

Abstract
Motivation
G-quadruplex structures in RNA molecules are known to have regulatory impacts in cells but are difficult to locate in the genome. The minimal requirements for G-quadruplex folding in RNA (G≥3N1-7 G≥3N1-7 G≥3N1-7 G≥3) is being challenged by observations made on specific examples in recent years. The definition of potential G-quadruplex sequences has major repercussions on the observation of the structure since it introduces a bias. The canonical motif only describes a sub-population of the reported G-quadruplexes. To address these issues, we propose an RNA G-quadruplex prediction strategy that does not rely on a motif definition.
Results
We trained an artificial neural network with sequences of experimentally validated G-quadruplexes from the G4RNA database encoded using an abstract definition of their sequence. This artificial neural network, G4NN, evaluates the similarity of a given sequence to known G-quadruplexes and reports it as a score. G4NN has a predictive power comparable to the reported G richness and G/C skewness evaluations that are the current state-of-the-art for the identification of potential RNA G-quadruplexes. We combined these approaches in the G4RNA screener, a program designed to manage and evaluate the sequences to identify potential G-quadruplexes.
Availability and implementation
G4RNA screener is available for download at http://gitlabscottgroup.med.usherbrooke.ca/J-Michel/g4rna_screener.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Ribonucleic acids (RNA) are versatile molecules which can both serve as a scaffold to transfer information and as a support to drive reactions or regulatory mechanisms (Kwok, 2016; Sharp, 2009). While most RNA functions are guided by their sequences, these polymers adopt three-dimensional structures, adding a supplementary layer of complexity. The structure is a powerful regulatory system of RNA since it can control the position, interactions and accessibility of the sequence it bears (Lai et al., 2013). Paradoxically, the structure of the RNA is dependent on its sequence as well as the presence of interactors and its environmental context.
Intramolecular G-quadruplexes (G4) are tetrahelical structures found in nucleic acids. G4 are highly dependent on the guanine (G) richness of the sequence and the presence of potassium cations in its vicinity (Agarwala et al., 2015; Rouleau et al., 2017). RNA G4, the focus of this study, was successfully observed in mammalian cells in accordance with the fact that potassium is the most abundant metallic ion in mammalian cells (Biffi et al., 2014). RNA G4 are known to modulate different mechanisms; G4 found in mRNA can regulate translation, localization, polyadenylation, splicing, etc (Agarwala et al., 2015); while G4 found in miRNA precursors can regulate their processing (Pandey et al., 2015). These functions are mainly attributed to the high stability of the G4 and its distinct shape (Agarwala et al., 2015).
G4 folding is dependent on the RNA sequence, requiring the stacking of at least two G-quartets. A G-quartet is a planar interaction of four guanine (G) residues through Hoogsteen pair bonding. The intramolecular stacking of G-quartets requires four series of consecutive G. The length of the G series determines the number of planes the G4 harbors (Malgowska et al., 2016). Each G series is separated from the next by a stretch of random nucleotide composition, which bulges out of the tetrahelix and forms three distinct loops. Canonical G4 can be described using the GXNL1GXNL2GXNL3GX motif where X is the length of G stretches, N is any nucleotide (A, U, C and G) and L1, L2, L3 are the lengths of the loops.
So far, potential G4 have been described by this motif and most identification strategies rely on it (Eddy and Maizels, 2006; Huppert and Balasubramanian, 2005; Kikin et al., 2006; Lorenz et al., 2013; Menendez et al., 2012). However, its usage is limited. The classical X = 3 and 1 ≤ L ≤ 7 fails to identify several unorthodox structures identified in recent years (Faudale et al., 2009; Jodoin et al., 2014; Martadinata and Phan, 2014; Warner et al., 2014). Adjusting the motif to accommodate these new structures by reducing X to 2 or raising the upper limit of L increases the number of hits drastically, likely introducing many false positives. In fact, the high diversity of sequences shown to fold into G4 exposes a challenge for their prediction. A partial solution to filter out sequences not folding into a G4 is to consider their flanking sequences. The presence of runs of cytosines (C) in the flanking sequences of a potential G4 can hinder its folding. The consecutive guanine over consecutive cytosine (cGcC) score was a first endeavor to address this issue (Beaudoin et al., 2014). Recently, G4Hunter (G4H), a tool providing a similar score, was used to assess the G4 propensity of the mitochondrial genome (Bedrat et al., 2016). It was designed for DNA but was shown to be usable on RNA, although non-exhaustively (Bedrat et al., 2016). Both the cGcC and G4H are limited by their consideration of G and C nucleotides alone. The absence of C in sequences strongly increases the cGcC score. The substitution of a single nucleotide to C decreases the score by ∼10-fold while one C alone is not considered to be relevant to interfere with G runs. Both the cGcC and G4H tools are not designed to cope with exceptions. Some G rich sequences were reported not to fold in G4, while on the other hand G4 presenting bulges broaden sequence requirements reducing the importance of consecutive G. To improve the identification of G4, we chose to consider both the required sequence, which is currently undefined, and its flanking sequences, using a machine learning approach trained considering unusual G4.
To undertake this endeavor, we first implemented the G4RNA database, which aims to host available RNA sequences investigated for G4 folding, whether the outcome of the experiment was positive or negative (Garant et al., 2015). Exploring the data manually failed to expose an intuitive way to classify or discriminate sequences. Thus, to learn from the rich G4RNA data, we chose to submit them to a machine learning algorithm to explore the ability of such an approach to classify and extrapolate this classification logic and ultimately identify potential G4 in human transcripts. Machine learning has often been used to identify sequence elements in genomic data (Libbrecht and Noble, 2015). Our hypothesis is that machine learning, with its use of combinatorial representations of variables to resolve complex situations, would be able to resolve the minimal features needed in a sequence to observe a G4 and/or the combination of features that would prevent the folding of a G4. Following extensive comparison of performance with comparable tools that do not rely on motif search, we are releasing G4RNA screener, a comprehensive software to cope with the need of potential G4 identification.
2 Results and discussion
The identification of potential G4 can be transposed computationally as a classification problem in which each sequence can be categorized as either a positive (G4) or negative (non-G4) case. We propose a new score based on abstract sequence similarity, computed by a simple artificial neural network (ANN) named G4NN which was trained on the sequences of the G4RNA database. G4NN was built to provide G4 detection without a definition of the motif and to minimize bias from experts’ assumptions. G4NN learns from available examples and considers irregular G4 just as it does canonical G4. The ANN takes the trinucleotide composition of a window as its input, which translates to an abstract representation of the sequence. By providing the composition of all trinucleotides, we do not bias the classifier in considering specific trinucleotides as more important than any other. The implementation set used for training consists of 149 G4 and 179 non-G4 sequences with folding outcome validated and 200 sequences randomly taken in the transcriptome (O’Leary et al., 2016) (Fig. 1). The architecture was optimized using a 5-fold cross-validation strategy while monitoring the receiver operating characteristic (ROC) curve to appreciate the classification power of G4NN. We reached an average area under the ROC curve (AUC) of 0.953 ± 0.013 and 0.928 ± 0.024 for the training sets and test sets respectively on our last iteration of optimization (Fig. 1).
Fig. 1.

Implementation of G4NN. (A) Sequence management to produce both the implementation set and independent test set. (B) Five-fold cross-validation strategy used to assess performance during optimization of the ANN architecture and learning parameters. ROC curve average AUC values for the optimal configuration are shown with standard deviation. (C) Illustration of the chosen architecture. (D) Classification performance of the implementation set when erroneous data are introduced by changing labels in the implementation set. The standard deviation is shown using error bars
Cross-validation usage facilitates the observation of over-training and over-fitting behavior but it does not prevent it. We chose to investigate further since the AUC values were very high and stable throughout the iterations. We validated that the classification power of G4NN relies on G4-related generalization by randomly switching an increasing number of the labels in the training set. The rationale behind this test is that an increasing number of randomly labelled examples should lead to a decrease in the accuracy of the predictor. G4NN lost its classification power linearly until half of the input data were randomly switched (G4, non-G4), at which point the classification is random (AUC = 0.5), as shown in Figure 1D.
Once G4NN was built, we compared our classification strategy to the previously used scoring systems by integrating them all in a new tool. With increasing reports of non-canonical G4 structures, requiring a recurrent redefinition of the G4 motif, we propose G4RNA screener to sift through RNA sequences and produce a profile of sequences as described by their cGcC, G4H and G4RNA score. The G4RNA screener also provides a means to compare the three available G4RNA predictors.
We first applied the G4RNA screener on the sequences from the G4RNA database using a sliding window of 60 nucleotides (nt) moving with steps of 10 nt to mimic a genuine search of potential G4s. Using the maximum value obtained for each score in the region of the known G4, we assessed their respective classification power as described by their ROC curve. Unsurprisingly G4NN displays a good performance on the implementation set sequences since this is the set on which it was trained. The G4H score provides a comparable performance, while its predecessor, the cGcC score, has a slightly lower prediction power (Fig. 2A). G4NN and cGcC have a similar pattern on the independent test set, which consists of sequences that were not included in the development of G4NN (Fig. 2B). We observe a lower classification power for G4H in the independent test set, showing that while evaluating similarly the G and C content, the cGcC and G4H scores can provide distinct insights on a sequence. Even though G4H was designed mainly for DNA (Bedrat et al., 2016) we confirm here its relevance for RNA, justifying its inclusion in G4RNA screener.
Fig. 2.
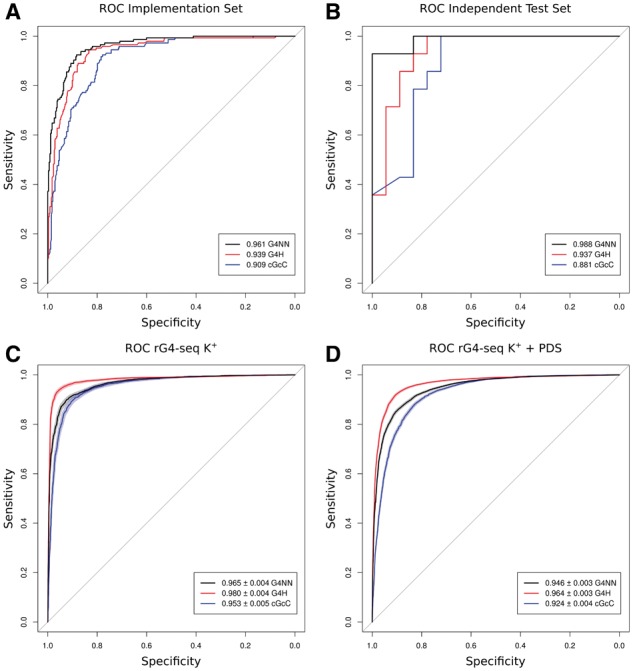
ROC curves of scores for each available dataset. (A, B) Classification performance on the implementation set (A) and independent test set (B) by G4NN (black), G4Hunter (red) and cGcC (blue). The AUC values are provided in legend. (C, D) Classification of the rG4-seq K+ (C) and rG4-seq K+ stabilized by PDS (D) datasets by G4NN (black), G4Hunter (red) and cGcC (blue). The AUC values provided in the legend are indicated with 95% confidence intervals (shading on the curves) computed by stratified bootstrap
In 2016, the rG4-seq method was introduced by Kwok and colleagues (Kwok et al., 2016), providing an approach to identify RNA G4s by high-throughput sequencing. The rG4-seq method is designed to capture the K+ dependent stalling of the reverse transcriptase when compared to Li+. The K+ dependent stalling is likely to directly precede a G4 since stalling occurs when the reverse transcriptase encounters a stable G4 (Kwok and Balasubramanian, 2015). The rG4-seq provides an interesting high-throughput independent dataset to compare the three RNA G4 predictors. We retrieved the hits from the Gene Expression Omnibus (GSE77282) and ran our tool on the sequences of the genes where a stalling was detected. We produced ROC curves using the maximum value of each score on the rG4-seq hits (Fig. 2C). G4NN yields good classification, however rG4-seq data was best classified by G4Hunter both with and without the usage of pyridostatin (PDS) as a G4 stabilizer (Fig. 2C, D). The lower performance of G4NN compared to G4H on this dataset is likely due to the fact that some rG4-seq identified G4 present important differences compared to the sequences on which G4NN was trained. This demonstrates the need to further characterize unusual G4 structures and include their sequences in a forthcoming training and update of G4NN.
To characterize further the three predictors, we plotted the sensitivity and specificity of each scoring methods (Fig. 3A–C). The sensitivity curve (descending curve) meets the specificity curve (increasing curve) at the score threshold that minimizes the number of false positives and false negatives for a given dataset. The G4NN score has an optimal threshold for G4RNA data at 0.5 which is consistent with the knowledge that it was trained to classify these sequences between 1 (G4) and 0 (not G4) (Fig. 3A). The G4H optimal threshold to classify G4RNA’s data is ∼0.9 (Fig. 3B) which is close to the recommended threshold of 1 for DNA (Bedrat et al., 2016).
Fig. 3.

Classification of datasets displayed using the G4NN, G4H and cGcC scores. (A–C) Specificity (positive slope) and sensitivity (negative slope) for the implementation set (black), rG4-seq K+ set (red) and rG4-seq K+ with PDS set (blue) as described by each score. The ninety-five percent confidence intervals computed by stratified bootstrap are provided in shading. (D–F) Datasets broken down and distributions of scores shown as boxplots. The implementation set (black) is split in three subsets; validated G4 from G4RNA, validated non-G4 from G4RNA and random transcriptomic sequences, all of which were used for the training of G4NN. rG4-seq K+ data (red) and rG4-seq K+ with PDS data (blue) are both split to show G4 hits and random transcriptomic sequences used to generate the curves. (G, H) Euler’s graph of either rG4-seq K+ hits or rG4-seq K+ with PDS hits (yellow) and the proportion predicted above the thresholds for each method; >0.5 G4NN (red), >0.9 G4H (blue), >4.5 cGcC (green)
We observed lower optimal thresholds to classify rG4-seq data than G4RNA data when considering each score, in accordance with the presence of non-G4 sequences in G4RNA that are very similar to G4 sequences. To illustrate this phenomenon, we drew the distribution of scores of each dataset, plotting G4 separately from non-G4 and from randomly chosen transcriptomic sequences (Fig. 3D–F). G4RNA data are more challenging to classify than rG4-seq data since their non-G4 sequences are more similar to G4 than the transcriptomic background. Non-G4 sequences were experimentally challenged and reported in the literature. Generally, they are either G4 sequences mutated at very specific positions to be used as negative control of folding, or sequences that were investigated but were shown not to adopt a G4 (Garant et al., 2015). G4NN is therefore the most efficient classifier when considering G4RNA data only (Fig. 3D–F, lanes 1 and 2) since the G and C content of non-G4 and G4 sequences are alike. G4NN can rely on the other nucleotides to have a more complete outlook of the sequence. rG4-seq provides only G4 sequences detected as hits, therefore their classification from all non-hits is efficiently performed using a lower threshold. It also explains the steep increase of the specificity of the ANN for rG4-seq hits compared to G4RNA data, reaching ∼90% of specificity with a score as low as 0.12 (Fig. 3A). The G4NN score is efficient at discarding random transcriptomic sequences by attributing a low score to them. The sensitivity of the G4NN score for rG4-seq data decreases in an almost linear manner which is consistent with the large diversity of sequences in rG4-seq hits (Fig. 3A).
We consider the validated non-G4 sequences from G4RNA to be relevant for the determination of thresholds when using the G4RNA screener as a tool to identify new potential G4. The contribution of each score to detect rG4-seq data was evaluated using Euler graphs with the following thresholds: 0.5 G4NN score, 0.9 G4Hunter score, 4.5 cGcC score (Fig. 3G, H). Since these thresholds are quite stringent as shown by the decrease of sensitivity, these graphs illustrate well the relationship between the scores. Considering rG4-seq K+ hits only (Fig. 3G), the three scores agree on the prediction of 53.2% sequences. Unsurprisingly, 95.0% of G4Hunter detected hits are also found in cGcC detected hits, consistent with these two predictors evaluating the G richness in a very similar way. rG4-seq hits detected by G4NN only are likely to be G4 sequences similar to the ones described previously but with a G/C skewness that would have been evaluated as unfavorable to G4 folding. In contrast, rG4-seq hits detected by G4Hunter and cGcC scores, but not by G4NN score, are likely to be G4 sequences rich in G but presenting a sequence different from the ones described so far. However, we are not able to evaluate the number of rG4-seq hits that are false signals, i.e. K+ dependent stalling not caused by G4 structure. For pyridostatin (PDS) stabilized rG4-seq hits, fewer hits are scored above the thresholds and fewer hits are shared between the scores (Fig. 3H). Our hypothesis is that the shared hits between K+ alone and K+ with PDS conditions are predicted the same way by the scores, they represent the predictable hits. While a large proportion of PDS dependent hits are sequences that are different from our understanding of potent G4 folding sequences, we lack knowledge about those ligand-dependent hits to properly address them. All three scores were designed to identify G4 occurrences in the absence of ligands, therefore we refrain from drawing conclusions.
3 Methods and implementation
3.1 Implementation data
G4RNA is a very inclusive database, useful to track all experiments performed on a particular sequence and all publications in which the sequence was investigated. It also holds sequences that present an experimental outcome that is either ambiguous or conflicting when compared to another experiment. However, such extensive data presents redundancy which must be taken into consideration to use as training data for a machine learning algorithm. Two filters were used; a uniqueness filter which retains the smallest sequence from duplicates and a length filter discarding all sequences longer than 300 nt. The length filter was required since some sequences in G4RNA are complete 5′ untranslated regions that are several hundred nucleotides long. We were concerned that long sequences would not be useful to determine the features associated with G4 since the G4 represents a short fraction of the overall sequence. The features associated with the G4 would be weakened by the much larger flanking sequence.
From the 590 sequences that were first available, 368 were conserved after filtering. The 40 sequences (∼11%) reported most recently in the literature were kept as an independent test to appreciate the final performance of our tool (Fig. 2B). The remaining 328 sequences, with an average length of 63 nt and median length of 57 nt, were used for implementation and optimization of the tool along with 200 sequences of 60 nt in length randomly obtained from the transcriptome (Fig. 1A). The random sequences were retrieved from RefSeq accessed through the UCSC RefGene database (O’Leary et al., 2016). They are essential in order for the tool to be trained on background sequences as well as the experimentally tested sequences. Overall, this dataset comprised 149 confirmed G4 folding sequences, 179 confirmed non-G4 sequences and 200 randomly chosen sequences assumed to be non-G4 and are referred to as the implementation set (Supplementary Table S1).
There are many similar sequences in the implementation set since most wild-type sequences experimentally challenged were compared to slightly mutated versions of the sequence. While similar data are usually discarded to reduce bias in the training of machine learning classifiers, we chose to keep sequences that were very similar since most of them are wild-type sequences with only few nucleotides changed in their mutated counterpart. The mutated sequences present minimalistic mutations to impede G4 folding. We believe that those sequences actually present critical information for our tool to learn.
3.2 Artificial neural network design and optimization
G4NN was implemented using the PyBrain library from the python programming language (Schaul et al., 2010). Sequences were provided to the algorithm as vectors of their tri-nucleotide content. These 64 combinations of nucleotides were the features used as input for the ANN. G4NN has a very simple architecture with a single hidden layer and full connection between nodes of each layer (Fig. 1C). The objective is to obtain a tool that would have generalization power rather than a deep learning architecture with greater classification power. The architecture and various learning parameters of the ANN were optimized using a 5-fold cross-validation strategy in an iterative process where the values of the parameters are gradually changing (Fig. 1B). To do so, the implementation set was split into five non-overlapping sets of equal size. Four of these sets were used as a training set and the fifth set was used as a test. The ANN is trained using half the data from the training set while the other half is used as validation to determine when training must end. The performance was evaluated on both the training set and the test set. The training using the implementation set was done five times, each time using a different combination of training and test sets (Fig. 1B). The final architecture and learning parameters were chosen where the classification performance was kept at its highest and the computational requirements were reasonable. The classification performance was monitored using the area under ROC curve (AUC) and computational requirement was monitored using memory usage, CPU usage and computing time to train.
This optimal ANN was achieved using full connections between the 64 input nodes and 35 hidden nodes through a switch sigmoid activation layer and the application of a sigmoid squashing function on the output layer. Its training was performed using a resilient back propagation algorithm and by using evenly the sequences for training and validation at random. Through iterations, weights between nodes were gradually changing until it reached convergence with minimal validation error. The average AUC from the cross-validation for the training values using the optimal architecture was 0.953 ± 0.013 (stdev) and the AUC for the corresponding test values was 0.928 ± 0.024 (Fig. 1B).
With such good classification power from the tool and knowing that there is similarity between G4 folding sequences and non-folding sequences, we investigated whether G4NN is overtrained or overfitted. Overtraining and overfitting happens when a classifier is able to classify all values without generalization, registering the training values individually with their outcome. In order to address the issue, we chose to purposely induce errors in the implementation set by gradually permuting the labels (G4 or non-G4) of the sequences at random and restarting the cross-validation procedure. Overtraining, overfitting or classification not related to G4 would be suggested if the architecture of the tool allows good classification of scrambled data, while a tool relying on generalization would not be able to classify correctly those erroneous data. Our neural network architecture lost its classification power as the proportion of permuted labels increased up to half of the data (Fig. 1D). This convinced us that the classification power of our tool was relying on G4 related generalization (Ojala and Garriga, 2009).
3.3 G4RNA screener
We wrapped our new G4NN together with the previously established cGcC scoring system and the newly described G4Hunter in a single tool, as the G4RNA screener. The program is written in python and is available at http://gitlabscottgroup.med.usherbrooke.ca/J-Michel/g4rna_screener along with its documentation and manual. The repository hosts the program in its most user friendly form, i.e. without the training and validation codes. Those are available in the more inclusive development repository http://gitlabscottgroup.med.usherbrooke.ca/J-Michel/g4rna_screener_dev. The G4RNA screener runs from the terminal, the input is passed as arguments and the results of the analysis are displayed in the standard output as a tab delimited values file by default (.tsv) or BEDGRAPH file (.bedgraph). It can easily be implemented as part of a large analysis pipeline.
G4RNA screener was used to mimic a genuine search for G4 on the implementation set and the independent test set. Using 60 nt long windows with steps of 10 nt, we analyzed all sequences and used the maximum value to compare with the label associated with the sequence. We then plotted the ROC curves of each dataset using the three scores available in G4RNA screener to observe their classification power (Fig. 2A, B).
3.4 Validation using rG4-seq high-throughput data
We performed the same genuine search for G4 previously described on the sequence of transcripts where a rG4-seq hit was detected. We used chromosomal positions of rG4-seq hits provided in the BED files available from the gene expression omnibus (GSE77282) and retrieved sequences corresponding to the position in transcripts from the RefSeq database through the UCSC table browser. We then used the maximal value obtained in windows overlapping the positions of the hits to plot ROC curves. Random transcriptomic sequences that did not overlap with rG4-seq hits were picked as negative values to plot the curves (Figs 2C, D and 3A–C) and their distributions are shown (Fig. 3D–F).
4 Conclusion
G4RNA screener provides a reliable way to identify potential RNA G4. It includes the tools developed so far that are not limited by a motif definition of the G4. G4NN, one of the tools included in G4RNA screener, is a novel machine learning approach trained on sequences that were investigated experimentally in previous studies. Its abstract representation of the sequence can be used along with previously developed G richness-based predictors to evaluate how a sequence relates to the known G4 of G4RNA database. G4NN could be trained again easily to keep pace with the new G4 that are described. The G4RNA screener repository is available at http://gitlabscottgroup.med.usherbrooke.ca/J-Michel/g4rna_screener.
Supplementary Material
Acknowledgements
The authors are grateful to members of their groups for helpful comments.
Funding
JMG is the recipient of a Natural Sciences and Engineering Research Council of Canada (NSERC) graduate scholarship. JPP holds the Chaire de recherche de l’Université de Sherbrooke en Structure et Génomique de l’ARN. MSS holds a Fonds de Recherche du Québec – Santé (FRQS) Research Scholar Junior 2 Career Award. Both JPP and MSS are members of the Centre de Recherche du CHUS. The project was supported by funding from the Fonds de recherche Nature et technologies (FQRNT) [to JPP] and from NSERC [to MSS].
Conflict of Interest: none declared.
References
- Agarwala P. et al. (2015) The tale of RNA G-quadruplex. Org. Biomol. Chem., 13, 5570–5585. [DOI] [PubMed] [Google Scholar]
- Beaudoin J.-D. et al. (2014) New scoring system to identify RNA G-quadruplex folding. Nucleic Acids Res., 42, 1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedrat A. et al. (2016) Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res., 44, 1746–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi G. et al. (2014) Visualization and selective chemical targeting of RNA G-quadruplex structures in the cytoplasm of human cells. Nat. Chem., 6, 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy J., Maizels N. (2006) Gene function correlates with potential for G4 DNA formation in the human genome. Nucleic Acids Res., 34, 3887–3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faudale M. et al. (2009) Photoactivated cationic alkyl-substituted porphyrin binding to g4-RNA in the 5′-UTR of KRAS oncogene represses translation. Chem. Commun. Camb. Engl., 48, 874–876. [DOI] [PubMed] [Google Scholar]
- Garant J.-M. et al. (2015) G4RNA: an RNA G-quadruplex database. Database, 2015, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppert L.J., Balasubramanian S. (2005) Prevalence of quadruplexes in the human genome. Nucleic Acids Res., 33, 2908–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jodoin R. et al. (2014) The folding of 5′-UTR human G-quadruplexes possessing a long central loop. RNA, 20, 1129–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikin O. et al. (2006) QGRS Mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res., 34, W676–W682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok C.K. (2016) Dawn of the in vivo RNA structurome and interactome. Biochem. Soc. Trans., 44, 1395–1410. [DOI] [PubMed] [Google Scholar]
- Kwok C.K., Balasubramanian S. (2015) Targeted Detection of G-Quadruplexes in Cellular RNAs. Angew. Chem. Int. Ed., 54, 6751–6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok C.K. et al. (2016) rG4-seq reveals widespread formation of G-quadruplex structures in the human transcriptome. Nat. Methods, 13, 841–844. [DOI] [PubMed] [Google Scholar]
- Lai D. et al. (2013) On the importance of cotranscriptional RNA structure formation. RNA, 19, 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libbrecht M.W., Noble W.S. (2015) Machine learning in genetics and genomics. Nat. Rev. Genet., 16, 321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R. et al. (2013) 2D meets 4G: G-quadruplexes in RNA secondary structure prediction. IEEE/ACM Trans. Comput. Biol. Bioinform., 10, 832–844. [DOI] [PubMed] [Google Scholar]
- Malgowska M. et al. (2016) Overview of the RNA G-quadruplex structures. Acta Biochim. Pol., 63, 609–621. [DOI] [PubMed] [Google Scholar]
- Martadinata H., Phan A.T. (2014) Formation of a stacked dimeric G-quadruplex containing bulges by the 5′-terminal region of human telomerase RNA (hTERC). Biochemistry (Mosc.), 53, 1595–1600. [DOI] [PubMed] [Google Scholar]
- Menendez C. et al. (2012) QGRS-H Predictor: a web server for predicting homologous quadruplex forming G-rich sequence motifs in nucleotide sequences. Nucleic Acids Res., 40, W96–W103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojala M., Garriga G.C. (2009) Permutation tests for studying classifier performance. In 2009 Ninth IEEE International Conference on Data Mining Presented at the 2009 Ninth IEEE International Conference on Data Mining, pp. 908–913.
- O’leary N.A. et al. (2016) Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res., 44, D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey S. et al. (2015) The RNA stem–loop to G-quadruplex equilibrium controls mature MicroRNA production inside the cell. Biochemistry (Mosc.), 54, 7067–7078. [DOI] [PubMed] [Google Scholar]
- Rouleau S. et al. (2017) RNA G-Quadruplexes as Key Motifs of the Transcriptome. Adv. Biochem. Eng. Biotechnol [DOI] [PubMed] [Google Scholar]
- Schaul T. et al. (2010) PyBrain. J. Mach. Learn. Res., 11, 743–746. [Google Scholar]
- Sharp P.A. (2009) The centrality of RNA. Cell, 136, 577–580. [DOI] [PubMed] [Google Scholar]
- Warner K.D. et al. (2014) Structural basis for activity of highly efficient RNA mimics of green fluorescent protein. Nat. Struct. Mol. Biol., 21, 658–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.