

Summary
While G protein-coupled receptors are often studied by analyzing antagonist radioligand: “cold” agonist inhibition curves using an independent site model, it is now clear that KL and KH values determined in these analyses are not reliable estimates of the affinities of the agonists for “free” and G protein-coupled forms of the receptor. Thus, such experiments cannot he used to contrast the characteristics of a given type of receptor in different tissues, i.e., to probe for the existence of receptor subtypes. Since treatment with N-ethylmaleimide treatment blocks receptor: Gi/Go protein interactions, such analyses on N-ethylmaleimide-pretreated membranes should allow direct assessment of the affinities of competing ligands for the free receptor or for multiple receptor subtypes.
As A1 adenosine receptors couple to Gi, and perhaps to Go, we have performed A1 adenosine receptor radioligand “competition” studies first on control, then on N-ethylmaleimide-pretreated bovine cardiac and cerebral cortical membranes. Results of experiments with the antagonist radioligand [3H]xanthine amine congener appeared to be confounded by ligand binding to A2 adenosine receptors present in the cardiac membrane preparations. Further experiments utilized the A1-specific radioligand [3H]1,3-dipropyl-8-cyclopentylxanthine. These experiments confirmed once more that the KL values determined by computer analysis of “competition” curves performed on control membranes are not reliable estimates of the affinities of the competing ligand for free receptors. Furthermore the results supported the hypothesis that similar analyses on NEM-treated membranes provide reliable estimates of the affinity(s) of competing ligands for free receptors. Lastly, the results suggest that cardiac membranes contain two subtypes of A1 adenosine receptors that are differentiated by 5′-modified but not N6-modified adenosine analogs. One of these receptor subtypes appears to be the same as the A1 receptor detected in cortical membranes.
Keywords: A1 adenosine receptor, N-ethylmaleimide pretreatment, Receptor subtypes
Introduction
Effects of exogenous adenosine on cardiac function were first reported almost 60 years ago (Drury and Szent-Györgi 1929). Several years ago we reported that the A1 adenosine receptor agonist radioligand [3H]R-phenylisopropyladenosine ([3H]R-PIA) binds to adenosine receptors in membranes prepared from newborn chick ventricle and that R-PIA causes the inhibition of isoproterenol-stimulated adenlyate cyclase activity in the same preparations (Hosey et al. 1984). These results suggested that the cardiac adenosine receptor is similar, if not identical to, the well characterized A1 receptor present in the CNS. This conclusion is supported by subsequent ligand binding studies using a variety of [125I]-labeled agonist radioligands in rat and bovine myocardial membranes (Linden et al. 1985; Lohse et al. 1985). The complete characterization of the cardiac adenosine receptor(s) using radioligand binding methodologies has been difficult because of the relatively low Bmax values of the agonist radioligands in cardiac membranes (Linden et al. 1985; Hosey et al. 1984; Lohse et al. 1985). More recently three antagonist radioligands ([3H]xanthine amine congener ([3H]XAC) (Jacobson et al. 1986), [3H]8-cyclopentyl-1,3-dipropylxanthine ([3H]CPX) (Bruns et al. 1987; Lohse et al. 1987) and [125I]3-(4-amino)phenethyl-l-propyl-8-cyclopentylxanthine ([125I]BW-A844U) (Patel et al. 1988) which bind to A1 receptors in the CNS with high affinities became available. This prompted us to perform antagonist radioligand: “cold” ligand competition experiments which we analyzed according to an independent site model with the computer program LIGAND (Munson and Rodbard 1980). Further extensive studies on the analysis of ligand binding to A1 receptors in bovine brain membranes (Leung et al. 1990) showed that this analysis is inappropriate and that much more complex analyses are necessary in order to adequately describe the behavior of the system. Because of the complexity of these analyses, and the relative paucity of A1 adenosine receptors in the heart, we felt that this approach was not practical for probing for possible differences between cardiac and brain A1 adenosine receptors. We therefore performed radioligand “competition” experiments on N-ethylmaleimide (NEM)-treated membranes in which all A1receptor: G protein interactions appear to be blocked (Leung et al. 1990). This effect of NEM to block high affinity agonist binding to many types of receptors that couple to Gi or Go appears to be due to a modification of the alpha subunits of these G proteins (Asano and Ogasawara 1986). Under these conditions we postulate that analyses based on the independent site model give valid estimates of the affinities of the various compounds for the free, uncoupled receptors. All of these experiments including the initital experiments on nonpretreated membranes are reported herein in the order in which they evolved. The results show how radioligand “competition experiments” on receptors in NEM-treated membranes can be used for the characterization of receptors that couple to Gi/Go type G proteins. Furthermore, the results suggest that cardiac membranes may contain a subtype of A1 adenosine receptor that is not detected in cortical membranes.
Materials and methods
Materials
[3H]XAC (118 Ci/mmol) was from DuPont NEN (Boston, Mass., USA). [3H]CPX (93.8 Ci/mmol) was from Amersham (Arlington Heights, IL, USA). N6-(3-iodo-4-aminobenzyl)adenosine (IABA) was a gift from Dr. J. Linden (Department of Physiology, University of Virginia School of Medicine, Charlottesville, VA, USA). 5′-N-ethylcarboxamide adenosine (NECA) and 5′-N-cyclopropylcarboxamide adenosine (NCCA) were gifts from Abbott Laboratories (North Chicago, IL., USA). R- and S-PIA were from Boehringer Mannheim (Indianapolis, IN, USA); CHA and CPX were from Research Biochemicals Inc. (Natick, Mass, USA); bacitracin was from Upjohn (Kalamazoo, Mi., USA). Other chemicals were from Sigma (St Louis, Mo., USA). Bovine tissues were obtained from a local slaughter house.
Preparation of bovine cardiac membranes
Bovine cardiac membranes were prepared by a modification of the method of Flockerzi et al. 1983. 200–300 g of bovine ventricle was suspended in 0.3 M sucrose containing 10 mM imidazole HC1 (pH 7.0), 5 mM MgSO4, 0.1 mM ethylenediaminetetraacetic acid (EDTA) and 100 μM phenylmethylsulphonyl floride (MSF), minced, and homogenized using a Polytron at setting 7 for 1 min. The homogenate was adjusted to 0.4 M sucrose and centrifuged at 28,000 g for 30 min. The supernatant was collected and diluted 1.5 fold with 150 mM NaCl, 20 mM Tris HC1 (pH 7.0, 25 °C) and 0.1 mM EDTA and centrifuged at 44,000 g for 30 min. The pellet was collected and suspended in 1 M sucrose containing 5 mM MgSO4, 0.1 mM EDTA and 10 mM imidazole HCl (pH 7.0). The membrane suspension was overlaid with 0.3 M sucrose containing the same buffer and centrifuged in a SW40 swinging bucket for 90 min at 178,000 g. The material at the 0.3 M and 1.0 M sucrose interface was collected, diluted 20 fold with 150 mM NaCl containing 10 mM Tris HCl (pH 7.0) and 1 mM EDTA. This suspension was then centrifuged for 30 min at 100,000 g in a TI40 rotor. The final pellet was suspended in 10 mM Tris HCl (pH 7.5) containing 0.1 mM EDTA and stored in liquid nitrogen until use.
Preparation of bovine cerebral cortical membranes
Cerebral cortex from bovine brain was suspended in 3–4 volumes of ice-cold 10% sucrose containing 20 mM Tris HCl (pH 7.0, 25 °C), 1 mM EDTA and 100 μM PMSF and homogenized with a Polytron homogenizer at setting 5 for 15 sec. The homogenate was centrifuged at 400 g for 10 min at 4 °c. The supernatant was removed and the pellet was resuspended in the same solution and recentrifuged for 10 min at 400 g. The supernatants from the two centrifugations were combined and centrifuged at 6500 g for 20 min. The pellet was resuspended in the 20 mM Tris buffer (pH 7.5, 25 °C) containing 1 mM EDTA and after 30 min recentrifuged at 6500 g for 20 min. The pellet was resuspended in the same buffer at a protein concentration of 10 mg/ml and stored in liquid nitrogen until use. Protein concentrations were determined by the method of Bradford (1976) using human gamma globulin as the standard.
Ligand binding assays
Ligand binding assays were performed in a volume of 50 to 500 μl (37 °C, 2 h) containing 20 mM Tris HCl (pH 7.5), 2 mM EDTA, 3 mM MgSO4, 5 U/ml adenosine deaminase, 0,1 mM benzamidine, 0.1 mg/ml bacitracin, 0.01 mg/ml soybean trypsin inhibitor, radioligand and other additions as indicated. Saturation isotherms were analyzed by least squares fits of Scatchard plots or by the nonlinear curve fitting program LIGAND (Munson and Rodbard 1980). Inhibition curves were analyzed by LIGAND. In all cases the data summarized in the tables are the result of simultaneously analyzing three identical experiments (on different membrane preparations). The SEMs given are estimates calculated by the computer program and are not meant to be used in statistical tests. The two site model was accepted over the one site model when the F value based on the “extra sum of squares” principle (Munson and Rodbard 1980) was significant at the P<0.05 level. NEM-pretreated membranes were prepared by incubating membranes for 10 min (37 °C) with 3 mM NEM + 0.1 mM Gpp(NH)p (same conditions listed above) followed by the addition of 3 mM DTT. The membranes were subsequently diluted and inhibition experiments in the presence of 0.1 mM Gpp(NH)p were performed. Bound and free ligand were separated by filtration through Whatman GF/A filters. Filter papers for experiments using [3H]XAC were presoaked in 0.3% polyethylenimine for 0.5 h. Filtration was performed on a modified cell harvester (Brandel, Gaithersburg, Md., USA). Filters were washed 3 times with 5 ml aliquots of ice-cold buffer (5 mM glycylglycine pH 7.5, 1 mM MgSO4). Theophylline (5 mM) was used to define non-specific binding.
Results
Binding of [3H]XAC to adenosine receptors in cardiac and cortical membranes
Our initial experiments were performed with [3H]XAC. The binding of [3H]XAC to adenosine receptors in both cardiac and cortical membranes required 2 h to reach equilibrium (data not shown). [3H]XAC bound to adenosine receptors in bovine cardiac membranes with high affinity and good specificity. The Kd calculated from linear Scatchard plots was 0.14±0.05 nM (mean±SEM, n = 9), the Bmax was 139±19 fmol/mg. In twelve similar experiments on cerebral cortical membranes the Kd and Bmax values calculated from linear Scatchard plots were 0.10±0.02 nM and 581±48 fmol/mg, respectively.
Inhibition of [3H]XAC binding by adenosine receptor agonists and antagonists
Table 1 summarizes the results of our initial series of experiments in which the affinities of a series of agonists and antagonists for adenosine receptors in cardiac and cerebral cortical membranes were estimated based on their ability to inhibit [3H]XAC binding. Panel A of Fig. 1 shows representative inhibition curves for R-PIA and NECA. As expected, the inhibition curves of the antagonists theophylline and XAC were monophasic and single Kd values were calculated. All the agonist inhibition curves were better fit by a a two site model (P < 0.05) and two Kd values for each compound were calculated. (We have designated these two Kd values as KH and KL as is common in the literature. This nomenclature is used because these Kd values are often interpreted as measures of the affinities of the agonists for coupled (KH) and uncoupled (KL) states of the receptor. As discussed below, this is not necessarily true). Note that the N6-modified analogs appeared to differentiate between the cardiac and cortical receptors (KLs for sites in cardiac membranes > KLS in cortical membranes) while the 5′-modified analogs (NECA and NCCA) behaved similarly in the two preparations.
Table 1.
Inhibition of [3H]XACa binding by adenosine receptor agonists and antagonists in bovine cortical and cardiac membranes. KH and KL are the dissociation constants for the high and low affinity states determined by LIGAND. %H is the percentage of high affinity binding sites. Values are means ± SEM for 3 experiments
| Compound | Cortex
|
Heart
|
||||
|---|---|---|---|---|---|---|
| KH, nM | KL, nM | %H | KH, nM | KL, nM | %H | |
| R-PIA | 0.07 ± 0.02 | 4.5 ± 2.1 | 81 ± 7 | 0.36 ± 0.16 | 4000 ± 1600 | 57 ± 7 |
| S-PIA | 1.30 ± 0.40 | 140.0 ± 40.0 | 83 ± 7 | 5.30 ± 2.20 | 5500 ± 2200 | 61 ± 6 |
| CHA | 0.14± 0.04 | 8.3 ± 3.1 | 76 ± 6 | 0.67 ± 0.44 | 5200 ±3200 | 66 ±11 |
| IABA | 0.21 ± 0.11 | 18.0 ± 12.9 | 78 ±10 | 0.40 ± 0.21 | 4700 ±2700 | 65 ± 9 |
| NECA | 13.0 ± 5.2 | 700 ± 400 | 74 ± 10 | 14.7 ± 16.0 | 900 ± 600 | 41 ± 14 |
| NCCA | 5.3 ± 5.5 | 1376 ± 886 | 51 ± 13 | 3.8 ± 7.6 | 1000 ± 800 | 41 ± 25 |
| Theophylline | 9600 ± 1400 | – | – | 32500 ± 7900 | – | – |
| XAC | 0.17 ± 0.01 | – | – | 0.20 ± 0.07 | – | – |
[3H]XAC was present in concentrations of 0.06 – 0.07 nM for cortical membranes and 0.15–0.25 nM for cardiac membranes
Fig. 1.
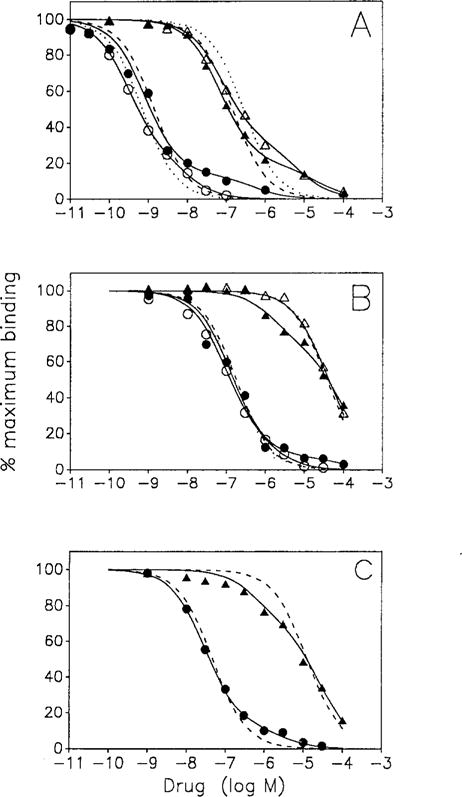
A–C. Inhibition curves for antagonist radioligand binding by R-RIA (circles) and NECA (triangles) to sites in cortical (open symbols) and cardiac (closed symbols) membranes. Panel A [3H]XAC, control membranes. Panel B [3H]XAC, NEM-pretreated membranes in presence of 0.1 mM Gpp(NH)p. Panel C [3H]CPX: NEM-pretreated membranes in presence of 0.1 mM Gpp(NH)p. Points shown are averages of duplicate determinations. Nonspecific binding as defined by theophylline (5 mM) is substracted from all data. The two site fits are shown as solid tines. One site fits are shown as – – – (heart) or ⋯ (Brain)
Inhibition of [3H]XAC binding by adenosine receptor agonists after NEM-pretreatment
The analyses presented in Table 1 are based on a model in which the antagonist binds to all species binding sites with equal affinity and the multiple “sites” for competing ligands are due to multiple independent, noninteracting sites. Extensive work from our laboratory subsequently performed on bovine brain membranes showed that antagonists recognize different states of the receptor with different affinities and that much more complex analyses must be performed in order to characterize the system (Leung et al. 1990). Furthermore, we realized that even if we could perform similar experiments on cardiac membranes, (which we probably could not do because of the paucity of receptors in the cardiac membranes) it was questionable if this approach would be useful for probing for differences between cardiac and brain A1 adenosine receptors. We therefore decided to interrupt all receptor: G protein interactions so that the system would be simplified and could be legitimately analyzed using the independent site model. We use NEM-treatment to uncouple all receptors as we had previously shown that this treatment, but not the addition of 0.1 mM (Gpp(NH)p alone, accomplishes this goal (Leung et al. 1990). As we were interested in the behaviors of N6- and 5′-modified adenosines we studied two N6-substituted analogs, R-PIA and IABA, and two 5′-substituted compounds, NECA and NCCA. Representative inhibition curves for R-PIA and NECA performed under these conditions are shown in panel B of Fig. 1. The analyses of these experiments are summarized in Table 2. In all cases the analyses of the inhibition curves performed on cortical membranes indicated the presence of a single site. The inhibition curves performed on cardiac membranes were better fit by a two site model. Note that we denote the two Kds as K1 and K2, not KH and KL, as these values are interpreted to be measures of the affinities of the agonists for separate binding sites. (It is obvious that the estimates of the lower affinity Kd values are very imprecise. This is partially due to the relatively high nonspecific binding of [3H]XAC as the error estimates in the experiments using [3H]CPX were much lower (see below), and to the error model used in these analyses. We have used the error model used in the original paper by Munson and Rodbard (1980), i.e., constant percent error in the dependent variable. While addition of a constant error term usually reduced inordinately large standard error estimates, the means were little affected and we have used the constant percent error model throughout). Several points should be noted. Firstly, the apparent higher affinity Kd values for the N6-substituted analogs were similar to the single values determined in the cortical membranes. Secondly, the percentage of sites with the higher affinity Kd was much higher for the N6-analogs as compared to the 5′-analogs. Lastly, the lower affinity site for the 5′-analogs appeared to correspond to the single site present in the cortical membranes, i.e., the site in the cardiac membranes detected by the 5′-analogs with higher affinity was not detected in the cortical membranes.
Table 2.
Dissociation constants of adenosine receptor agonists for binding to sites present in NEM-pretreated cortical and cardiac membranes estimated by inhibition of [3H]XACa binding
| Compound | Cortexb
|
Heart
|
|
|---|---|---|---|
| Kd, nM | K1, nM (% total) |
K2, nM (% total) |
|
| R-PIA | 26 ± 6 | 17 ± 9 (86%) | 28 248 ± 30322 (14%) |
| IABA | 55 ± 10 | 98 ± 44 (90%) | 15480 ± 17421 (10%) |
| NECA | 6329 ±347 | 124 ± 80 (36%) | 16155 ± 3836 (64%) |
| NCCA | 6329 ± 336 | 107 ± 68 (33%) | 12121 ± 2159 (67%) |
[3H]XAC was present in concentrations of 0.6 – 0.7 nM for both cortical and cardiac membranes. The Kd values of [3H]XAC under these conditions was 0.20 ± 0.0.02 nM (Bmax 101 ± 14 fmol/mg, N = 3) for cardiac membranes and 0.09 ± 0.01 nM (Bmax 780 ± 75 fmol/mg, N=3) for cortical membranes
Inhibition curves were monophasic giving a single Kd value
Inhibition of [3H]CPX binding by adenosine receptor agonists after NEM-pretreatment
The presence of two sites in NEM-pretreated cardiac membranes could be explained by the presence of A2 receptors in these membranes if these receptors have sufficient affinity for [3H]XAC to be detected under the conditions employed. That this could be the case was suggested by the report that [3H]XAC binds to A2 adenosine receptors in rabbit striatum (Ji et al. 1991). Inhibition experiments with [3H]XAC and CPX (an antagonist with reportedly greater A1-selectivity (Lohse et al. 1987) in control membranes modeled to a single site in cortical membranes (Kd 0.09 ± 0.02 nM, n = 3) while those in cardiac membranes modeled to two sites (K1 0.15 ± 0.04 nM (89% of total sites); K2 2045 ± 1439 nM, n = 3). These results suggested that the cardiac membranes may contain a small population of A2 adenosine receptors that, under the experimental conditions employed, bind [3H]XAC with high affinity and adenosine receptor agonists with low affinity. We therefore performed a final series of experiments on NEM-pretreated membranes using [3H]CPX. Representative inhibition curves for R-PIA and NECA are shown in Panel C of Fig. 1. The analyses of these experiments are summarized in Table 3. Under these conditions the estimates of the lower affinity Kd values for the 5′-modififed analogs were more precise and more closely approximated those in the cortical membranes (Table 2). The lower affinity site detected by the N6-modified analogs was either absent (IABA) or still present albeit with a greatly reduced Kd (R-PIA). Most importantly, this last series of experiments confirmed the presence of a site in the cardiac membranes that bind 5′ analogs with high affinity.
Table 3.
Dissociation constants of adenosine receptor agonists for binding to sites present in NEM-preteated cardiac membranes estimated by inhibition of [3H]CPXa binding
| Compound | K1, nM (% total) |
K2, nM (% total) |
|---|---|---|
| R-PIA | 5 ± 3 (84%) | 152 ± 96 (16%) |
| IABA | 25 ± 4 (100%) | – |
| NECA | 89 ± 44 (30%) | 4016 ± 398 (70%) |
| NCCA | 110 ± 66 (24%) | 4049 ± 354 (76%) |
The Kd for [3H]CPX was 0.12 ± 0.01 nM (Bmax 100 ± 8 fmol/mg; N = 3). 0.6 nM [3H]CPX was used in the inhibition experiments
Discussion
Previous work from this and other laboratories has suggested that the adenosine receptors in the heart have ligand binding characteristics similar (Linden et al. 1985; Hosey et al. 1984, Lohse et al. 1985) but perhaps not identical to (Leung et al. 1988) the A1 receptor found in the CNS. The experiments reported herein were performed to further compare and contrast the A1 receptors present in bovince cardiac and cerebral cortical membranes.
Equilibrium binding studies showed that [3H]XAC binds to adenosine receptors in cardiac membranes and cerebral cortical membranes with high affinity and good specificity. Although the number of sites in the cardiac membranes was only about 25% of that in the cerebral cortical membranes, the specific binding at about the Kd was more than twice nonspecific binding. Under the conditions employed the total and nonspecific cpm were in the range of 600–1000 and 200–400, respectively. It thus appeared that this ligand would be useful for the characterization of cardiac adenosine receptors. The Kds for [3H]XAC binding to adenosine receptors in cardiac and cortical membranes were similar. (It should be noted that these experiments were performed before the utility of [3H]CPX for studying cardiac A1 adenosine receptors was reported (Liang 1989; Lohse et al. 1987; Tawfik-Schlieper et al. 1989).
Previous reports on neuronal A1 receptors have suggested that agonists bind to these receptors with multiple affinities while antagonists bind to all receptors with a single affinity (Bruns et al. 1987; Jacobson et al. 1986; Lohse et al. 1984; Yeung and Green 1983). Similar results are obtained with other receptors that couple to their effectors via G proteins (reviewed in Leung et al. 1990). While mechanistically the multiple agonist affinity states are believed to be due to the agonist differentiating between G-protein coupled and “free” receptors such that some form of a ternary complex model (Wreggett and DeLean 1984) is operational, computer programs such as LIGAND (Munson and Rodbard 1980) which are based on an independent site model are commonly used to analyze inhibition curves generated in studies on these receptors. The results in Table 1 summarize data obtained with this approach using the antagonist radioligand[3H]XAC. As expected the antagonists inhibited [3H]XAC with a single apparent affinity while the inhibition curves of the agonists were better fit by a two site model. These data suggested that the cardiac and cortical A1 receptors differ in that the KL values of the N6-analogs but not 5′-modified analogs are significantly larger in the heart as compared to the brain.
After performing the experiments and subsequent analyses summarized in Table 1 we became concerned about the appropriateness of the model used in these analyses. Work from other laboratories had shown that analyses with the independent site model does not always give valid estimates of the affinities of the competing ligand for the coupled and uncoupled forms of the receptor proposed to exist with the ternary complex model (Wreggett and DeLean 1984; Abramson et al. 1987). This led us to further studies on bovine cortical A1 receptors which have been reported elsewhere (Leung and Green 1989; Leung et al. 1990). These studies suggested that: (1) the KL determined from experiments such as those summarized in Table 1 is not the Kd of the agonist for the free receptor, (2) agonists form two types of high affinity complexes with receptors in cortical membranes (this observation was equally well explained by the presence of more than one type of receptor, or by the presence of one type of receptor that couples to more than one type of G-protein), and (3) antagonist ligands differentiate between free and coupled receptors, the Kd for the binding to free receptors being more favorable. Furthermore, these experiments suggested that the affinities of unlabeled compounds for the free A1 receptor can be determined from inhibition curves of antagonist radioligand binding to receptors in NEM-pretreated membranes. We therefore performed a second series of inhibition experiments using [3H]XAC and NEM-pretreated membranes (Table 2). A high concentration of Gpp(NH)p was also included in an effort to assure that all coupling of A1 receptors was blocked. Under these conditions all of the curves for agonist inhibition of ligand binding to receptors in cortical membranes were monophasic. The apparent Kd values, which we interpret as estimates of the affinities of the agonists for free cortical A1 receptors, were all greater than the KL values determined in the untreated membranes (Table 1). It should also be noted that these results suggested the presence of a single type of A1 adenosine receptor, they do not eliminate the possibility that the cortical membranes contain multiple subtypes of A1 adenosine receptors that recognize the compounds studied with similar affinities.
Agonist inhibition of [3H]XAC binding to NEM-pretreated cardiac membranes remained complex. The majority of the binding (79%-90%) of the compounds with N6-substitutions (R-PIA and IABA) bound with apparent Kd values similar to those determined in the NEM-pretreated cortical membranes. Interestingly, it was the lower affinity component of the binding of the 5′-modified analogs (NECA and NCCA) that more closely approximated the Kd values determined in the NEM-pretreated cortical membranes.
The significance of the quantitatively minor site in cardiac membranes identified by the N6-modified analogs was unclear. As [3H]XAC had been demonstrated to bind to A2 receptor in rabbit striatum (Ji et al. 1991), one possibility was that the low concentration of [3H]XAC used binds to an A2 receptor present in the cardiac membranes preparations. This would not necessarily imply that cardiac cells per se contain A2 adenosine receptors as they could easily arise from membranes from other cell types in the “cardiac” membrane preparations (Schrader and Londos 1983; Schutz et al. 1986). [3H]XAC/CPX inhibition curves suggested that this may be the case in that a minor proportion of the sites identified by [3H]XAC in the cardiac membranes was inhibited only by high concentrations of CPX. We therefore performed a final series of experiments with [3H]CPX, and two N6- and two 5′-modified analogs (Table 3). Under these conditions one N6-analog, IABA, exhibited a single Kd, while the second, R-PIA, still exhibited an apparent second minor low affinity site. It is unclear whether or not this low affinity site is “real” or is an artifact of the analysis. More importantly, the Kds of the 5′-modified analogs for the higher affinity site were unchanged while the apparent Kds for binding to the lower affinity site decreased to more closely approximate those determined in the cortical membranes. Thus, while it appears that the data obtained with [3H]XAC and [3H]CPX differed quantitatively, perhaps due to [3H]XAC binding to a minor population of A2 receptors in the cardiac membranes preparations, the results obtained with the two antagonist radioligands did not differ qualitatively. These results therefore suggest that the cortical and cardiac membranes both contain an A1 receptor subtype that binds N6 but not 5′-substituted analogs with high affinity, and that the cardiac membranes but not the cortical membranes contain an additional A1 receptor subtype that binds both N6 and 5′-substituted analogs with high affinity. The estimated affinities of the 5′-analogs for the putative A1 subtypes in cardiac membranes are calculated using a single Kd for [3H]CPX. While these values would be differentially affected if, in fact, [3H]CPX binds to the putative receptor subtypes with different affinities, Scatchard plots of [3H]CPX binding to NEM-pretreated cardiac membranes were linear suggesting that [3H]CPX does not significantly differentiate between the putative receptor subtypes.
It should be noted that the utility of using NEM treatment to block receptor: G (specifically Gi/Go) protein interactions so that ligand binding to free, uncoupled receptors can be unambiguously studied is predicated on the assumption that NEM does not modify the receptor itself so as to modify its ligand binding characteristics. This assumption appears to be valid for A1 adenosine receptors as NEM does not affect the binding of [3H]CPX to A1 receptors purified from rat testes (Nakata 1990) or chicken brain (R. D. Green, unpublished data). While it could be argued that we have not tested the effect of NEM on purified cardiac A1 receptors, such an effect would require that the cardiac receptor differ from that in the brain, a requirement in agreement with the proposed existence of subtypes of cardiac A1 receptors.
Acknowledgments
These studies were supported by a grant from the National Science Foundation (BNS-8719594).
Abbreviations
- [125I]ABA
[125I]N6-p-aminobenzyl)adenosine
- [3H]CPX
[3H]8-cyclopentyl-1,3-dipropylxanthine
- [3H]R-PIA
[3H]N6-R-phenylisopropyladenosine
- [3H]XAC
[3H]xanthine amine congener
- CHA
N6-cyclohexyladenosine
- EDTA
ethylenediaminetetraacetic acid
- [125I]BW-A844U
[125I]3-(4-amino)phenethyl-1-propyl-8-cyclopen-tylxanthine
- NCCA
5′-N-cyclopropylcarboxamide adenosine
- NECA
5′-N-ethylcarboxamide adenosine
- PMSF
phenylmethylsulphonyl fluoride
- NEM
N-ethylmaleimide
References
- Abramson SN, McGonigle P, Molinoff PB. Evaluation of models for analysis of radioligand binding data. Mol Pharmacol. 1987;31:103–111. [PubMed] [Google Scholar]
- Asano T, Ogasawara N. Uncoupling of γ-aminobutyric acid B receptors from GTP-binding proteins by N-ethylmaleimide: Effect of ethylmaleimide on purified GTP-binding proteins. Mol Pharmacol. 1986;29:244–249. [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Bruns RF, Fergus JH, Badger EW, Bristol JA, Santay LA, Hartman JD, Hays SJ, Huang CC. Binding of the A1-selective adenosine antagonist 8-cyclopentyl-l,3-dipropylxanthine to rat brain membranes. Naunyn-Schmiedeberg’s Pharmacol. 1987;335:59–63. doi: 10.1007/BF00165037. [DOI] [PubMed] [Google Scholar]
- Drury AN, Szent-Györgi A. The physiological activity of adenine compounds with special reference to their actions upon the mammalian heart. J Physiol (Lond) 1929;68:213–237. doi: 10.1113/jphysiol.1929.sp002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flockerzi V, Mewes R, Ruth P, Hofmann F. Phosphorylation of purified bovine cardiac sarcolemmma and potassium-stimulated calcium uptake. Eur J Biochem. 1983;135:131–142. doi: 10.1111/j.1432-1033.1983.tb07628.x. [DOI] [PubMed] [Google Scholar]
- Hosey MM, McMahon KK, Green RD. Inhibitory adenosine receptors in the heart: Characterization by ligand binding studies and effects of β-adrenergic receptor stimulated adenylate cyclase and membrane protein phosphorylation. J Mol Cell Cardiol. 1984;16:931–941. doi: 10.1016/s0022-2828(84)80029-2. [DOI] [PubMed] [Google Scholar]
- Jacobson KA, Ukena D, Kirk KL, Daly JW. (3H]-Xanthine amine congener of l,3-dipropyl-8-phenyIxanthine: an antagonist radioligand for adenosine receptors. Proc Natl Acad Sci USA. 1986;83:4089–4093. doi: 10.1073/pnas.83.11.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X-D, Stiles GL, Jacobson KA. [3H]XAC (xanthine amine congener) is a radioligand for A2-adenosine receptors in rabbit striatum. Neurochem Internat. 1991;18:207–213. doi: 10.1016/0197-0186(91)90187-i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung E, Kwatra MM, Hosey MM, Green RG. Characterization of cardiac A1-adenosine receptors by ligand binding and photoaffinity labelling. J Pharmacol Exp Ther. 1988;244:1150–1156. [PubMed] [Google Scholar]
- Leung E, Green RD. Density gradient profiles of A1 adenosine receptors labeled by agonist and antagonist radioligands before and after detergent solubilization. Mol Pharmacol. 1989;36:412–419. [PubMed] [Google Scholar]
- Leung E, Jacobson KA, Green RD. Analysis of agonist: antagonist interactions at A1 adenosine receptors. Mol Pharmacol. 1990;38:72–83. [PMC free article] [PubMed] [Google Scholar]
- Liang BT. Characterization of the adenosine receptor in cultured embryonic chick atrial myocytes: Coupling to modulation of contractility and adenylate cyclase activity and identification by direct ligand binding. J Pharmacol Exp Ther. 1989;249:775–784. [PubMed] [Google Scholar]
- Linden J, Patel A, Sadek S. [125I]Aminobenzyladenosine, a new radioligand with improved specific binding to adenosine receptors in the heart. Circ Res. 1985;56:279–284. doi: 10.1161/01.res.56.2.279. [DOI] [PubMed] [Google Scholar]
- Lohse MJ, Lenschow V, Schwabe U. Two affinity states of Ri adenosine receptors in brain membranes. Naunyn-Schmiedeberg’s Arch Pharmacol. 1984;26:1–9. [PubMed] [Google Scholar]
- Lohse MJ, Ukena D, Schwabe U. Demonstration of Ri-type adenosine receptors in bovine myocardium by ligand binding. Naunyn-Schmiedeberg’s Arch Pharmacol. 1985;328:310–316. doi: 10.1007/BF00515559. [DOI] [PubMed] [Google Scholar]
- Lohse MJ, Klotz K-N, Lindenborn-Fotinos J, Reddington M, Schwabe U, Olsson RA. 8-cyclopentyl-l,3-dipropylxanthine (DPCPX)-a selective high affinity agonist radioligand for A1 adenosine receptors. Naunyn-Schmiedeberg’s Arch Pharmacol. 1987;336:204–210. doi: 10.1007/BF00165806. [DOI] [PubMed] [Google Scholar]
- Munson PJ, Rodbard D. LIGAND: a versatile computerized approach for characterization of ligand-binding systems. Anal Biochem. 1980;107:220–239. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- Nakata H. A1 receptor of rat testes membranes. J Biol Chem. 1990;265:671–677. [PubMed] [Google Scholar]
- Patel A, Craig RH, Daluge SM, Linden J. 125I-BW-A844U, an antagonist radioligand with high affintiy and selectivity for adenosine A1 receptors, and 125I-Azido-BW-A844U, a photoaffinity label. Mol Pharmacol. 1988;33:585–591. [PubMed] [Google Scholar]
- Schrader J, Londos C. Is there a highly purified cardiac sarcolemma preparation? Naunyn-Schmiedeberg’s Arch Pharmacol. 1983;322:R14. (abstr) [Google Scholar]
- Schutz W, Freissmuth M, Hausleithner V, Tuisl E. Cardiac sarcolemmal purity is essential for the verification of adenylate cyclase inhibition via A1-adenosine receptors. Naunyn-Schmiedeberg’s Arch Pharmacol. 1986;333:156–162. doi: 10.1007/BF00506519. [DOI] [PubMed] [Google Scholar]
- Tawfik-Schlieper H, Klotz K-N, Kreye VAW, Schwabe U. Characterization of the K+-channel-coupled adenosine receptor in guinea pig atria. Naunyn-Schmiedeberg’s Arch Pharmacol. 1989;340:684–688. doi: 10.1007/BF00717745. [DOI] [PubMed] [Google Scholar]
- Wreggett KA, De Lean A. The ternary complex model: Its properties and application to ligand interactions with the D2-dopamine receptor of the anterior pituitary gland. Mol Pharmacol. 1984;26:214–227. [PubMed] [Google Scholar]
- Yeung S-MH, Green RD. Agonist and antagonist affinities for inhibitory adenosine receptors are reciprocally affected by 5′-guanylyl-imidodiphosphate and N-ethylmaleimide. J Biol Chem. 1983;258:2334–2339. [PubMed] [Google Scholar]