in Methods. (B) HEK 293 cells stably expressing NPRA-GFP were transfected to express myc-Raptor (wild type) or myc-Raptor (S791A) and treated with ANP or BNP. The residual faint band seen in the ANP and BNP treated Raptor S791A immunoprecipitate is a non-specific protein that is recovered with the myc-antibody beads. Phosphorylation of Raptor was performed as described in Methods.
3.3. Natriuretic peptides activate mTORC1 in vivo
We next assessed whether NPRA-PKG signaling could activate mTORC1 in vivo. Mice were administered 5 mg/kg BNP and treated with or without 4 mg/kg rapamycin for 7 days, following which inguinal (iWAT) and gonadal white adipose tissue (gWAT) were collected to examine phosphorylation of mTORC1 downstream targets. In iWAT, NP treatment increased the phosphorylation of S6K1 and S6; both of which were blocked by treatment with the mTORC1 inhibitor, rapamycin (Figure 3A). In gWAT, BNP did not alter S6K1 or S6 phosphorylation, but basal levels of phosphorylation were eliminated by rapamycin (Figure 3B). Since NPRC, the natriuretic peptide clearance receptor, is more abundant in gWAT than iWAT (Figure S1), dampened ability of NPs to signal through NPRA might have contributed to a lack of clear mTORC1 activation in response to BNP in this depot. We suspect that these basal levels of phosphorylation are due to residual sympathetic nervous system (SNS) activity, perhaps due to the mice being housed at room temperature, a condition in which there is residual sympathetic activation of BAT, instead of thermoneutrality. In this case rapamycin may be inhibiting low level PKA-mTORC1 signaling [12].
Figure 3.

B-type natriuretic peptide activates mTORC1 in inguinal adipose tissue. Female mice administered BNP (5 mg/kg/day) with or without RAPA (4 mg/kg/day). Western blots for S6K1 and S6 in (A) iWAT or (B) gWAT, N = 3 mice per group.
We previously reported that BNP administration increased the expression of brown adipocyte markers, such as UCP1, in iWAT [9]. In another cohort of mice similarly treated with BNP with or without rapamycin, we found that BNP treatment increased Ucp1 expression in iWAT by about 7-fold, which was blunted by rapamycin (Figure 4A). Similarly, BNP increased PGC-1α gene (Ppargc1a) expression, which was blocked by rapamycin (Figure 4A). Interestingly, BNP treatment appeared to reduce the size of adipocytes in iWAT and this effect was also blocked by rapamycin (top panel, Figure 4B). Consistent with the gene expression data, we observed increased immunohistochemical staining of UCP1 in iWAT, in response to BNP and these UCP1 positive cells were largely absent from tissues of mice that were administered rapamycin along with BNP (lower panel, Figure 4B). These data indicate that PKG is able to activate mTORC1 in adipose tissues in vivo, which is necessary for BNP-PKG-evoked browning of iWAT in vivo.
Figure 4.

Rapamycin blocks BNP-evoked increase of inguinal adipose tissue thermogenic gene expression. Male mice administered B-type natriuretic peptide (BNP; 5 mg/kg/day) with or without the mTORC1 inhibitor rapamycin (RAPA; 4 mg/kg/day). (A) Gene expression levels of Ucp1 and Ppargc1a in iWAT. (B) H&E staining (top) and UCP1 staining (bottom) of iWAT. Values represent mean ± SEM: *, P ≤ 0.05; **, P ≤ 0.01. N = 6 (vehicle), 8 (BNP), and 4 (BNP + RAPA). Images shown are representative of those analyzed from 4 mice per treatment group and are of same magnification; see scale bar.
4. Discussion
mTOR is best known as a crucial mediator of growth in response to nutrients and anabolic signals such as insulin and growth factors [21], [22]. Catabolism is the opposite state, in which breakdown of tissue mass provides energy resources. Signaling from the SNS, which regulates the “fight or flight” response, is generally catabolic as it promotes the breakdown of molecules for utilization as an energy source [23]. Given the opposing roles of insulin and SNS signaling in anabolism and catabolism, it would be expected that these two pathways would have opposing effects on mTORC1. Indeed, the sympathetic-cAMP pathway has been shown to disrupt insulin-evoked activation of mTORC1 in adipocytes [24], [25]. However, we recently discovered that the βAR-cAMP-PKA pathway itself can lead to mTORC1 activation and is necessary for the induction of adipose tissue browning and iBAT development [12]. Therefore, mTORC1 appears to have an important role in the catabolic process of adipose tissue browning and the dissipation of chemical energy by thermogenesis.
Targeting of G-protein-coupled receptors, such as the βARs by the sympathetic nervous system, to increase cAMP and PKA activity is a well-established mechanism to promote increases in brown adipose tissue thermogenic activity and the appearance of thermogenically active UCP1-positive, mitochondria-rich adipocytes in white adipose depots, referred to as ‘browning’ [26]. However, more recently it is appreciated that cGMP-regulated pathways have similar effects in adipose tissue (reviewed in [27], [28]). We previously showed that ANP and BNP, which increase intracellular cGMP and PKG activity through their membrane-bound guanylyl cyclase-coupled receptor NPRA, comprise a signaling pathway in adipocytes that parallels that of the SNS and βARs [1], [29]. Given that the cyclic nucleotides cAMP and cGMP lead to PKA and PKG phosphorylation of closely conserved amino acid motifs [30], we asked whether mTORC1 was also essential for cGMP-PKG evoked induction of thermogenic adipocytes. Indeed, the present studies demonstrate that activation of mTORC1 is essential for NP-cGMP stimulation of thermogenic markers in adipocytes. We induced browning in vivo by a short-term BNP treatment in mice, which reduced adipocyte cell size and increased thermogenic gene expression (Figure 4). Both the reduction in adipocyte size and induction of thermogenic genes was blocked when the mice were co-treated with the mTORC1 inhibitor, rapamycin. These results confirm two of our prior observations: first, NP's promote adipose tissue browning [9], [18], and second, mTORC1 activation is important for this adipose tissue browning [12]. The present studies link together these two previously disparate observations, demonstrating that mTORC1 activation is also a necessary component for NP-evoked adipose tissue browning.
Several studies have suggested that cGMP produced by soluble guanylyl cyclases (sGCs), which are activated by nitric oxide (NO) may also play a role in adipose browning [31], [32], [33], [34], [35]. A few studies have also linked sGCs to mTOR signaling; however, the underlying signaling mechanism is not so clear. For example, NO-dependent cGMP has been noted to activate mTOR in human and rat vascular smooth muscle cells [36] and in primary human small airway epithelial cells [37]. However, in both of these models, AKT was also activated in response to increasing cGMP [36], [37]. This is in stark contrast to what we have shown in adipocytes. Whether the stimulus is natriuretic peptide-dependent PKG activity, as in our present studies, or our earlier studies with PKA [12], Akt is clearly not involved. In a few cases in which an NO/cGMP activation of mTOR has been invoked, such as in melanoma cells [38], it was proposed to be independent of guanylyl cyclase; instead by nitrosylating and inactivating tuberous sclerosis complex (TSC), which is a component of the canonical signaling cascade for mTOR activation downstream of Akt [39]. The present studies are the first, to our knowledge, to demonstrate that activation of membrane-bound guanylyl cyclases leads to the activation of mTOR. All together, these studies demonstrate that mTORC1 is an important effector of PKG signaling, not only in adipocytes, but apparently in many cell types. Clearly there is more work to be done to fully understand what role the sGCs play in the adipocyte, and the signaling pathways involved.
5. Conclusion
Our studies demonstrate that there is a signaling cascade linking cGMP to mTORC1 activation and that this activation of mTORC1 is necessary for NP-induced browning of adipocytes. This was previously unexpected given the catabolic nature of thermogenic adipocytes and the key role mTORC1 plays in anabolic signaling. However, after we demonstrated that PKA directly activates mTOR and Raptor [12], a similar role for PKG appeared likely. These studies further demonstrate the importance of mTORC1 in the browning process, implicating it in both PKA- and PKG-mediated pathways. These results also have broader significance, as mTORC1 may likely be an important signaling component to other PKG-dependent pathways.
Acknowledgments
The authors wish to thank Drs. George Kyriasis and Katalin Karolyi for help with immunohistochemistry images and Mr. Lorenzo Thomas for administrative assistance. This work was supported by NIH R01 DK103056.
Footnotes
Supplementary data related to this article can be found at https://doi.org/10.1016/j.molmet.2017.12.017.
Conflicts of interest
The authors have no conflicts of interest to disclose.
Appendix A. Supplementary data
The following is the supplementary data related to this article:
References
- 1.Collins S. A heart-adipose tissue connection in the regulation of energy metabolism. Nature Reviews Endocrinology. 2014;10:157–163. doi: 10.1038/nrendo.2013.234. [DOI] [PubMed] [Google Scholar]
- 2.Volpe M., Rubattu S., Burnett J. Natriuretic peptides in cardiovascular diseases: current use and perspectives. European Heart Journal. 2014;35:419–425. doi: 10.1093/eurheartj/eht466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zois N.E., Bartels E.D., Hunter I., Kousholt B.S., Olsen L.H., Goetze J.P. Natriuretic peptides in cardiometabolic regulation and disease. Nature Reviews Cardiology. 2014;11:403–412. doi: 10.1038/nrcardio.2014.64. [DOI] [PubMed] [Google Scholar]
- 4.de Bold A.J., Borenstein H.B., Veress A.T., Sonnenberg H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Sciences. 1981;28:89–94. doi: 10.1016/0024-3205(81)90370-2. [DOI] [PubMed] [Google Scholar]
- 5.Coué M., Moro C. Natriuretic peptide control of energy balance and glucose homeostasis. Biochimie. 2016;124:84–91. doi: 10.1016/j.biochi.2015.05.017. [DOI] [PubMed] [Google Scholar]
- 6.Galitzky J., Sengenes C., Thalamas C., Marques M.A., Senard J.M., Lafontan M. The lipid-mobilizing effect of atrial natriuretic peptide is unrelated to sympathetic nervous system activation or obesity in young men. The Journal of Lipid Research. 2001;42:536–544. [PubMed] [Google Scholar]
- 7.Lafontan M., Moro C., Sengenes C., Galitzky J., Crampes F., Berlan M. An unsuspected metabolic role for atrial natriuretic peptides: the control of lipolysis, lipid mobilization, and systemic nonesterified fatty acids levels in humans. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25:2032–2042. doi: 10.1161/01.ATV.0000183728.14712.d8. [DOI] [PubMed] [Google Scholar]
- 8.Sengenes C., Berlan M., De Glisezinski I., Lafontan M., Galitzky J. Natriuretic peptides: a new lipolytic pathway in human adipocytes. The FASEB Journal. 2000;14:1345–1351. [PubMed] [Google Scholar]
- 9.Bordicchia M., Liu D., Amri E.Z., Ailhaud G., Dessi-Fulgheri P., Zhang C. Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. Journal of Clinical Investigation. 2012;122:1022–1036. doi: 10.1172/JCI59701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao W., Daniel K.W., Robidoux J., Puigserver P., Medvedev A.V., Bai X. p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Molecular and Cellular Biology. 2004;24:3057–3067. doi: 10.1128/MCB.24.7.3057-3067.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Collins S., Bordicchia M. Heart hormones fueling a fire in fat. Adipocyte. 2013;2:104–108. doi: 10.4161/adip.22515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu D., Bordicchia M., Zhang C., Fang H., Wei W., Li J.L. Activation of mTORC1 is essential for β-adrenergic stimulation of adipose browning. Journal of Clinical Investigation. 2016;126:1704–1716. doi: 10.1172/JCI83532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tran C.M., Mukherjee S., Ye L., Frederick D.W., Kissig M., Davis J.G. Rapamycin blocks induction of the thermogenic program in white adipose tissue. Diabetes. 2016;65:927–941. doi: 10.2337/db15-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu J., Bostrom P., Sparks L.M., Ye L., Choi J.H., Giang A.H. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150:366–376. doi: 10.1016/j.cell.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodriguez A.M., Elabd C., Delteil F., Astier J., Vernochet C., Saint-Marc P. Adipocyte differentiation of multipotent cells established from human adipose tissue. Biochemical and Biophysical Research Communications. 2004;315:255–263. doi: 10.1016/j.bbrc.2004.01.053. [DOI] [PubMed] [Google Scholar]
- 16.Martin F.L., Sangaralingham S.J., Huntley B.K., McKie P.M., Ichiki T., Chen H.H. CD-NP: a novel engineered dual guanylyl cyclase activator with anti-fibrotic actions in the heart. PLoS One. 2012;7:e52422. doi: 10.1371/journal.pone.0052422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sarbassov D.D., Ali S.M., Kim D.H., Guertin D.A., Latek R.R., Erdjument-Bromage H. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Current Biology. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 18.Wu W., Shi F., Liu D., Ceddia R.P., Gaffin R., Wei W. Enhancing natriuretic peptide signaling in adipose tissue, but not in muscle, protects against diet-induced obesity and insulin resistance. Science Signaling. 2017;10:eaam6870. doi: 10.1126/scisignal.aam6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smolenski A., Bachmann C., Reinhard K., Hönig-Liedl P., Jarchau T., Hoschuetzky H. Analysis and regulation of vasodilator-stimulated phosphoprotein serine 239 phosphorylation in vitro and in intact cells using a phosphospecific monoclonal antibody. Journal of Biological Chemistry. 1998;273:20029–20035. doi: 10.1074/jbc.273.32.20029. [DOI] [PubMed] [Google Scholar]
- 20.Breuleux M., Klopfenstein M., Stephan C., Doughty C.A., Barys L., Maira S.M. Increased AKT S473 phosphorylation after mTORC1 inhibition is rictor dependent and does not predict tumor cell response to PI3K/mTOR inhibition. Molecular Cancer Therapeutics. 2009;8:742–753. doi: 10.1158/1535-7163.MCT-08-0668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haissaguerre M., Saucisse N., Cota D. Influence of mTOR in energy and metabolic homeostasis. Molecular and Cellular Endocrinology. 2014;397:67–77. doi: 10.1016/j.mce.2014.07.015. [DOI] [PubMed] [Google Scholar]
- 22.Lee P.L., Jung S.M., Guertin D.A. The complex roles of mechanistic target of rapamycin in adipocytes and beyond. Trends in Endocrinology and Metabolism. 2017;28:319–339. doi: 10.1016/j.tem.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Romijn J.A., Fliers E. Sympathetic and parasympathetic innervation of adipose tissue: metabolic implications. Current Opinion in Clinical Nutrition and Metabolic Care. 2005;8:440–444. doi: 10.1097/01.mco.0000172586.09762.55. [DOI] [PubMed] [Google Scholar]
- 24.Mullins G.R., Wang L., Raje V., Sherwood S.G., Grande R.C., Boroda S. Catecholamine-induced lipolysis causes mTOR complex dissociation and inhibits glucose uptake in adipocytes. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:17450–17455. doi: 10.1073/pnas.1410530111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scott P.H., Lawrence J.C., Jr. Attenuation of mammalian target of rapamycin activity by increased cAMP in 3T3-L1 adipocytes. Journal of Biological Chemistry. 1998;273:34496–34501. doi: 10.1074/jbc.273.51.34496. [DOI] [PubMed] [Google Scholar]
- 26.Pfeifer A., Hoffmann L.S. Brown, beige, and white: the new color code of fat and its pharmacological implications. Annual Review of Pharmacology and Toxicology. 2015;55:207–227. doi: 10.1146/annurev-pharmtox-010814-124346. [DOI] [PubMed] [Google Scholar]
- 27.Lafontan M., Moro C., Berlan M., Crampes F., Sengenes C., Galitzky J. Control of lipolysis by natriuretic peptides and cyclic GMP. Trends in Endocrinology and Metabolism. 2008;19:130–137. doi: 10.1016/j.tem.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 28.Hoffmann L.S., Larson C.J., Pfeifer A. cGMP and brown adipose tissue. In: Herzig S., editor. Metabolic control. Springer International Publishing; Cham: 2016. pp. 283–299. [Google Scholar]
- 29.Collins S., Sarzani R., Bordicchia M. Coordinate control of adipose ‘browning’ and energy expenditure by β-adrenergic and natriuretic peptide signalling. International Journal of Obesity Supplements. 2014;4:S17–S20. doi: 10.1038/ijosup.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tegge W., Frank R., Hofmann F., Dostmann W.R.G. Determination of cyclic nucleotide-dependent protein kinase substrate specificity by the use of peptide libraries on cellulose paper. Biochemistry. 1995;34:10569–10577. doi: 10.1021/bi00033a032. [DOI] [PubMed] [Google Scholar]
- 31.Hoffmann L.S., Etzrodt J., Willkomm L., Sanyal A., Scheja L., Fischer A.W.C. Stimulation of soluble guanylyl cyclase protects against obesity by recruiting brown adipose tissue. Nature Communications. 2015;6:7235. doi: 10.1038/ncomms8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nisoli E., Clementi E., Tonello C., Sciorati C., Briscini L., Carruba M.O. Effects of nitric oxide on proliferation and differentiation of rat brown adipocytes in primary cultures. British Journal of Pharmacology. 1998;125:888–894. doi: 10.1038/sj.bjp.0702131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nisoli E., Clementi E., Paolucci C., Cozzi V., Tonello C., Sciorati C. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- 34.Sansbury B.E., Cummins T.D., Tang Y., Hellmann J., Holden C.R., Harbeson M.A. Overexpression of endothelial nitric oxide synthase prevents diet-induced obesity and regulates adipocyte phenotype. Circulation Research. 2012;111:1176–1189. doi: 10.1161/CIRCRESAHA.112.266395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan H., Aziz E., Shillabeer G., Wong A., Shanghavi D., Kermouni A. Nitric oxide promotes differentiation of rat white preadipocytes in culture. The Journal of Lipid Research. 2002;43:2123–2129. doi: 10.1194/jlr.m200305-jlr200. [DOI] [PubMed] [Google Scholar]
- 36.Doronzo G., Viretto M., Russo I., Mattiello L., Di Martino L., Cavalot F. Nitric oxide activates PI3-K and MAPK signalling pathways in human and rat vascular smooth muscle cells: influence of insulin resistance and oxidative stress. Atherosclerosis. 2011;216:44–53. doi: 10.1016/j.atherosclerosis.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 37.Park H.-S., Park J.-W., Kim H.-J., Choi C.W., Lee H.-J., Kim B.I. Sildenafil alleviates bronchopulmonary dysplasia in neonatal rats by activating the hypoxia-inducible factor signaling pathway. American Journal of Respiratory Cell and Molecular Biology. 2013;48:105–113. doi: 10.1165/rcmb.2012-0043OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lopez-Rivera E., Jayaraman P., Parikh F., Davies M.A., Ekmekcioglu S., Izadmehr S. Inducible nitric oxide synthase drives mTOR pathway activation and proliferation of human melanoma by reversible nitrosylation of TSC2. Cancer Research. 2014;74:1067–1078. doi: 10.1158/0008-5472.CAN-13-0588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarbassov D.D., Ali S.M., Sabatini D.M. Growing roles for the mTOR pathway. Current Opinion in Cell Biology. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.