

Abstract
The HIV-1 gp41 envelope glycoprotein promotes fusion of the virus and cell membranes through the formation of a trimer-of-hairpins structure, in which the amino- and carboxyl-terminal regions of the gp41 ectodomain are brought together. Synthetic peptides derived from these two regions (called N and C peptides, respectively) inhibit HIV-1 entry. In contrast to C peptides, which inhibit in the nanomolar range, N peptides are weak inhibitors with IC50 values in the micromolar range. To test the hypothesis that the weak inhibition of N peptides results from their tendency to aggregate, we have constructed chimeric variants of the N-peptide region of gp41 in which soluble trimeric coiled coils are fused to portions of the gp41 N peptide. These molecules, which present the N peptide in a trimeric coiled-coil conformation, are remarkably more potent inhibitors than the N peptides themselves and likely target the carboxyl-terminal region of the gp41 ectodomain. The best inhibitors described here inhibit HIV-1 entry at nanomolar concentrations.
To infect a cell, HIV type 1 (HIV-1) must fuse its membrane with the host cell membrane and release its contents into the cell. The fusion process is mediated by the envelope glycoprotein (Env) on the surface of the virus. Env is composed of two subunits: gp120, a surface subunit that binds host cell receptors, and gp41, a transmembrane subunit that ultimately inserts into the host cell membrane and promotes the fusion event. The ectodomain of gp41 contains two helical regions, one near the amino terminus (N helix) and one near the carboxy terminus (C helix). In the fusogenic conformation of gp41, the N and C helices interact with each other in an antiparallel manner, like a hairpin, bringing the amino and carboxy termini of the ectodomain together. This facilitates the juxtaposition of the virus and cell membranes, leading to membrane fusion (1).
The fusogenic structure of gp41 is composed of three gp41 hairpins (i.e., trimer-of-hairpins). The N helices from three gp41 ectodomains form a central three-stranded coiled coil and are surrounded by three antiparallel C helices that bind to conserved grooves on the coiled-coil surface (2–5). Synthetic peptides derived from the N- and C-helix regions of gp41 (called N and C peptides, respectively) inhibit HIV-1 viral infection (3, 6–10). C peptides are potent inhibitors of HIV-1 infectivity with activity at nanomolar concentrations (3, 6, 8, 10, 11), and one, T20, has shown promising results in anti-HIV-1 human clinical trials (12). C peptides likely inhibit formation of the fusogenic six-helix bundle in a dominant-negative manner by binding the N-helix region of gp41 (Fig. 1A) (3, 9, 13–16). Recently, efforts to target a hydrophobic pocket on the N helix of gp41 led to the identification of cyclic D peptides that inhibit HIV-1 entry, likely in the same dominant-negative manner (17, 18).
Figure 1.

(A) Working model of HIV-1 membrane fusion and its inhibition (for review, see ref. 1). Before exposure to cellular receptors, Env exists in a native state (“Native”) on the surface of the virus. After interaction with CD4 and the coreceptor, a conformational change allows gp41 to insert its amino-terminal fusion-peptide domain into the cell membrane, forming a transient prehairpin intermediate (“Prehairpin”). In the prehairpin intermediate, the N-peptide region (gray) and possibly the C-peptide region (yellow) are exposed and vulnerable to inhibitors [e.g., C peptides (3, 6, 7, 9, 10), D peptides (18), and 5-Helix (21)]. Although 5-Helix is depicted as targeting the prehairpin intermediate, it is unknown whether it targets the native state, the prehairpin intermediate, or both (see ref. 1). All of these inhibitors work in a dominant-negative manner by preventing formation of the trimer-of-hairpins (see text). In the absence of inhibitors, the prehairpin intermediate slowly resolves to the trimer-of-hairpins structure that juxtaposes the virus and cell membranes and leads to fusion. (B) Representation of two possible mechanisms for N-peptide inhibitory activity. N peptides may target a vulnerable C-helix region of gp41 (yellow). Alternatively, the N peptides (blue) could intercalate with the N helices of gp41 (gray) to form a heterotrimeric coiled coil and interfere with the coiled-coil formation of gp41.
N peptides can also inhibit HIV-1 entry (3, 7). Two possible mechanisms for their inhibitory activity can be envisioned (Fig. 1B). First, the N peptides may target an exposed C-helix region of gp41 (3). Alternatively, synthetic N peptides could intercalate with the N helices of gp41, thereby forming a heterotrimeric coiled coil and interfering with the coiled-coil formation of gp41 (7, 19).
N peptides, which aggregate in isolation (3, 20), are far less potent than C peptides. It has been proposed that N peptides are poor inhibitors because their aggregation interferes with their ability to efficiently target gp41 via either mechanism described above (3, 21). However, if an N peptide could be properly sequestered into a nonaggregating trimeric coiled-coil conformation, it should be able to effectively bind and sequester the C helix of gp41 and inhibit HIV-1 entry via the first mechanism described above.
Indeed, recent work has shown that the C-helix region of gp41 is a useful target for inhibiting HIV-1 entry (21). A protein designed to have a very high affinity for C helix is an extremely effective, broad-spectrum antiviral compound, with a low nanomolar IC50 value. This molecule, 5-Helix, consists of five of the six helices of the gp41 trimer-of-hairpins joined by short peptide linkers (21). 5-Helix forms the trimer-of-hairpins structure, exposing one C-helix-binding site, and its antiviral activity depends on its ability to bind C helix.
In this study, we designed nonaggregating trimeric coiled-coil N peptides and characterized their inhibitory activity. Each peptide is a chimeric molecule consisting of a designed trimeric coiled coil (22, 23) fused to a portion of the gp41 N helix. One of these peptides, IQN17, was previously shown to form a soluble, stable, trimeric coiled coil (18). These peptides sustain the N helix in a coiled-coil conformation and, strikingly, they have dramatically increased antiviral potency over the corresponding N peptides themselves. The inhibitory activity of the chimeric N peptides is correlated to their stability. The most potent chimeric N peptide described here, IQN23, inhibits HIV-1 entry with an IC50 value of 15 nM.
Materials and Methods
Peptide Synthesis and Purification.
All peptides were chemically synthesized on a PE Biosystems 431A peptide synthesizer upgraded with feedback monitoring. The standard Fmoc/HBTU [fluoroenylmethyl chloroformate/2-(1H-benzotriazol-1-yl)-1,1,3,3,tetramethyluronium hexafluorophosphate] chemistry (24) was modified with DMSO/NMP resin swelling and acetic anhydride capping after every couple. The peptides were cleaved from the PE Biosystems Pal resin with Reagent K (25). Each peptide has an acetylated amino terminus and a carboxyl-terminal amide.
The sequences of N17, N23, N36, IQN17, IQN23, IQN36, IIN17, IQ22N17, II22N17, IQ15N17, II15N17, IZN17, IZN23, and IZN36 are shown in Fig. 2. The first 29 residues of IQN17 are derived from a designed trimeric coiled coil, GCN4-pIQI′ (18), and the final 17 residues are derived from the N-helix region of gp41 from the HXB2 strain of HIV-1. The sequence of GCN4-pIQI′ is Ac-RMKQIEDKIEEIESKQKKIENEIARIKKLIGERY-NH2. The N17 peptide is 17 aa long and consists of only the gp41 residues of IQN17. IQN17–2Pro contains a Gln→Pro mutation at position 16 of IQN17 and an Ile→Pro mutation at position 37. IQN17 G572D contains a Gly→Asp mutation at position 36 of IQN17. Longer chimeric N peptides were made by inserting additional residues from the HXB2 gp41 N-peptide region, taking care to keep the coiled-coil heptad repeat register intact. These peptides are IQN23 and IQN36. The N23 and N36 peptides contain only the gp41 residues of IQN23 and IQN36, respectively. Sequential heptads were removed from the amino terminus of IQN17 to yield two shorter peptides: IQ22N17 and IQ15N17. More stable versions of the IQN17, IQ22N17, and IQ15N17 peptides were made by changing the glutamine residues at position 16 of IQN17, position 9 of IQ22N17, and position 2 of IQ15N17 to isoleucines. These peptides are called IIN17, II22N17, and II15N17, respectively. Also, additional chimeric N peptides were made in which a different designed trimeric coiled coil was placed amino-terminal to the gp41-derived residues. The coiled coil, IZm, was based on a design described by Suzuki et al. (23). Alterations were made at some of the e and g positions of the Suzuki sequence to avoid similarity to GCN4-pIQI′. Also, one Ile→Gln substitution was made at an a position. These additional chimeric peptides are called IZN17, IZN23, and IZN36. The sequence of IZm is Ac-YGGIKKEIEAIKKEQEAIKKKIEAIEKEIEA-NH2.
Figure 2.

HIV-1 gp41 structure and chimeric N peptides. (A) Schematic of the primary structure of HIV-1 HXB2 gp41, including the sequence of residues 539–581. The N-peptide (gray) and C-peptide regions (yellow) are indicated. The residues corresponding to the N peptides discussed in this study are indicated. (B) Model of IQN17, which represents the general structure of the chimeric N peptides discussed in this study. Each chimeric N peptide consists of an amino-terminal designed trimeric coiled coil (purple) fused to a sequence from the N-helix region of gp41 (gray). (C) The peptide sequences of the chimeric N peptides. The designed trimeric coiled-coil sequences (purple for GCN4-pIQI′, red for IZm) are aligned, as are the gp41 sequences (black). The a positions of the heptad repeats are indicated by a larger font.
After cleavage from the resin, each peptide was desalted over a Sephadex G-25 column (Amersham Pharmacia) and lyophilized. The peptide was then resuspended in 5% acetic acid and purified over a Vydac C18 (Hesperia, CA) preparative column on a reverse-phase high-performance liquid chromatography apparatus (Waters). The peptide was eluted from the column with a water–acetonitrile gradient in the presence of 0.1% trifluoroacetic acid and lyophilized. The molecular weights of each peptide were confirmed by using matrix-assisted laser desorption ionization-time-of-flight mass spectrometry (PerSeptive Biosystems, Framingham, MA).
For all experiments, peptide stock solutions (typically 200–800 μM) were prepared by resuspending lyophilized peptide in water. The precise concentrations of the peptide stocks were determined by using tyrosine and tryptophan absorbance at 280 nm in 6 M GuHCl (26). The stock solutions were then diluted to the desired concentration in the appropriate buffer for each experiment. After resuspension in water, N17 and N23 solutions were cloudy. The aggregates were pelleted at high speed for 10 min, and the concentration of the supernatant was determined.
CD.
All CD measurements were performed on either an Aviv 62 DS or 62A DS (Aviv Associates, Lakewood, NJ) CD spectrometer. Standard scans were performed on 10 μM solutions of peptide in PBS [50 mM sodium phosphate, 150 mM sodium chloride (pH 7.4)] from 200 to 260 nm in a 1-cm pathlength cuvette with a 5-second averaging time. Percent helicity as recorded at 222 nm was calculated according to Chen et al. (27). Thermal denaturation scans of 10 μM peptide solutions in PBS were recorded at 222 nm. The peptide was heated at 2-degree intervals starting at 4 or 20°C, with an equilibration time of 1.5 min and an averaging time of 60 seconds. The melting temperatures, or midpoints of the cooperative thermal unfolding transitions (Tm), were estimated from the thermal dependence of the ellipticity at 222 nm. For most of the peptides with a melting temperature of 100°C or higher in PBS, thermal denaturation experiments were performed in PBS in the presence of 2 M guanidine hydrochloride.
Sedimentation Equilibrium.
All measurements were recorded on a Beckman XL-A (Beckman Coulter) analytical ultracentrifuge equipped with an An-60 Ti rotor (Beckman Coulter). Peptide stock solutions were diluted to 100–200 μM and dialyzed overnight against PBS. After dialysis, the concentration was redetermined, and the peptides were diluted to 20 μM by using the dialysis buffer. The samples were centrifuged at speeds ranging from 19,000 to 26,000 rpm.
HIV-1 Infectivity Assay.
Inhibitory activity of the chimeric N peptides was determined by using an HIV luciferase assay (28). Virus was made by cotransfecting 293T cells with an HIV-1 genome containing a frame-shift mutation in env and a luciferase gene replacing nef (NL43LucR-E-) along with pCMVHXB2, an expression vector with the HXB2 gp160 gene. Because its genome lacks env, the resultant virus is viable only for one round of infection. The cellular debris was removed by low-speed centrifugation. The remaining viral supernatant was used to infect HOS-CD4/fusion cells (N. Landau, National Institutes of Health AIDS Reagent Program) in the presence of 2-fold dilutions of the experimental peptides. Two days postinfection, the cells were lysed, and luciferase activity was monitored on a Wallac AutoLumat LB953 luminometer (Gaithersburg, MD). IC50 values (the peptide concentration at which half of the viral infection is inhibited) were calculated by fitting the data to a Langmuir equation [y = k/(1 + [peptide]/IC50)], where y = luciferase activity and k is a scaling constant.
Results
Synthetic Peptides from the N-Helix Region of gp41 Weakly Inhibit Viral Infectivity.
Previously, a peptide containing 36 amino acids derived from the N-helix region of gp41, called N36, was identified by protein dissection of the ectodomain of gp41 (2, 20). Here, we examined the HIV-1 inhibitory activity of N36 and two shorter N peptides: N17 and N23. N17 comprises the residues that form the hydrophobic pocket of the N-helix trimer that, in the fusogenic gp41 structure, binds to three hydrophobic residues of the C-helix region. N23 and N36 contain the residues of N17 with additional amino-terminal N-helix residues (Fig. 2A). N36 aggregates in solution (20), and both N17 and N23 are extremely insoluble (see Materials and Methods). These peptides are not very helical in isolation, as determined by CD (Table 1). The inhibitory potencies of N17, N23, and N36 are low, with IC50 values of 13, 29, and 2 μM, respectively (Table 1).
Table 1.
Biophysical data and HIV-1 inhibitory activity of chimeric N peptides
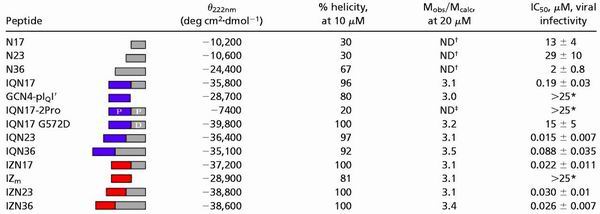 |
CD scans were performed on 10 μM peptide solutions in PBS. The CD signal at 222 nm (θ222nm) and the percent helicity are reported. Sedimentation equilibrium studies were performed on 20 μM peptide solutions in PBS. The results are reported as a ratio of the experimental molecular weight to the calculated molecular weight for a monomer (Mobs/Mcalc). Inhibition of viral entry was measured in a single-round infectivity assay. The results indicate the mean IC50 value (the peptide concentration at which half the viral infection is inhibited) and SEM for no less than two experiments, each performed in duplicate. ND, not determined.
No activity seen up to 25 μM.
Aggregated (see Materials and Methods and ref. 21).
Unfolded, as determined by CD.
The Pocket-Forming Region of the N Peptide Inhibits as a Coiled-Coil Trimer.
In the fusogenic trimer-of-hairpins structure, the N peptides form a trimeric coiled coil and bind to three helical C peptides (2–5, 20). If synthetic N peptides could be presented in a trimeric coiled-coil conformation rather than an aggregated state, they might bind the C-helix region of gp41 better than aggregated N peptides. Because the C helix has been shown to be an effective antiviral target (21), coiled-coil N peptides also may have higher inhibitory potency than aggregated N peptides. Previously, such a soluble peptide was designed (18). IQN17 is a chimeric peptide that presents a portion of the N helix comprising a hydrophobic pocket in a trimeric coiled-coil conformation. In IQN17, a designed trimeric coiled coil (GCN4-pIQI′) was fused to the amino terminus of 17 residues of the N helix (N17, as seen above), creating a continuous coiled coil 45 aa in length (Fig. 2 B and C).
IQN17 is a fully helical and discretely trimeric species at 10–20 μM (ref. 18 and Table 1). IQN17 is extremely stable, with a melting temperature of ≈100°C at 10 μM (Table 1), and guanidine denaturation studies predict it maintains its trimeric state even at low nanomolar concentrations (unpublished results). Additionally, with an IC50 value of 190 nM, IQN17 is almost two orders of magnitude more potent than N17 (Table 1). The GCN4-pIQI′ portion of the molecule alone as well as an unfolded IQN17 (containing two proline residues in the hydrophobic core) do not show inhibitory activity (tested up to 25 μM). In addition, a derivative of IQN17 that introduces a charge into the hydrophobic pocket, IQN17 (G572D) has little inhibitory activity, with an IC50 value of 15 μM. Therefore, the inhibitory activity of the hydrophobic pocket region of the N peptide is greatly enhanced in a trimeric coiled-coil conformation.
GCN4-pIQI′ was also fused to the amino terminus of N23 and N36 to make IQN23 and IQN36, respectively (Fig. 2C). Both of these peptides are extremely helical (Table 1). IQN23 is discretely trimeric, whereas IQN36 is a slightly aggregated trimer (Table 1). Both IQN23 and IQN36 are dramatically better inhibitors than the corresponding N peptides themselves, with IC50 values of 15 and 88 nM, respectively (Table 1).
The Inhibitory Potency of IQN17 Is Correlated to Its Stability.
If the N-peptide inhibitors must be trimeric to inhibit HIV-1 entry, one might expect the thermodynamic stability of the peptides to be correlated to their inhibitory activity. In particular, a less stable IQN17 derivative that is dissociated at the IC50 concentration of IQN17 should be a less potent inhibitor. A series of peptides was made to determine the effect of stability of the trimeric coiled-coil structure on the inhibitory potency of the chimeric N peptides.
The GCN4-pIQI′ trimeric coiled coil contains one glutamine per monomer (at position 16) that packs into the interior of the coiled coil. This glutamine was shown to decrease stability compared with a coiled coil with an all-isoleucine core (22, 29). The glutamine in the IQN17 core (at position 16) was changed to an isoleucine to make a more stable peptide, IIN17 (Fig. 2C). Shorter less stable IQN17 derivatives were made by sequentially removing groups of seven residues from the amino terminus of IQN17. These two IQN17 derivatives contain 22 and 15 aa of the GCN4-pIQI′ region (IQ22N17 and IQ15N17, respectively) (Fig. 2C). The corresponding isoleucine derivatives of these peptides (II22N17 and II15N17) were studied as well.
In each peptide pair (IQN17 and IIN17, IQ22N17 and II22N17, and IQ15N17 and II15N17), the isoleucine derivative is the more stable of the two peptides, as determined by thermal denaturation (Table 2). The isoleucine derivative in both of the two pairs of shorter IQN17 derivatives (IQ22N17 and II22N17 and IQ15N17 and II15N17) has more potent inhibitory activity that is statistically significant (Table 2). However, there is little, if any, difference in the inhibitory activity of the three most stable peptides that have melting temperatures of 100°C or above (IQN17, IIN17, and II22N17). Therefore, the inhibitory activity of the peptides correlates to the stability of the coiled-coil structures, until the point where a highly stable coiled-coil structure is reached.
Table 2.
Stability of chimeric N peptides is correlated to inhibition
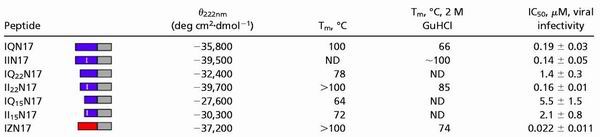 |
CD scans were performed on 10 μM peptide solutions in PBS. The CD signal at 222 nm (θ222nm) is reported. Thermal denaturation CD scans were performed on 10-μm peptide solutions in PBS and in PBS with 2 M guanidine hydrochloride. The midpoint of thermal denaturation (Tm) was estimated from the thermal dependence of the CD signal at 222 nm. Inhibition of viral entry was measured in a single-round infectivity assay. The results indicate the mean IC50 value (the peptide concentration at which half the viral infection is inhibited) and SEM for no less than two experiments, each performed in duplicate. ND, not determined.
An Alternate Chimeric N Peptide, IZN17, Is a More Potent Inhibitor.
We studied an additional series of chimeric N peptides in which the GCN4-pIQI′ portion of the molecule was replaced with a different designed trimeric coiled coil. This coiled coil, called “IZm” for modified isoleucine zipper, is based on a design described by Suzuki et al. (23) and is helical and trimeric in solution (Table 1). The resulting chimeric peptides are termed IZN17, IZN23, and IZN36 (Fig. 2C). Each of the peptides is helical, as determined by CD (Table 1 and Fig. 3A). IZN17 (Fig. 3B) and IZN23 are discrete trimers, and IZN36 is an aggregated trimer, as determined by sedimentation equilibrium (Table 1). The IC50 values of IZN17 (Fig. 3C), IZN23, and IZN36 in the HIV-1 infectivity assay are 22, 30, and 26 nM, respectively (Table 1).
Figure 3.

IZN17: biophysical data and inhibitory activity. (A) The mean of two CD spectra of 10 μM solutions of IZN17 in PBS (pH 7.4), 4°C. IZN17 is fully helical, with an approximate ellipticity of −37,200 deg cm2⋅dmol− at 222 nm. (B) Representative sedimentation equilibrium data of a 20 μM solution of IZN17 in PBS (pH 7.4), 4°C. The slope of the data as plotted is proportional to the molecular mass of the peptide oligomer. Lines indicate the expected slopes for dimeric, trimeric, and tetrameric oligomeric states. The deviation in the data from the line representing the trimeric state is plotted (Upper). IZN17 is a discrete trimer under the experimental conditions. (C) Inhibition of viral infectivity by IZN17 as determined by an HIV-1 luciferase assay. The data represent the mean +/− SEM of five separate experiments. The approximate IC50 value of IZN17 is 22 nM.
Interestingly, IZN17 is close to an order of magnitude more potent than IQN17 in the viral infectivity assay. Although it is not entirely clear why this is the case, there are two potential reasons that may contribute to the increase in potency. First, IZN17 is more stable than IQN17, as determined by thermal denaturation (Table 2). However, this is not likely to contribute significantly to the increased potency of IZN17, because IIN17 is much more stable than IZN17 and yet a less potent inhibitor. Second, IZN17 contains two additional residues from the gp41 N-helix region (residues 20 and 21 of IZN17, corresponding to residues 560 and 561 of HXB2) because of a coincidence in sequence between IZm and gp41. The additional gp41 residues could provide an increase in binding affinity to the C-helix region of gp41.
Discussion
Previously studied N peptides are weak inhibitors of HIV-1 entry, with IC50 values in the micromolar range (3, 7). Our results demonstrate that N peptides can be potent inhibitors when presented in a trimeric coiled-coil conformation. In the best example, addition of a designed coiled coil to the amino terminus of N23 resulted in a remarkable decrease in the IC50 value of three orders of magnitude (Table 1). In addition, the chimeric N peptides likely inhibit HIV-1 entry in a dominant-negative manner by binding to the C-helix region of gp41, as seen in the x-ray structure of fusogenic gp41 (2, 4, 5). Recently, Louis et al. described the design of another soluble N-helix trimeric coiled coil, NCCG-gp41, which is stabilized by both fusion to a thermostable subdomain of gp41 and intersubunit disulfide bonds (30). NCCG-gp41 inhibits HIV-1 entry at nanomolar concentrations, in agreement with our results.
The chimeric N peptides studied here inhibit HIV-1 entry with a wide range of IC50 values, from 15 nM to 5 μM. Several factors appear to influence their potency. First, increased thermal stability of the coiled coil, within a range, correlates with increased potency. For example, II22N17 is much more resistant to thermal denaturation than IQ22N17 and is almost an order of magnitude more potent. However, the effects of stability on potency decline once a certain level of stability is obtained. For example, the IC50 values of IQN17, IIN17, and II22N17 are similar despite large differences in thermal stability. These observations are likely because of the association state of the peptides. As mentioned, guanidine denaturation studies indicate that IQN17 is fully trimeric at low nanomolar concentrations (unpublished results). Because IIN17 and II22N17 are more stable than IQN17 according to thermal denaturation studies, they should also be fully trimeric at low nanomolar concentrations. Therefore, at the IC50 concentration slightly below 200 nM, IQN17, IIN17, and II22N17 are likely completely associated, and the IC50 value of IQN17 likely represents a maximum inhibitory potency. The less stable peptides with significantly lower inhibitory potencies (IQ22N17, IQ15N17, and II15N17) are likely at least partially unfolded at 200 nM, leading to a decrease in inhibitory potency.
Second, increasing the length of the gp41 portion can increase the inhibitory activity of the chimeric N peptides (e.g., IQN23 is a much better inhibitor than IQN17). However, adding additional gp41 residues does not always result in a better inhibitor (e.g., IQN36 is less potent than IQN23, possibly because of the increased aggregation of IQN36).
Third, the identity of the designed coiled coil can alter the inhibitory activity of the attached N peptide (e.g., IZN17 is a much better inhibitor than IQN17). It is unclear why the identity of the designed coiled coil affects the inhibitory activity of the chimeric N peptides. In the case of IZN17, it is slightly more stable than IQN17 and also contains two additional residues amino terminal to N17 that are identical to the gp41 N-peptide sequence (residues 20 and 21 of IZN17). These two factors may explain at least some of the increased potency of IZN17. However, it is also possible that because the residues in GCN4-pIQI′ and IZm differ, they differentially affect the binding of the N-peptide region of the molecule to the C-helix region of gp41. For example, in the context of N17 binding, IZm may contribute favorable interactions with gp41 or GCN4-pIQI′ may make unfavorable contacts.
The range of potencies of the panel of chimeric N peptides studied here suggests it may be possible to design additional chimeric N peptides with increased potency. For example, changing the identity or location of the designed coiled coil (such as placing it on the carboxy terminus of the N peptide rather than the amino terminus) and using different regions of the N helix may result in better inhibitors. It is possible that the peptides studied here, or similar ones not yet designed, would provide a useful additional therapy for HIV-1 patients. Indeed, some of these chimeric N peptides are of similar size and potency to synthetic C peptides currently in human clinical trials (12).
Finally, the chimeric N peptides likely present an accurate structure of the N-helix region of gp41 present transiently during the entry process. Indeed, cyclic D peptides that bind to IQN17 have been shown to inhibit HIV-1 entry (18). If an antibody that recognized this structure could be identified, it may have neutralizing activity (see also refs. 18, 31). Therefore, molecules such as the chimeric N peptides may serve as useful components of potential vaccines for raising an anti-HIV-1 neutralizing antibody response.
Acknowledgments
We thank members of the Kim lab, including Michael Burgess and Ben Sanford for peptide synthesis, Heng Chhay for antiviral assays, Lily Hong for technical assistance, Sam Sia for advice on the design of IZm, and Leslie Gaffney for editorial assistance. We also thank members of the Kim laboratory for critically reviewing the manuscript. This research was supported by the National Institutes of Health (GM44162).
Abbreviation
- HIV-1
HIV type 1
References
- 1.Eckert D M, Kim P S. Annu Rev Biochem. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- 2.Chan D C, Fass D, Berger J M, Kim P S. Cell. 1997;89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 3.Lu M, Blacklow S C, Kim P S. Nat Struct Biol. 1995;2:1075–1082. doi: 10.1038/nsb1295-1075. [DOI] [PubMed] [Google Scholar]
- 4.Tan K, Liu J, Wang J-H, Shen S, Lu M. Proc Natl Acad Sci USA. 1997;94:12303–12308. doi: 10.1073/pnas.94.23.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weissenhorn W, Dessen A, Harrison S C, Skehel J J, Wiley D C. Nature (London) 1997;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 6.Jiang S, Lin K, Strick N, Neurath A R. Nature (London) 1993;365:113. doi: 10.1038/365113a0. [DOI] [PubMed] [Google Scholar]
- 7.Wild C, Oas T, McDanal C, Bolognesi D, Matthews T. Proc Natl Acad Sci USA. 1992;89:10537–10541. doi: 10.1073/pnas.89.21.10537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wild C T, Greenwell T, Matthews T. AIDS Res Hum Retroviruses. 1993;9:1051–1053. doi: 10.1089/aid.1993.9.1051. [DOI] [PubMed] [Google Scholar]
- 9.Wild C T, Greenwell T, Shugars D, Rimsky-Clarke L, Matthews T. AIDS Res Hum Retroviruses. 1995;11:323–325. doi: 10.1089/aid.1995.11.323. [DOI] [PubMed] [Google Scholar]
- 10.Wild C T, Shugars D C, Greenwell T K, McDanal C B, Matthews T J. Proc Natl Acad Sci USA. 1994;91:9770–9774. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan D C, Chutkowski C T, Kim P S. Proc Natl Acad Sci USA. 1998;95:15613–15617. doi: 10.1073/pnas.95.26.15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kilby J M, Hopkins S, Venetta T M, DiMassimo B, Cloud G A, Lee J Y, Alldredge L, Hunter E, Lambert D, Bolognesi D, et al. Nat Med. 1998;4:1302–1307. doi: 10.1038/3293. [DOI] [PubMed] [Google Scholar]
- 13.Chan D C, Kim P S. Cell. 1998;93:681–684. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- 14.Chen C-H, Matthews T J, Mcdanal C B, Bolgnesi D P, Greenberg M L. J Virol. 1995;69:3771–3777. doi: 10.1128/jvi.69.6.3771-3777.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malashkevich V N, Chan D C, Chutkowski C T, Kim P S. Proc Natl Acad Sci USA. 1998;95:9134–9139. doi: 10.1073/pnas.95.16.9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rimsky L T, Shugars D C, Matthews T J. J Virol. 1998;72:986–993. doi: 10.1128/jvi.72.2.986-993.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cole J L, Garsky V M. Biochemistry. 2001;40:5633–5641. doi: 10.1021/bi010085w. [DOI] [PubMed] [Google Scholar]
- 18.Eckert D M, Malashkevich V N, Hong L H, Carr P A, Kim P S. Cell. 1999;99:103–115. doi: 10.1016/s0092-8674(00)80066-5. [DOI] [PubMed] [Google Scholar]
- 19.Weng Y, Weiss C D. J Virol. 1998;72:9676–9682. doi: 10.1128/jvi.72.12.9676-9682.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu M, Kim P S. J Biomol Struct Dyn. 1997;15:465–471. doi: 10.1080/07391102.1997.10508958. [DOI] [PubMed] [Google Scholar]
- 21.Root M J, Kay M S, Kim P S. Science. 2001;291:884–888. doi: 10.1126/science.1057453. [DOI] [PubMed] [Google Scholar]
- 22.Eckert D M, Malashkevich V N, Kim P S. J Mol Biol. 1998;284:859–865. doi: 10.1006/jmbi.1998.2214. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki K, Hiroaki H, Kohda D, Tanaka T. Protein Eng. 1998;11:1051–1055. doi: 10.1093/protein/11.11.1051. [DOI] [PubMed] [Google Scholar]
- 24.Fields C G, Lloyd D H, Macdonald R L, Otteson K M, Noble R L. Peptide Res. 1991;4:95–101. [PubMed] [Google Scholar]
- 25.King D S, Fields C G, Fields G B. Int J Pep Protein Res. 1990;36:255–266. doi: 10.1111/j.1399-3011.1990.tb00976.x. [DOI] [PubMed] [Google Scholar]
- 26.Edelhoch H. Biochemistry. 1967;6:1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- 27.Chen Y, Yang J T, Chau K H. Biochemistry. 1974;13:3350–3359. doi: 10.1021/bi00713a027. [DOI] [PubMed] [Google Scholar]
- 28.Chen B K, Saksela K, Andino R, Baltimore D. J Virol. 1994;68:654–660. doi: 10.1128/jvi.68.2.654-660.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harbury P B, Zhang T, Kim P S, Alber T. Science. 1993;262:1401–1407. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- 30.Louis J M, Bewley C A, Clore G M. J Biol Chem. 2001;276:29485–29499. doi: 10.1074/jbc.C100317200. [DOI] [PubMed] [Google Scholar]
- 31.LaCasse R A, Follis K E, Trahey M, Scarborough J D, Littman D R, Nunberg J H. Science. 1999;283:357–362. doi: 10.1126/science.283.5400.357. [DOI] [PubMed] [Google Scholar]