

Abstract
The ability of bacteria to regulate gene expression in response to changes in cell density is termed quorum sensing. This behavior involves the synthesis and recognition of extracellular, hormone-like compounds known as autoinducers. Here we report the structure of an autoinducer synthase, LuxS from Bacillus subtilis, at 1.6-Å resolution (Rfree = 0.204; Rwork = 0.174). LuxS is a homodimeric enzyme with a novel fold that incorporates two identical tetrahedral metal-binding sites. This metal center is composed of a Zn2+ atom coordinated by two histidines, a cysteine, and a solvent molecule, and is reminiscent of active sites found in several peptidases and amidases. Although the nature of the autoinducer synthesized by LuxS cannot be deduced from the crystal structure, features of the putative active site suggest that LuxS might catalyze hydrolytic, but not proteolytic, cleavage of a small substrate. Our analysis represents a test of structure-based functional assignment.
Certain species of bacteria alter their behavior in response to intercellular signals known as autoinducers (AIs). This phenomenon, quorum sensing, allows bacteria to sense their population density through detection of the relative concentration of autoinducer in the surrounding environment and to respond accordingly through appropriate changes in gene expression (1, 2). Quorum sensing has been observed in a broad range of bacteria and is known to regulate such important processes as competence, sporulation, motility, biofilm formation, and virulence. In general, the AIs of Gram-negative bacteria are of the acyl-homoserine lactone (AHL) class, and those of Gram-positive species tend to be peptides or peptide derivatives (3, 4).
In Vibrio harveyi, two quorum-sensing systems have been described, both of which modulate the expression of the genes responsible for bioluminescence. The first responds to AI-1 (hydroxybutanoyl-l-homoserine lactone), a member of the AHL family of autoinducers (5, 6). The chemical structure of AI-2, the signal for the second system, has not been reported, but AI-2 is not believed to be an AHL (7). The gene responsible for AI-2 production, luxS, has been cloned and sequenced, as have the genes responsible for its detection and for the subsequent signaling cascade that leads to bioluminescence (8–10). LuxS shares no sequence similarity with any known class of AHL synthases.
Other species, such as Escherichia coli and Salmonella typhimurium, also produce AI-2 (or a closely related signal), as suggested by their ability to induce bioluminescence through the AI-2 pathway in luxS- V. harveyi (8, 11–13). Indeed, psi-blast database searches reveal that homologs of luxS can be identified in the genomes of most bacterial species for which complete sequence data are available. Such cross-species signaling has not generally been observed in studies of quorum sensing. Thus, LuxS-dependent signaling might be a novel form of interspecies communication whereby a particular strain is able to estimate the total bacterial population in an environment harboring multiple species (2).
A link between quorum sensing and virulence has been established for a number of pathogenic bacteria, suggesting that interference with these signaling circuits might be therapeutically useful (14–16). Several studies have attempted to determine whether a role exists for LuxS in pathogenesis (12, 13, 17, 18). Of particular interest is recent work demonstrating that enterohemorrhagic and enteropathogenic E. coli control the expression of several virulence factors in a LuxS-dependent fashion (17). Given its wide phylogenetic distribution and a connection to pathogenesis, LuxS might represent a novel target for broad-spectrum therapeutics. In an attempt to obtain information about the reaction catalyzed by LuxS, we determined the crystal structure of LuxS from Bacillus subtilis.
Materials and Methods
Cloning and Mutagenesis.
Using Pfu DNA polymerase (Stratagene), the luxS gene (formerly ytjB) was PCR amplified from B. subtilis genomic DNA with primers that incorporated 5′ NdeI and 3′ NotI sites. These sites were used for cloning of the PCR products into expression vector pET29a(+) (Novagen). The resulting vector, pLuxSH6, encoded a product with a C-terminal hexahistidine tag, but the purified protein did not yield crystals. A vector lacking the affinity tag, pLuxS, was constructed with the QuickChange Site-Directed Mutagenesis Kit (Stratagene) by the introduction of a TAA stop codon immediately downstream of the luxS coding region.
Protein Expression and Characterization.
E. coli BL21(DE3) cells transformed with pLuxS were grown at 37°C in Luria–Bertani broth supplemented with 50 mg liter−1 kanamycin. Expression was induced when cultures reached an OD600 of 1.0 by the addition of isopropyl-β-d-galactopyranoside to 0.5 mM. After eight additional hours of growth, the cultures were harvested and resuspended in 30 mM sodium phosphate (pH 7.2) and 2.0 mM tri(carboxyethyl)phosphine. The cells were sonicated, and the clarified extract was loaded on a High-Q anion exchange column (Bio-Rad). Elution with a potassium chloride gradient (20–400 mM) yielded fractions enriched for LuxS. These were pooled and subjected to an additional anion exchange step in an alternate buffer (30 mM Tris/0.5 mM EDTA/2.0 mM tri(carboxyethyl)phosphine, pH 8.0). The resulting protein was homogeneous LuxS, as judged by SDS/PAGE and confirmed by N-terminal amino acid sequencing (Biomedical Research Core Facility, University of Michigan, Ann Arbor). Selenomethionine-substituted LuxS was prepared according to published protocols, with E. coli B834(DE3) as the host strain (19). Metal analysis was performed by Ted J. Huston (Department of Geological Sciences, University of Michigan) on a FinniganMAT Element-1 inductively coupled plasma–high-resolution mass spectrometer.
Crystallization and Structure Determination.
Crystals were grown at 22°C by the hanging drop vapor diffusion method. Equal volumes of protein (16 mg ml−1 LuxS/20 mM Tris, pH 8.0/2 mM tri(carboxyethyl)phosphine) and precipitant (1.8 M ammonium sulfate/100 mM Tris, pH 8.0), equilibrated against 1.0 ml of precipitant, produced diffraction-quality crystals within 1 week. Crystals grown under these conditions belong to space group P6522, with a = b = 62.73 Å and c = 150.1 Å. Derivatives were prepared by soaking crystals for 1–2 days in a holding solution (1.8 M ammonium sulfate/0.1 M Tris, pH 8.0) supplemented with ≈2 mM heavy atom reagent (Table 1).
Table 1.
Heavy atom derivatives and refinement statistics
| Data | Resolution, Å | Reflections
|
Rsym* | Sites | Phasing power† centric/acentric | Rcullis‡ | |
|---|---|---|---|---|---|---|---|
| Measured/unique | % complete | ||||||
| Native | 20. –1.60 | 413,208/23,431 | 98.0/92.8 | 0.063 | |||
| TMLA-1§ | 20. –1.70 | 207,803/18,561 | 92.2 | 0.062 | 1 | 0.78/0.85 | 0.69 |
| SeMet | 20. –1.90 | 213,577/14,003 | 95.1 | 0.078 | 4 | 0.87/1.02 | 0.63 |
| Sm(Ac)3 | 20. –1.80 | 251,896/16,360 | 95.7 | 0.068 | 3 | 0.54/0.67 | 0.74 |
| K2PtCl4 | 20. –2.00 | 201,680/12,785 | 99.3 | 0.062 | 1 | 0.42/0.43 | 0.80 |
| TMLA-2 | 20. –1.60 | 409,741/23,364 | 97.7 | 0.069 | 2 | 0.82/0.89 | 0.68 |
| FMA§ | 20. –1.80 | 343,634/16,616 | 98.2 | 0.094 | 5 | 1.09/1.19 | 0.68 |
| Refinement statistics | |||
| Protein non-hydrogen atoms | 1428 | R-factor | 0.174 |
| Solvent atoms (with glycerol) | 193 | Free R-factor (2,203 reflections) | 0.204 |
| Average B, protein, Å2 | 24.3 | Bond distances, r.m.s. deviation, Å | 0.016 |
| Average B, waters, Å2 | 37.6 | Bond angles, r.m.s. deviation, ° | 1.82 |
| B, zinc atom, Å2 | 26.2 |
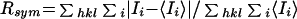 ,
where Ii is the intensity of a measurement of
reflection i, and 〈Ii〉 is the
mean intensity for reflection i.
,
where Ii is the intensity of a measurement of
reflection i, and 〈Ii〉 is the
mean intensity for reflection i.
Phasing power =
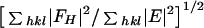 ,
where FH is the heavy atom structure factor, and
E is the r.m.s. lack of closure. Values shown are for all
data to 1.6 Å.
,
where FH is the heavy atom structure factor, and
E is the r.m.s. lack of closure. Values shown are for all
data to 1.6 Å.
Rcullis = r.m.s. lack of closure/r.m.s. isomorphous difference.
TMA, trimethyl lead acetate; FMA, fluorescein mercuric acetate.
For data collection, crystals were soaked for 1 min in 30% (vol/vol) glycerol, 2.0 M ammonium sulfate, 0.1 M Tris (pH 8.0) and then flash-cooled by direct transfer into a 140 K nitrogen stream. Intensities were measured with a Rigaku RAXIS IV phosphor-imaging plate detector with incident CuKα x-rays from a rotating anode x-ray source collimated by the Yale mirror system. Indexing, integration, and scaling were performed with the HKL package (20). Phases were determined by multiple isomorphous replacement with the use of six heavy atom derivatives (Table 1). Potential derivatives were identified by statistical analysis (20) and by inspection of difference Patterson maps generated in ccp4 (21). All steps of structure solution were performed in solve (22). The electron density map calculated with the use of MIR phases with all data to 1.60 Å was exceptionally clear (〈m〉 was 0.54 for all data and 0.63 for data from 20.0 Å to 2.06 Å), and the polypeptide (residues 2–157) was built with the program o (23). Refinement was performed with cns (24), with all available data [no I/σ(I) cutoff], with the default bulk solvent correction. Multiple rounds of refinement, including simulated annealing, resulted in an R of 0.174 and an Rfree of 0.204 (Table 1). For inspection of the model, composite simulated annealing omit maps were periodically calculated during refinement. No explicit restraints were applied to the zinc atom or its ligands. The asymmetric unit includes one protein monomer, one zinc atom, one glycerol molecule, a cysteine sulfonic acid at position 84, and 187 solvent molecules. The stereochemical quality of the model is excellent, as assessed by the program procheck (25), and all residues lie in allowed regions of the Ramachandran map.
Results and Discussion
Structure of LuxS.
The structure determination reveals an α + β fold featuring a four-stranded antiparallel β sheet partially surrounded by five helices (Fig. 1A). The topology of this sheet is 1-2–4-3, with a 310 helix and helix α1 packing adjacent (and roughly parallel) to strands β1 and β3. Helices α2, α3, and α4 are nearly perpendicular to α1 and pack against the same face of the β sheet as α1. Despite this rather simple arrangement of secondary structure elements, the fold of LuxS appears to be novel. The likelihood of this uniqueness had been evident before structural analysis from the failure to find matches with the use of either sequence similarity-based searches (26) or fold recognition methods (27). Two analyses made with the use of the coordinates of LuxS have now confirmed its novelty. First, a search of the structure database with the dali server (28) failed to yield significant matches (the largest Z score was 5.2). Comparisons with known structures with the use of the tops algorithm (29, 30) identified several domains containing features similar to those of LuxS. All of these domains were α/β layered structures, including four-stranded antiparallel β sheets with the topology 1–2-4–3, but they differed from LuxS in the arrangements of the helices surrounding the central sheets.¶
Figure 1.

The structure of LuxS. (A) A stereodrawing of the monomer fold, oriented to show the topology of the sheet and the zinc-binding site. The assignments of residues to various secondary structures are shown in C. Zn is represented as a large sphere, and the protein ligands, His-54, His-58, and Cys-126, are drawn in ball-and-stick mode. The crystallographic 2-fold axis relating the monomer units is approximately vertical in this view. A and panel B were composed with the RIBBONS program (32). (B) The dimer of LuxS in two orientations, viewed down the 2-fold axis (Left) and perpendicular to the 2-fold axis (Right). The helices are labeled and sheet strands are numbered according to their order in the sequence. (C) Sequence alignments of LuxS proteins from selected organisms. Five representatives of the 26 sequences used for alignment are included here. Residues that are invariant in all 26 sequences are labeled in boldface; those that are conserved in at least 80% of the sequences are highlighted in gray. Metal ligands are denoted with circles, and oxidized cysteine is indicated by a triangle.
As observed in the crystal, LuxS forms a homodimer, with the subunits related by crystallographic symmetry. The central part of the dimer interface is a β-sandwich composed of the sheets from each monomer. Portions of the other elements of secondary structure also contribute to dimer formation (Fig. 1B). A number of lines of evidence are consistent with the dimeric species being functionally relevant and not being an artifact of crystallization. First, the dimer interface is extensive, burying 2,125 Å2 of solvent-accessible area per monomer. This finding compares favorably with homodimers in general, which in one analysis were found to bury a mean surface area of 1,685 Å (31). Second, dynamic light scattering measurements indicate that LuxS exists as a dimer in solution (data not shown). Finally, as explained below, the pattern of conserved residues, when mapped onto the structure, suggests participation of both chains in each of the two active sites.
Active Site Identification and Features.
In the context of the structure, the conserved residues of LuxS indicate its probable active center. Based on our alignment of 26 LuxS sequences, 23 residues are absolutely conserved (Fig. 1C). Most of these residues cluster near the dimer interface to form two identical active sites composed of residues from both polypeptide chains.
The presence of a metal cofactor at the center of these conserved clusters strongly supports our identification of the active site. This metal ion was initially assumed to be zinc, an assignment confirmed by metal analysis, and was found to be ligated by three protein side chains and a solvent molecule. The residues coordinating the metal, His-54, His-58, and Cys-126, are all donated by a single monomer chain. His-54 and His-58, which bind the metal through their Nɛ atoms, are from adjacent turns of the central region of α1. The cysteine ligand is located in the extended loop between α2 and α3 (Fig. 2). These residues, and the water molecule that completes the coordination sphere, bind the zinc with tetrahedral geometry. The bond lengths are as follows: Nɛ54—Zn, 2.06 Å, Nɛ58—Zn, 2.13 Å, Sγ126—Zn, 2.27 Å, and O(HOH)—Zn, 1.93 Å. The bond angles at zinc vary from 102° to 116°. The geometry of this site and the presence of a solvent ligand immediately suggested that the metal functions in a catalytic rather than a structural role (33). Moreover, the sequence and structural motifs associated with the metal-binding site were reminiscent of those observed in several peptidases and amidases, suggesting that LuxS might function as a hydrolase in the production of AI-2.
Figure 2.

A stereoview of the Zn-ligand cluster and the putative active site of LuxS. Helix α1 bearing the HXXEH motif is at the Left; the chain from A118 to A132 underpins the zinc-binding site, covering the Zn ion (white) and its ligands. Invariant residues are drawn in ball-and-stick mode; A and B designate the two chains of the dimer. The conserved Asp-37 of the B chain lies beneath Arg-39 and is not labeled; Asn-44 is above Arg-39B at the border of the picture. The N-terminal 310 helix that may control entry to the active site is dark blue. In this view the substrate-binding cavity lies below the water bound to zinc and nestles against the strands of the β-sheet from the B chain, seen at the back. Important interatomic distances in the active site region are as follows: Cys-84 Sγ—Zn, 4.86 Å; Arg-39 NH1—Cys-84 Sγ, 4.04 Å; Glu-57 Oɛ2—Zn, 4.69 Å.
The active site cavity is remarkably polar (Fig. 2). A number of conserved residues from the symmetry-related chain contribute to the lining of the cavity that presumably accommodates the substrate (Figs. 2 and 3). The “B” chain residues Ser-6, His-11, Arg-39, Asn-44, and Cys-84 are potentially important for substrate binding or catalysis.
Figure 3.

The active site cavity in LuxS. This view is rotated ≈180° about the vertical from Fig. 2 for better display of the cavity and channel to solvent, which are outlined by the red mesh. Helix α1 with the HXXEH motif is now to the Right; strands from the B chain sheet (partly clipped) cover the cavity, and the N-terminal 310 helix (dark blue) is toward the reader. The cavity surface was generated with SURFNET (44).
Cys-84, with its sulfur positioned 4.86 Å from the Zn2+, is an unusual feature of the structure. This residue was found to be oxidized to a sulfonic acid (or a mixture of sulfonic and sulfinic acids), as evidenced by the three lobes of density arranged around the Sγ atom in a tetrahedral fashion. It is unclear whether an oxidized form of cysteine might be of significance to the mechanism or regulation of LuxS, or is simply an artifact of its ex vivo treatment. Interestingly, the oxidation of this residue is probably facilitated by a close (4.04 Å) interaction with Arg-39; positively charged environments tend to stabilize cysteine thiolates, which are, in turn, more susceptible to oxidation (34). Because the corresponding cysteine and arginine residues are conserved among all known LuxS sequences, the oxygen sensitivity of Cys-84 might be a general property of these enzymes.
A catalytic role for cysteine sulfenate has been demonstrated for several proteins. These include NADH peroxidase (35), NADH oxidase (36), peroxiredoxins (34, 37), and transcription factors believed to use cysteine oxidation as a redox-sensitive regulatory mechanism (38). Although oxidation of a cysteine in LuxS might represent an intriguing regulatory mechanism (39), it should be noted that adventitious cysteine oxidations are not uncommon. At least 60 such modifications have been recorded in the Protein Data Bank, as determined with the use of the Hetero-compound Information Centre-Uppsala (HIC-Up) server (40).
A catalytic role for a reduced Cys-84 thiolate, which would be an excellent nucleophile, cannot be excluded. LuxS possesses the elements of the Cys-His pair found in the active sites of papain (41) and in certain families of amidotransferases (42), but His-11 and Cys-84 of LuxS are not arranged with the geometry that appears to be characteristic for these enzymes.
The Active Site Cavity and AI-2.
The putative substrate-binding site of LuxS is buried and irregularly shaped, as shown in Fig. 3. The calculated volume (43) of the cavity adjoining zinc is only 90 Å3, suggesting that the reactive moieties of the LuxS substrate and the product, AI-2, must be small. Entry from the surface of the enzyme could be afforded by a partially hydrophobic tunnel so narrow that the active site appears to be inaccessible in the static structure. However, one wall of the access tunnel is formed by the N-terminal region of the symmetry-related chain. This region is the most mobile segment of the polypeptide, as judged by its elevated B-factors (the 〈B〉 for the atoms of residues 2–4 is 58.5 Å2, compared with 24.3 Å2 for the full polypeptide). It would be reasonable to assume that the N terminus functions as a flexible gate to permit substrate access.
LuxS and Zinc-Dependent Hydrolases.
The geometry and composition of the LuxS metal site invited comparison with several zinc-containing hydrolytic enzymes. One large class of zinc metalloenzymes (which includes thermolysin, carboxypeptidase A, and sonic hedgehog) can be defined by the presence of a tetrahedral ligand set composed of two histidines, a water, and a glutamate (33, 45). The sequences that bind the metal ion seem to relate LuxS to the subclasses of zinc hydrolases that are characterized by the metal-binding motif HEXXH. Indeed, despite the large differences in the overall folds of the proteins, the metal-ligand set of thermolysin, a prototypical member of this group, can be superimposed on the metal-binding center of LuxS (Fig. 4). This exercise is striking in that it results in the alignment of Glu-57 of LuxS with a conserved catalytic glutamate from thermolysin. In thermolysin, this residue accepts a proton from the zinc-bound water molecule, generating the hydroxide that mediates hydrolysis through a nucleophilic attack on the substrate. The analogous residue of LuxS, Glu-57, is similarly positioned and is absolutely conserved, supporting the notion that LuxS might be a hydrolytic enzyme.
Figure 4.

Comparison of the LuxS zinc-binding site with thermolysin. A molecular superposition based on the metal-ligand clusters of thermolysin (52) and LuxS shows the resulting positions of secondary structure elements from the two enzymes. The backbone ribbons of LuxS are yellow; ribbons from thermolysin are silver. The Zn ligands of LuxS are in standard atom colors; residues from LuxS are labeled in blue, and those from thermolysin, in gray. Helices carrying the HEXXH and HXXEH motifs run in opposite directions but correspond well in the two structures, matching the catalytic glutamate of thermolysin with the conserved Glu-57 from LuxS. The other substructures of the proteins are not equivalent. In thermolysin a long horizontal groove, delimited by strands at its lower edge and by two helices at its upper edge, is accessible to polypeptide substrates. In LuxS the substrate cavity is closed off by loops and by sheet strands from both chains (Figs. 2 and 3).
Comparisons of LuxS with Peptidases and Amidases.
Because quorum sensing in Gram-positive organisms typically depends on peptide-based autoinducers, the similarity of the LuxS metal-binding site to metalloproteases like thermolysin encouraged closer comparisons of the active site of LuxS with zinc-dependent peptidases. At the same time, the unusual set of metal ligands in LuxS suggested possible analogies to two amidases, peptide deformylase and T7 lysozyme.
Despite the similarities between the metal-ligand clusters of LuxS and numerous zinc-dependent proteases, the architecture of the active site of LuxS differs markedly from that of these enzymes. For example, the superposition of thermolysin on LuxS reveals very disparate shapes and features in their active sites. There are no matches of surfaces or secondary structures apart from the alignment of the helices bearing the zinc-binding motifs (Fig. 4). In thermolysin, as in many proteases, a long open groove supports endopeptidase activity by accommodating residues on both sides of the catalytic metal and the scissile bond. In contrast, the closed and capped active site in LuxS precludes internal residues of a peptide from binding at the zinc ion.
An unusual feature of LuxS is the presence of an inverted metal-binding motif, HXXEH. Like thermolysin, which carries the more common motif HEXXH, LuxS displays its inverted HXXEH motif on a helix. Notably, superpositions of the metal centers (Fig. 4) show that in LuxS the helix direction, like the sequence, is inverted with respect to its orientation in thermolysin. The inverted motif is also the hallmark of an intriguing set of metalloproteases, the inverzincin or M16 family, which includes a variety of bacterial and mammalian endopeptidases (33). No structures of inverzincin proteases have been determined, and the shared motifs raise the possibility that a core region of the LuxS fold might be related to the inverzincin family, despite the lack of obvious sequence similarity.
Metal binding by LuxS differs from thermolysin in that it employs Cys-126 in place of the glutamate ligand of the thermolysin metal center. This substitution appears to be rare among metallohydrolases. To our knowledge, only three other crystal structures have revealed a ligand cluster composed of two histidines, a water molecule, and cysteine: T7 lysozyme (46), peptide deformylase (47), and threonyl-tRNA synthetase (48). Consistent with the hypothesis that this ligand set is associated with hydrolytic reactions, the former two enzymes are amidases. The role of this site in the threonyl-tRNA synthetase, however, appears to be in substrate discrimination rather than catalysis (49).
Like thermolysin, T7 lysozyme possesses an open cleft that binds the N-acetylmuramic acid substrate. It further differs from LuxS in that the histidine ligands to zinc, His-17 and His-122, are situated in loops rather than along a single helix. In peptide deformylase, an HEXXH motif contributes the catalytic glutamate and two of the histidine ligands to the metal ion (Fe2+), but in contrast to LuxS, the third ligand, cysteine, lies upstream in the sequence. Superpositions of the structures of LuxS and peptide deformylase, based on the coordinates of the metal ligands, argue against a close relationship between the folds of these proteins.
Conclusions
In LuxS, the zinc ion that is presumed to be essential for catalysis is located near the floor of a small, enclosed substrate cavity lined with a number of potential catalytic groups. The architecture of the active site precludes the binding of a peptide substrate for endoproteolytic cleavage. Indeed, the size and shape of this active site make even exopeptidase activity implausible for LuxS. For these reasons, we surmise that LuxS may catalyze a hydrolytic cleavage, as suggested by the nature of the metal center and the HXXEH motif, but it is not a protease. Whether hydrolysis is the sole function of LuxS is unclear, inasmuch as additional features are also present in the active center.
Examination of the structure thus indicates that AI-2 is unlikely to be a peptide or a peptide derivative. The other major autoinducer compounds, AHLs, are also unlikely products of LuxS. AHLs are synthesized through the condensation of S-adenosylmethionine with intermediates in fatty acid biosynthesis (50), but the active site is not large enough to accommodate these reactants.
One of the goals of the emerging field of structural genomics is to assign biochemical function on the basis of macromolecular structure (51). The present study represents an example of such an approach. Although we were not able to deduce the chemical nature of AI-2, we have proposed, solely on the basis of structural features, that LuxS catalyzes cleavage of a substrate that is not closely related to the peptide or AHL classes of autoinducers.
Acknowledgments
This research was supported by a grant from the National Institutes of Health (GM 16429) and by a National Institutes of Health Training Program in Molecular Biophysics (GM 08270).
Abbreviations
- AI
autoinducer
- AHL
acylhomoserine lactone
Note
After the submission of this manuscript, the structures of LuxS from three other species were reported by H. A. Lewis et al. (53). These authors have modeled S-ribosylhomocysteine, the presumed substrate, into the active site. Conversion of this compound to AI-2, now identified as 4,5-dihydroxy-2–3-pentanedione, is an intriguing reaction that is unusual for a zinc-dependent enzyme and involves elimination and ring opening rather than net hydrolysis.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: Atomic coordinates have been submitted to the Protein Data Bank, www.rcsb.org (PDB ID code 1IE0).
The PDB identifiers for these proteins were 1dco, 1etd, 1xxa, 1atx, and 1prx. The first three are transcription factors, the fourth is a neurotoxin, and the last is a peroxidase.
References
- 1.Fuqua W C, Winans S C, Greenberg E P. J Bacteriol. 1994;176:269–275. doi: 10.1128/jb.176.2.269-275.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bassler B L. Curr Opin Microbiol. 1999;2:582–587. doi: 10.1016/s1369-5274(99)00025-9. [DOI] [PubMed] [Google Scholar]
- 3.Fuqua C, Greenberg E P. Curr Opin Microbiol. 1998;1:183–189. doi: 10.1016/s1369-5274(98)80009-x. [DOI] [PubMed] [Google Scholar]
- 4.Dunny G M, Leonard B A. Annu Rev Microbiol. 1997;51:527–564. doi: 10.1146/annurev.micro.51.1.527. [DOI] [PubMed] [Google Scholar]
- 5.Cao J G, Meighen E A. J Biol Chem. 1989;264:21670–21676. [PubMed] [Google Scholar]
- 6.Bassler B L, Wright M, Showalter R E, Silverman M R. Mol Microbiol. 1993;9:773–786. doi: 10.1111/j.1365-2958.1993.tb01737.x. [DOI] [PubMed] [Google Scholar]
- 7.Surette M G, Bassler B L. Proc Natl Acad Sci USA. 1998;95:7046–7050. doi: 10.1073/pnas.95.12.7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Surette M G, Miller M B, Bassler B L. Proc Natl Acad Sci USA. 1999;96:1639–1644. doi: 10.1073/pnas.96.4.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bassler B L, Wright M, Silverman M R. Mol Microbiol. 1994;13:273–286. doi: 10.1111/j.1365-2958.1994.tb00422.x. [DOI] [PubMed] [Google Scholar]
- 10.Bassler B L, Wright M, Silverman M R. Mol Microbiol. 1994;12:403–412. doi: 10.1111/j.1365-2958.1994.tb01029.x. [DOI] [PubMed] [Google Scholar]
- 11.Bassler B L, Greenberg E P, Stevens A M. J Bacteriol. 1997;179:4043–4045. doi: 10.1128/jb.179.12.4043-4045.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forsyth M H, Cover T L. Infect Immun. 2000;68:3193–3199. doi: 10.1128/iai.68.6.3193-3199.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joyce E A, Bassler B L, Wright A. J Bacteriol. 2000;182:3638–3643. doi: 10.1128/jb.182.13.3638-3643.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rumbaugh K P, Griswold J A, Hamood A. Microbes Infect. 2000;2:1721–1731. doi: 10.1016/s1286-4579(00)01327-7. [DOI] [PubMed] [Google Scholar]
- 15.Novick R P, Muir T W. Curr Opin Microbiol. 1999;2:40–45. doi: 10.1016/s1369-5274(99)80007-1. [DOI] [PubMed] [Google Scholar]
- 16.Finch R G, Pritchard D I, Bycroft B W, Williams P, Stewart G S. J Antimicrob Chemother. 1998;42:569–571. doi: 10.1093/jac/42.5.569. [DOI] [PubMed] [Google Scholar]
- 17.Sperandio V, Mellies J L, Nguyen W, Shin S, Kaper J B. Proc Natl Acad Sci USA. 1999;96:15196–15201. doi: 10.1073/pnas.96.26.15196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Day W A, Maurelli A T. Infect Immun. 2001;69:15–23. doi: 10.1128/IAI.69.1.15-23.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doublie S. Methods Enzymol. 1997;276:523–530. [PubMed] [Google Scholar]
- 20.Otwinowski Z, Minor W. Methods Enzymol. 1996;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 21.CCP, 4 Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 22.Terwilliger T C, Berendzen J. Acta Crystallogr D. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones T A, Zou J Y, Cowan S W, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 24.Brünger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 25.Laskowski R A, MacArthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 26.Altschul S F, Madden T L, Schaffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fischer D. Pac Symp Biocomput. 2000;5:119–130. [PubMed] [Google Scholar]
- 28.Holm L, Sander C. J Mol Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 29.Gilbert D, Westhead D, Nagano N, Thornton J. Bioinformatics. 1999;15:317–326. doi: 10.1093/bioinformatics/15.4.317. [DOI] [PubMed] [Google Scholar]
- 30.Westhead D R, Slidel T W, Flores T P, Thornton J M. Protein Sci. 1999;8:897–904. doi: 10.1110/ps.8.4.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones S, Thornton J M. Prog Biophys Mol Biol. 1995;63:31–65. doi: 10.1016/0079-6107(94)00008-w. [DOI] [PubMed] [Google Scholar]
- 32.Carson M. Methods Enzymol. 1997;277:493–505. [PubMed] [Google Scholar]
- 33.Lipscomb W N, Strater N. Chem Rev. 1996;96:2375–2433. doi: 10.1021/cr950042j. [DOI] [PubMed] [Google Scholar]
- 34.Choi H J, Kang S W, Yang C H, Rhee S G, Ryu S E. Nat Struct Biol. 1998;5:400–406. doi: 10.1038/nsb0598-400. [DOI] [PubMed] [Google Scholar]
- 35.Yeh J I, Claiborne A, Hol W G J. Biochemistry. 1996;35:9951–9957. doi: 10.1021/bi961037s. [DOI] [PubMed] [Google Scholar]
- 36.Mallett T C, Claiborne A. Biochemistry. 1998;37:8790–8802. doi: 10.1021/bi9803630. [DOI] [PubMed] [Google Scholar]
- 37.Hirotsu S, Abe Y, Okada K, Nagahara N, Hori H, Nishino T, Hakoshima T. Proc Natl Acad Sci USA. 1999;96:12333–12340. doi: 10.1073/pnas.96.22.12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Claiborne A, Yeh J I, Mallett T C, Luba J, Crane E J I, Charrier V, Parsonage D. Biochemistry. 1999;38:15407–15416. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- 39.Denu J M, Tanner K G. Biochemistry. 1998;37:5633–5642. doi: 10.1021/bi973035t. [DOI] [PubMed] [Google Scholar]
- 40.Kleywegt G J, Jones T A. Acta Crystallogr D. 1998;54:1119–1131. doi: 10.1107/s0907444998007100. [DOI] [PubMed] [Google Scholar]
- 41.Kamphuis I G, Kalk K H, Swarte M B, Drenth J. J Mol Biol. 1984;179:233–256. doi: 10.1016/0022-2836(84)90467-4. [DOI] [PubMed] [Google Scholar]
- 42.Tesmer J J G, Klem T J, Deras M, Davisson V J, Smith J L. Nat Struct Biol. 1996;3:74–86. doi: 10.1038/nsb0196-74. [DOI] [PubMed] [Google Scholar]
- 43.Liang J, Edelsbrunner H, Woodward C. Protein Sci. 1998;7:1884–1897. doi: 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laskowski R A. J Mol Graphics. 1995;13:323–330. doi: 10.1016/0263-7855(95)00073-9. [DOI] [PubMed] [Google Scholar]
- 45.Karlin S, Zhu Z Y. Proc Natl Acad Sci USA. 1997;94:14231–14236. doi: 10.1073/pnas.94.26.14231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng X, Zhang X, Pflugrath J W, Studier F W. Proc Natl Acad Sci USA. 1994;91:4034–4038. doi: 10.1073/pnas.91.9.4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chan M K, Gong W, Rajagopalan P T, Hao B, Tsai C M, Pei D. Biochemistry. 1997;36:13904–13909. doi: 10.1021/bi9711543. [DOI] [PubMed] [Google Scholar]
- 48.Sankaranarayanan R, Dock-Bregeon A C, Romby P, Caillet J, Springer M, Rees B, Ehresmann C, Ehresmann B, Moras D. Cell. 1999;97:371–381. doi: 10.1016/s0092-8674(00)80746-1. [DOI] [PubMed] [Google Scholar]
- 49.Sankaranarayanan R, Dock-Bregeon A C, Rees B, Bovee M, Caillet J, Romby P, Francklyn C S, Moras D. Nat Struct Biol. 2000;7:461–465. doi: 10.1038/75856. [DOI] [PubMed] [Google Scholar]
- 50.More M I, Finger L D, Stryker J L, Fuqua C, Eberhard A, Winans S C. Science. 1996;272:1655–1658. doi: 10.1126/science.272.5268.1655. [DOI] [PubMed] [Google Scholar]
- 51.Skolnick J, Fetrow J S, Kolinski A. Nat Biotechnol. 2000;18:283–287. doi: 10.1038/73723. [DOI] [PubMed] [Google Scholar]
- 52.Holland D R, Hausrath A C, Juers D, Matthews B W. Protein Sci. 1995;4:1955–1965. doi: 10.1002/pro.5560041001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lewis H A, Furlong E B, Laubert B, Eroshkina G A, Batiyenko Y, Adams J M, Bergseid M G, Marsh C D, Peat T S, Sanderson N E, et al. Structur. 2001. e 9, 527–538. [DOI] [PubMed] [Google Scholar]