

Abstract
Regulation of microRNAs (miRNA) has been extensively investigated in diseases; however, little is known about the roles of miRNAs in cleidocranial dysplasia (CCD). The aim of the present study was to investigate the potential involvement of miRNAs in CCD. In vitro site-directed mutagenesis was performed to construct three mutant Runx2 expression vectors, which were then transfected into LS8 cells and MC3T3-E1 cells, to determine the impact on amelogenesis and osteogenesis, respectively. miRCURY LNA miRNA microarray identify miR-185-5p as a miRNA target commonly induced by all three Runx2 mutants. Real-time quantitative PCR was applied to determine the expression of miR-185-5p and Dlx2 in samples. Dual-luciferase reporter assays were conducted to confirm Dlx2 as a legitimate target of miR-185-5p. The suppressive effect of miR-185-5p on amelogenesis and osteogenesis of miR-185-5p was evaluated by RT-PCR and western blot examination of Amelx, Enam, Klk4, and Mmp20 gene and protein expression, and by Alizarin Red stain. We found that mutant Runx2 suppressed amelogenesis and osteogenesis. miR-185-5p, induced by Runx2, suppressed amelogenesis and osteogenesis. Furthermore, we identified Dlx2 as direct target of miR-185-5p. Consistently, Dlx2 expression was inversely correlated with miR-185-5p levels. This study highlights the molecular etiology and significance of miR-185-5p in CCD, and suggests that targeting miR-185-5p may represent a new therapeutic strategy in prevention or intervention of CCD.
Introduction
Cleidocranial dysplasia (CCD; OMIM# 119600) is a rare congenital disorder which follows an autosomal dominant mode of inheritance. The main causes of CCD are heterozygous mutations (e.g., gene insertions and deletions) in the RUNT-related transcription factor 2 (RUNX2) gene, which is located on chromosome 6p21. The clinical manifestations of CCD are mainly related to the bones and teeth, presenting as abnormal clavicles, patent sutures and fontanelles, short stature, supernumerary teeth, delayed tooth eruption, enamel hypoplasia, and the absence of cellular cementum formation1–3.
The RUNX2 protein has several functional domains: a Runt domain, a nuclear localization signal (NLS), a Q/A domain, a proline-serine-threonine rich (PST) domain, a VWRPY motif, and a nuclear matrix targeting sequence (NMTS). The Runt domain interacts with CBFβ and actively binds to highly conserved DNA sequences4. Mutational analysis from patients with CCD identified many mutations in the Runt domain, indicating the importance of this domain to RUNX2 function5. The RUNX2 NLS is located at the C-terminal end of the Runt domain and plays a critical role in the nuclear transportation of the protein. The Q/A domain, a stretch of polyglutamine and polyalanine repeats, is located in the N-terminus of the protein, and the length of the Q or A regions is important for the transcriptional activity of RUNX2. The PST domain comprises the C-terminal half of the protein in which these three amino acids are enriched. Deletion of the PST domain by nonsense mutation or alternative splicing results in loss of function of the RUNX2 protein and causes CCD6. The VWRPY motif is highly conserved, and is located in the last five amino acids of the C-terminal end of all known runt-related proteins. In Drosophila, VWRPY interacts with groucho and leads to the transcriptional repression of runt7. No mutations have been reported in this domain. The NMTS is a highly conserved 26 amino acid domain that determines the subnuclear localization of the RUNX2 protein; this domain has binding affinity with several important nuclear proteins, including Smads, p300, and histone deacetylase8,9.
Mutations in RUNX2 have a high penetrance and extreme variability, ranging from isolated dental anomalies to the full manifestation of the CCD disease, exemplified by a poorly ossified cranium and the absence of clavicles. A number of patients with typical features of CCD do not have an identifiable mutation in the coding region, the splice sites, or the promoter region of RUNX2. The phenotype may vary greatly among individuals even in the same family with the same mutation, and no clear phenotype–genotype correlation has been established5,10. The precise biochemical and molecular mechanisms by which mutations in RUNX2 lead to CCD remain unclear.
MicroRNAs (miRNAs) are endogenous, small, noncoding RNAs, about 22 nucleotides in length, that regulate gene expression by binding to the 3′ untranslated region (3′UTR) of mRNAs to repress translation or induce mRNA cleavage11. miRNAs are involved in the regulation of a variety of biological events, including organ development, tumorigenesis, cell proliferation, and apoptosis12.
RUNX2 may regulate the expression of miRNAs that participate in the development of bones and teeth. Disruptions in the normal regulation of these miRNAs, such as through mutated RUNX2, may result in a corresponding adjustment effect, leading to the development of CCD and the manifestation of clinical symptoms. In this study, we report that altered expression of miRNAs associated with over-expression of mutant Runx2 affects amelogenesis and osteogenesis and we further clarify the role of miR-185-5p in the processes of amelogenesis and osteogenesis. We focus on three RUNX2 point mutations that were identified from CCD patients; R190W13x, R225Q14, and A362V4. These three mutations are located in the RUNX, NLS, and PST domains, respectively, and were selected to transfer into LS8 and MC3T3-E1 cells for the study (Supplementary Figures).
Results
Mutant Runx2 regulates amelogenesis and osteogenesis
We transfected cells with pcDNA vector as the mock group and wild-type Runx2 plasmid as the positive control group. GFP expression was observed in almost all infected cells (Fig. 1a, b). We examined the expression of two important enamel matrix proteins, amelogenin and enamelin, in murine LS8 cells after over-expression of Runx2 mutants (R190W, R225Q, A362V). The mRNA levels of amelogenin and enamelin were decreased in Runx2 mutant groups compared with the control group. Similarly, western blot analysis demonstrated decreased protein level of amelogenin and enamelin in cells with over-expression of mutant Runx2 (Fig. 1c, d).
Fig. 1. Mutant Runx2 decreases amelogenesis, and osteogenesis.

a, b Images of GFP expression in infected LS8 a and MC3T3-E1 b were photographed by fluorescent microscope. The mRNA and protein levels of enamelin c, amelogenin d, ALP e and Ocn f were decreased in mutant Runx2 groups compared with the control group; mRNA levels indicated by gel analysis of RT-PCR reactions; protein levels indicated by western blot. ALP staining shows the ALP activities in cells transfected with mutant Runx2 were significantly suppressed compared to ALP activity in control cells (G, top); these are in agreement with the results of Alizarin red staining (G, bottom)
Expression of ALP and osteocalcin (OCN) were reduced by transfection with mutant Runx2, both mRNA and protein levels were significantly decreased in cells with mutant Runx2 (Fig. 1e, f). Reduction in ALP activity was in agreement with the results of Alizarin red staining (Fig. 1g).
miR-185-5p is upregulated by mutant Runx2
miRNA microarray identified the miRNAs that were differentially expressed between LS8 or MC3T3-E1 cells transfected to over-express mutant or wild type Runx2. mmu-miR-185-5p (miR-185-5p) was the only miRNA that was significantly upregulated in all three mutant Runx2 transfection groups vs. control across both cell lines (data not shown).
The results of the miRNA microarray were validated by quantitative RT-PCR analysis of miR-185-5p expression in mutant and control Runx2 groups (Fig. 2). The data indicated that expression of miR-185-5p was up-regulated in LS8 and MC3T3-E1 cells transfected with all the three mutant Runx2 compared to the control group.
Fig. 2. miR-185-5p is upregulated by mutant Runx2.
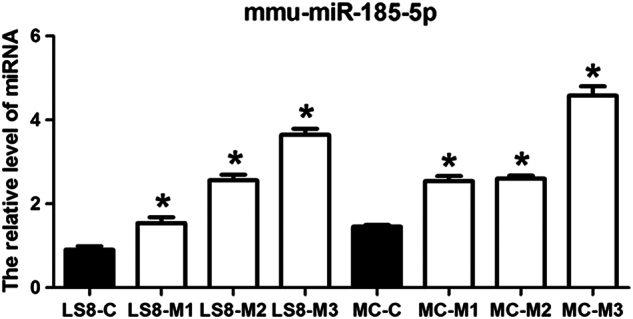
miR-185-5p is upregulated in LS8 and MC3T3-E1 cells transfected with mutant Runx2. miR-185-5p expression levels are indicated relative to U6 RNA
miR-185-5p expression changes during ameloblast differentiation and osteogenesis
To determine whether miR-185-5p expression relates to ameloblast differentiation and osteogenesis, expression kinetics of miR-185-5p were examined during ameloblast differentiation of LS8 cells and osteoblast differentiation in MC3T3-E1 cells. The expression of miR-185-5p was down-regulated during ameloblast differentiation over a 4-day time course of retinoic acid and dexamethasone (RA/DEX) treatment in LS8 cells (Fig. 3a). miR-185-5p expression was also reduced during osteoblast differentiation induced by 21-day treatment of MC3T3-E1 cells with GIBCO® STEMPRO (Fig. 3b). miR-185-5p expression was lowest on 4th day in LS8 cells undergoing ameloblast differentiation and in 14 days in MC3T3-E1 cells undergoing osteoblast differentiation (Fig. 3a, b). These results suggest that miR-185-5p may be involved in the ameloblast differentiation of LS8 cells induced by RA/DEX and the preosteoblast differentiation of MC3T3-E1 cells induced by STEMPRO.
Fig. 3. Dynamics of miR-185-5p expression during ameloblast differentiation in LS8 cells or osteoblast differentiation in MC3T3-E1 cells.

The expression level of miR-185-5p was down-regulated during the ameloblast differentiation of LS8 cells a and osteoblast differentiation in MC3T3-E1 cells b
miR-185-5p inhibits ameloblastic differentiation of LS8 cells
To determine whether miR-185-5p could affect ameloblast differentiation, miR-185-5p mimics or miRNA control (miR-NC) were transfected into LS8 cells, followed by RA/DEX treatment for 3 days. Then, the mRNA and protein levels of Amelx, Enam, Klk4, and Mmp20 were investigated. Expression of Amelx, Enam, Klk4, and Mmp20 was significantly lower in cells transfected with miR-185-5p mimic compared with those transfected with miR-NC (Fig. 4). Additionally, miR-185-5p transfection inhibited mRNA and protein expression of Amelx, Enam, Klk4, and Mmp20 in a dose-dependent manner, suggesting that miR-185-5p may act as a negative regulator of early amelogenesis induced by RA/DEX stimulation.
Fig. 4. miR-185-5p negatively regulates ameloblast differentiation.

LS8 cells were transfected with 50 or 100 nM miR-185-5p mimic or miR-NC for 24 h and then ameloblast differentiation was stimulated with RA/DEX for another 2 days. The cells transfected with control miR-NC was designated as negative control. The mRNA and protein expression of Amelx a,b, Enam c,d, Klk4 e,f, and Mmp20 g,h were detected by quantitative RT-PCR and western blot, respectively. Data are representative images or expressed as mean ± S.E. of each group of cells from three separate experiments, and the values of the control cells are designated as 1. * p < 0.05
miR-185-5p inhibits osteoblast differentiation
To address the functional activity of miR-185-5p in osteoblast differentiation, we examined the effects of miR-185-5p over-expression in preosteoblast MC3T3-E1 cells. MC3T3-E1 cells were transfected with miR-185-5p mimic or miRNA control for 48 h until cells cultures became confluent, after which time osteoblast differentiation was induced by STEMPRO treatment for 7 days.
ALP activity increases in a time-dependent manner in MC3T3-E1 cells after induction of osteoblast differentiation. Following osteoblast differentiation, the ALP activities in the transfected cells were investigated. ALP activity was significantly lower in MC3T3-E1 cells transfected with miR-185-5p compared with ALP activity in miR-NC transfected cells (Fig. 5a, b). Additionally, miR-185-5p inhibited ALP expression levels in a dose-dependent manner (Fig. 5c, d). These results suggested that miR-185-5p acts as a negative regulator of early osteogenesis in MC3T3-E1 cells induced by STEMPRO stimulation.
Fig. 5. miR-185-5p negatively regulates osteogenic differentiation.

a ALP activity in 100 nM miR-185-5p-transfected cells. b ALP mRNA expression in 50 or 100 nM miR-185-5p-transfected cells. c ALP protein expression in 50 or 100 nM miR-185-5p-transfected cells. d ALP staining in 50 or 100 nM miR-185-5p-transfected cells. Microscopic view of the effects of miR-185-5p on matrix mineralization in MC3T3-E1 cells. Shown is a representative microscopic view at a magnification of 200× of cells transfected with miR-185-5p or miR-NC after 28 days of culture. e Quantification of Alizarin Red S stain via extraction with cetylpyridinium chloride. f Levels of Ocn mRNA were determined using qRT-PCR and are shown as fold induction relative to control. g Ocn protein expression was determined by western blot and expressed as densitometry of Ocn/β-actin. Data are shown as means ± SD. *p < 0.05 vs. miR-NC, n = 3. Data are representative images or expressed as mean ± S.E. of each group of cells from three separate experiments, and the values of the control cells are designated as 1. *p < 0.05
To elucidate the role of miR-185-5p during terminal osteogenic differentiation, MC3T3-E1 cells were transfected with miR-185-5p or miR-NC, and osteoblastic differentiation was induced by STEMPRO for 28 days. Alizarin Red S stain and expression of OCN were measured at 24 or 28 days. Compared with control cells, transfection of miR-185-5p inhibited mineralized nodule formation in 28-day cultures (Fig. 5d, e). OCN mRNA and protein levels were also reduced by transfection of miR-185-5p, reflecting decreased differentiation at both time points (Fig. 5f, g). These findings indicate that miR-185-5p negatively regulates osteogenic differentiation.
Dlx2 is a direct target of miR-185-5p
To identify the miR-185-5p target genes in amelogenesis and osteogenesis, we searched for candidate genes using the miRNA target prediction database MirBase. miR-185-5p was predicted to target Dlx2, which is a key downstream mediator of enamel and bone formation15–17. There is one predicted miR-185-5p binding site in the 3′ UTR of Dlx2 (Fig. 6a). During the ameloblast differentiation, mRNA and protein levels of Dlx2 was barely detected in the LS8 cells without RA/DEX treatment. The expression level of Dlx2 was gradually upregulated during the amelogenic induction (Fig. 6b). The same expression pattern of Dlx2 was observed in the MC3T3-E1 cells (Fig. 6c). Knockdown of Dlx2 in LS8 inhibited Mmp20 and Klk4 after 2 days amelogenic induction (Fig. 6d). Knockdown of Dlx2 in MC3T3-E1 cells had no obvious effect on the expression of Alp in the uninduced group (Fig. 6e). However, after 7 days of osteogenic induction with STEMPRO, knockdown of Dlx2 could inhibit Alp expression obviously (Fig. 6f).
Fig. 6. iR-185-5p inhibits Dlx2 expression.

ma Schematic of the miR-185-5p putative target site in mouse Dlx2 3′ UTR. b Expression level of Dlx2 during the amelogenic differentiation of LS8 cells. c Expression change of Dlx2 during the osteogenic differentiation of MC3T3-E1 cells. d Knockdown of Dlx2 in LS8 cells treated with RA/DEX for 48 h inhibited the expression of Mmp20 and Klk4. e Knockdown Dlx2 in MC3T3-E1 cells at day 0 of osteogenic induction had no obvious effect on the expression of Alp. f Expression of Alp on day 7 of osteogenic induction was inhibited by si-Dlx2 in MC3T3-E1 cells. g Putative miR-185-5p-binding sequence in the 3′ UTR of Dlx2 mRNA. Mutations were generated in the Dlx2 sequence in the complementary site for the seed region of miR-185-5p as indicated. h Normalized luciferase activity from Dlx2 3′ UTR reporter vector containing a wild-type or mutant miR-185-5p. i and j Relative Dlx2 mRNA and protein expression, determined by RT-PCR and western blot, respectively, in LS8 cells or MC3T3-E1 cells transfected with 50 nM miR-NC, miR-185-5p mimic, or miR-185-5p inhibitor. RNA and protein were harvested 2 days after transfection. Cells transfected with control miR-NC were designated as negative control. Data are representative images or expressed as mean ± S.E. of each group of cells from three separate experiments, and the values of the control cells are designated as 1. *p < 0.05, **p < 0.01
To examine whether miR-185-5p could directly regulate Dlx2 expression, HEK293 cells were transfected with luciferase reporter constructs under transcriptional control of the wild-type Dlx2 3′ UTR or mutant Dlx2 3′UTR, together with the miR-185-5p mimic or non-targeting control (miR-NC) (Fig. 6g). miR-185-5p significantly inhibited the activity of the wild-type reporter gene, whereas mutation in the seed site completely abolished repression by exogenous miR-185-5p (Fig. 6h), demonstrating that miR-185-5p regulates Dlx2 gene expression by directly binding to the predicted seed site within its 3′ UTR.
To determine whether Dlx2 is regulated by miR-185-5p in amelgogenic or osteogenic differentiation, LS8 cells or MC3T3-E1 cells were transfected with miR-185-5p mimics or inhibitors. Dlx2 protein expression decreased when miR-185-5p increased, indicating that miR-185-5p is an important negative regulator of Dlx2 (Fig. 6i, j). Furthermore, quantitative RT-PCR assays demonstrated that Dlx2 mRNA did not change in miR-185-5p transfected cells (Fig. 6i, j), indicating that miR-185-5p regulates Dlx2 gene expression on the basis of translational repression rather than mRNA degradation.
miR-185-5p inhibits amelogenesis and osteogenesis through Dlx2 repression
To determine if miR-185-5p regulates amelogeneis and osteogenesis through Dlx2, miR-185-5p mimics were co-transfected with either wild-type Dlx2 over-expression vector or a 3′ UTR-mutant Dlx2 over-expression vector into LS8 cells and MC3T3-E1 cells. Overexpression of wild-type 3′-UTR Dlx2 over-expression plasmids only marginally alleviated the inhibitory effects of miR-185-5p on Amelx and Ocn expression, while over-expression of 3′ UTR-mutant Dlx2 significantly abolished the repression of Amelx and Ocn caused by miR-185-5p mimics (Fig. 7a–c). These data demonstrate that miR-185-5p inhibits amelogenesis and osteogenesis by repressing Dlx2. Furthermore, inhibition of miR-185-5p could partially rescue the phenotypes caused by mutant Runx2 (Fig. 7d, e).
Fig. 7. miR-185-5p inhibits amelogenesis and osteogenesis by suppressing Dlx2 expression.

a Overexpression of mutant 3′-UTR-Dlx2 in LS8 could abolish the negative effect of miR-185-5p on the expression of Amelx. b-c Overexpression of Dlx2 in MC3T3-E1 alleviated the inhibitory effect caused by miR-185-5p mimics. Alizarin Red S stain was quantified b and protein level of Ocn c was determined after treatment with STEMPRO for 21 days. d, e Inhibition of miR-185-5p could partially rescue the inhibitory effect of mutant Runx2 on expression of Amelx and Alp (1: Runx2 WT + inhibitor NC, 2: Runx2 WT + miR-185-5p inhibitor, 3: Runx2 M1 + inhibitor NC, 4: Runx2 M1 + miR-185-5p inhibitor, 5: Runx2 M2 + inhibitor NC, 6: Runx2 M2 + miR-185-5p inhibitor, 7: Runx2 M3 + inhibitor NC, 8: Runx2 M3 + miR-185-5p inhibitor)
Discussion
CCD (OMIM119600) is a dysplasia inherited as an autosomal dominant trait and it has a prevalence of 1 in 100,000 as reported18. The most common manifestations of CCD are related to skeletal dysplasia including delayed closure of the anterior fontanelle, supernumerary teeth, enamel hypoplasia, and some other skeletal abnormalities3,19.
CCD is mainly caused by mutations in RUNX220. RUNX2 is a member of the Runt-related transcription factors, which bind to transcriptional targets as multi-protein complexes with CBFβ interacting with the Runt domain21. RUNX2 increases or inhibits transcriptional activity of target genes, depending on the specific cell type, as well as the particular target gene. RUNX2 transcriptionally regulates several genes, which are important in the development and differentiation of skeletal tissues22. The majority of mutations identified in RUNX2 that are associated with CCD are known to disrupt the ability of RUNX2 to bind DNA23. There are researches confirmed that Runx2 could regulate the transcription of Ambn, Dmp1, Amgn, which are responsible for the ameloblastic differentiation24,25. In the present study, we demonstrated three Runx2 mutants (R190W, R225Q, A362V) could inhibit expression of markers for amelogenic and osteogenic differentiation, such as Amelx, Enam, Alp, and Ocn.
Gene expression is regulated by a wide variety of factors, including proteins and RNAs, and at multiple levels of control. In the developmental context, transcription factor-mediated regulation serves as an ON/OFF switch for consequent gene expression26. miRNAs form an additional regulatory circuit that is responsible for regulating gene expression and mediating cellular developmental processes. Additionally, miRNA expression occurs in a tissue-specific and developmental stage-specific manner, due to transcriptional regulation of miRNAs, complex signaling networks, regulatory sequences present in promoters, and chromatin modifications27,28.
To explore how miRNAs profile change after Runx2 mutated during the development of bone and teeth, we selected three-point mutations present in patients with CCD and constructed vectors for over-expression of these Runx2 mutants. Using miRNA expression microarrays, we identified that miR-185-5p was upregulated by the three Runx2 mutants in both LS8 and MC3T3-E1 cell lines compared with wild type Runx2 groups. As the most common manifestations of CCD are related to abnormal development of bone and tooth, we further aimed to verify the roles of miR-185-5p in the development of bone and dental enamel.
The kinetics of miR-185-5p in LS8 cells stimulated by RA/DEX or MC3T3-E1 cells stimulated by STEMPRO suggests that it may play an important role in ameloblast and osteoblast differentiations (Fig. 5). We further demonstrated that miR-185-5p may inhibit ameloblast and osteoblast differentiations by regulating the protein level of Dlx2, which is a critical factor in ameloblast and osteoblast differentiations.
To determine whether miR-185-5p is directly involved in ameloblast differentiation, we investigated the action of miR-185-5p in the process of RA/DEX-induced LS8 ameloblastogenesis. miR-185-5p over-expression inhibited RA/DEX-induced ameloblastogenesis, as evidenced by decreased levels of Amelx, Enam, Klk4, and Mmp20. miR-185-5p over-expression also decreased protein, but not mRNA, levels of the ameloblast differentiation transcription factor Dlx2. To investigate the effects of miR-185-5p on osteogenic differentiation, we first examined the efficiency and specificity of mimics and inhibitors of miR-185-5p. miR-185-5p over-expression led to decreased ALP activity, mRNA, and protein levels of ALP compared with the control cells. These data suggest that miR-185-5p is a negative regulator of osteoblast differentiation in preosteoblast cell lines.
Using miRBASE, we identified a potential binding site of miR-185-5p in the 3′UTR of Dlx2. Dlx2 plays an important role in ameloblast and osteoblast differentiations15–17,29–32. Studies have revealed that Dlx2 is an orthologue of the Drosophila distal-less gene, which plays a central role in patterning of jaws17,33,34. During tooth development Dlx2 is expressed in the dental epithelial cells throughout each stage of tooth germ development35. Homozygous mutants of Dlx2 exhibit abnormalities in craniofacial and neuronal development36. Dlx1/Dlx2 mutants displayed poorly organized dental epithelial cells and lost the palisade pattern of ameloblasts17. During late stages of tooth morphogenesis, ameloblasts derived from the inner enamel epithelium secrete amelogenin, which is a key enamel protein and required for late stage tooth development37. The promoter region of amelogenin contains several binding sites for FoxJ1 and Dlx2 transcription factors. Le ´ zot et al. reported that there were five DLX homeoprotein-binding sites in the promoter of Amelx. They identified two elements had inhibitory effect while the other three could drive transcriptional activation of Amelx in CHO-KI cells35. Meanwhile, Venugopalan et al. showed that endogenous Dlx2 could activate amelogenin (Amelx) promoter by binding to its promoter at a Dlx2 element located at -847 to -856 bp in LS8 cells29. In our study, we found that the miR-185-5p-Dlx2 axis could negatively regulate the expression of Amelx in LS8 cells, and our results are consistent with Venugopalan’s studies.
Several miRNAs have been reported to target Dlx2 and to regulate Dlx2 expression in different physiologic contexts. For example, miR-26a regulates osteogenic differentiation of human adipose tissue-derived stem cells by targeting the DLX2 transcription factor. In addition, miR-199a* was found to impair early chondrocyte differentiation by directly targeting Dlx2.
This present study reports the novel regulation of Dlx2 expression by miR-185-5p in ameloblast and osteoblast differentiations. We identified one potential binding site for miR-185-5p in the Dlx2 3′ UTR using bioinformatic analysis, and we demonstrated direct evidence that Dlx2 is a bona fide target for miR-185-5p (Fig. 6). Mutation of the binding site partially eliminated the repressive effect of exogenous miR-185-5p on luciferase reporter activity under transcriptional control of the Dlx2 3′UTR. Furthermore, mutation of this binding site also abolished the inhibitory function of endogenous miR-185-5p on Dlx2 expression, supporting the importance of this site for regulation of Dlx2 expression. In addition to the role of miR-185-5p in osteogenesis, it would be interesting to investigate its role in osteoclast differentiation or in chondrocyte differentiation, as Dlx2 is also involved in chondrogenesis.
Although we found that miR-185-5p is associated with osteogenic differentiation, it remains to be determined whether it is differentially expressed in cartilage, bone, or both. Future in vivo experiments in mouse models are necessary to evaluate the function of miR-185-5p in more details.
In conclusion, our study showed that miR-185-5p served as an important regulator of amelogenesis and osteoblast differentiation and may contribute to ameloporosis. We demonstrated that miR-185-5p acts as a negative regulator of osteoblast differentiation through suppression of the Dlx2 transcription factor. These findings suggest that the miR-185-5p-Dlx2 axis plays a role in the abnormal development of bone and teeth, which is present in CCD. Furthermore, this study provides new insights into the regulation of amelogenesis and osteoblast differentiations.
Materials and methods
Construction of Runx2-mutant expression vectors
The expression vector pcDNA3.1-Runx2 with c-Myc epitope tag was purchased from OriGene Technologies, Inc. In vitro site-directed mutagenesis was performed to construct the Runx2 mutants M1 (pcDNA3.1- R197WRunx2), M2 (pcDNA3.1- R232QRunx2), and M3 (pcDNA3.1- A369VRunx2) using the QuikChange Lightning Site-Directed Mutagenesis Kit (Stratagene Corp., La Jolla, CA, USA). The sequences of mutantconstructs were verified by sequencing of the whole vectors (Supplementary Figures).
Cell culture, stimulation, and transfection
LS8 cells (kindly provided by Dr. Malcolm L. Snead, USC, Los Angeles, CA) were maintained in high-glucose Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, NY, USA), containing 100 U/ml penicillin, 100 mg/ml streptomycin, and 10% fetal bovine serum (Gibco, Grand Island, NY, USA) at 37 °C in 5% CO2 humidified atmosphere. Ameloblast differentiation in LS8 cells was induced by supplementing growth medium with 20 mg/ml retinoic acid (RA, U.S. Pharmacopeia) and 10−7 M dexamethasone (DEX, Sigma)38,39.
MC3T3-E1 cells were obtained from the Chinese Academy of Sciences Cell Bank (Shanghai, China) and were maintained in α modification of Eagle’s minimal essential medium (α-MEM, Invitrogen) containing 100 units/ml penicillin and 100 µg/ml streptomycin. To induce osteoblast differentiation, MC3T3-E1 cells (1 × 105/well) were cultured in six-well plates overnight and treated with GIBCO® STEMPRO for various durations (refer to section “Results”).
Additional osteoblast differentiation experiments involved overnight culture of MC3T3-E1 (1 × 105) cells in 24-well plates and transfected with 50 or 100 nM miR-NC or miR-185-5p mimics (Genepharma) using Lipofectamine 2000 (Invitrogen). Two days later, these cells were stimulated with GIBCO® STEMPRO for varying times. MC3T3-E1 cells were harvested for protein and mRNA or were fixed with 95% ethanol (v/v) for histochemical examination of ALP activity.
RNA isolation and RNA quality control
Total RNA was isolated from cells using TRIzol® reagent (Invitrogen life technologies) and further purified using a RNeasy mini kit (Qiagen, Denmark) according to the manufacturer’s instructions. The concentration and quality of RNA from cells were measured using a NanoDrop spectrophotometer and verified by gel electrophoresis. MiRNA expression profiles were determined and generated from each of two groups (wild-type group and mutant group).
MiRNA labeling and miRNA array hybridization
RNA of sufficient quality from eight group samples (n = 3, total 24 samples) was submitted to KangChen-Biotech (Shanghai, China). RNA groups above mentioned by applying the miRCURY locked nucleic acid (LNA) microarray platform (Exiqon, Denmark). All procedures were carried out according to manufacturer’s protocol. Briefly, 1 μg total RNA was labeled with the Hy3™ or Hy5™ fluorophores, using miRCURY™ Array Power Labeling kit (Exiqon, Denmark). After stopping the labeling procedure, briefly spin the reaction and leave it at 4 °C. The two samples from the Hy3™ and Hy5™ labeling reactions are combined on ice. The samples were hybridized on a hybridization station using miRCURY™ LNA miRNA array (v.18.0) containing Tm-normalized probes for 847 human miRNAs. Microarrays with labeled samples were hybridized at 56 °C for overnight using a heat-shrunk hybridization bag and washed using miRCURY Array Wash buffer kit (Exiqon, Denmark). After hybridization, the chip slides were washed, dried, and scanned immediately. Each miRNA spot was replicated for four times on the same slide and two microarray chips have been used for each group.
Scanning and data analysis
Scanning was performed with an Axon GenePix 4000B microarray scanner (Axon Instruments, Union City, CA, USA). GenePix Pro 6.0 software (Axon) was used to read the raw intensity of the image. In order to produces the best within-slide normalization to minimize the intensity dependent differences between the dyes, signal intensities for each spot were scanned and calculated by subtracting local background (based on the median intensity of the area surrounding each spot) from total intensities using locally weighted scatter plot smoothing (Lowess) Normalization (MIDAS, TIGR Microarray Data Analysis System). After normalization, obtained average values for each miRNA spot were used for statistics. The ratio of green signal to red signal was calculated. The threshold value we used to screen up-regulated and down-regulated miRNAs is fold change > 2.00 and fold change < 0.50. Hierarchical clustering for differentially expressed miRNAs was generated using standard correlation as a measure of similarity.
Real-time quantitative PCR
Real-time quantitative PCR was performed to confirm the array results. Reverse transcriptase reactions contained 600 ng of purified total RNA, 20 nM stem-loop RT primer (data not shown), 1 × RT buffer, 0.125 mM each of dATP, dGTP, dCTP, and dTTP, 1 U/µl reverse transcriptase and 0.6 U/µl RNase Inhibitor. Using the Gene Amp PCR System 9700 (Applied Biosystems, USA), 20 µl reactions were performed with the following thermal cycling parameters: 30 min at 16 °C, 42 min at 42 °C, 5 min at 85 °C, and then held at 4 °C. Each reaction mixture for real-time quantitative PCR contained 1 × PCR buffer, 1.5 mM MgCl2, 0.25 mM each of dATP, dGTP, dCTP, and dTTP, 1 U DNA polymerase, 0.4 µM of each primer (data not shown), 0.25 × SYBR Green I, 1 µl cDNA, and deionized water to a total volume of 25 µl. Reactions were run with the following thermal cycling parameters: 95 °C for 5 min followed by 35 cycles of 95 °C for 10 s and 60 °C for 60 s. The threshold cycle (Ct) is defined as the fractional cycle number at which the fluorescence passes the fixed threshold, and each sample was normalized on the basis of its endogenous U6 RNA content. The experiment was conducted in triplicate.
Reagents
Antibodies: Anti-Dlx2 (catalog no. 1649-1) was purchased from Epitomics, Inc. (Burlingame, CA). Anti-ALP (catalog no. 9516) was purchased from Cell Signaling Technology (Danvers, MA). Anti-Ocn was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
The ALP assay kit, LabAssay™ ALP, was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan).
miR-185-5p mimic, mimic control (miR-NC), and miR-185-5p inhibitor were synthesized in Genepharma (Shanghai, China).
Dual-luciferase reporter assay
HEK293 cells were transfected using Lipofectamine 2000 (Invitrogen) with either WT or mutant pGL3-Dlx2 constructs (200 ng) and miR-185-5p or miR-NC for 48 h. As a positive control, the modified pGL3 control vector was used without a 3UTR insert. Cells treated with Lipofectamine 2000 reagent alone served as negative controls.
The Dual Luciferase Reporter Assay System (Promega) was used to quantify luminescence signals using a luminometer (Glomax; Promega). Firefly luciferase levels were normalized to the Renilla luciferase values from the co-transfected phRL-null vector (Promega).
Construction of Dlx2 expression vectors
Complementary DNAs for the mouse Dlx2 genes were obtained by an RT-PCR technique using the Prime-Script™ PT reagent kit (TaKaRa). Total RNAs prepared from mouse MC3T3-E1 cells were used for the RT-PCR. PCR products were digested with BglII and NotI (Promega), purified from agarose gels, and subcloned into pcDNA3.1 (Clontech). Each cDNA was confirmed by DNA sequencing. Plasmid DNA was transfected into cells using Lipofectamine 2000 (Invitrogen). Mutant-specific primers were used to generate Dlx2 mutant constructs (MT- pcDNA3.1-Dlx2) by using KOD-Plus (Toyobo Co.).
Quantitative real-time RT-PCR
After differentiation of LS8 cells into ameloblasts was induced with RA/DEX for 48 h, total cellular RNA was extracted with the use of the E.Z.N.A.® total RNA kit (OMEGA Bio-Tec, USA) according to the manufacturer’s protocol. Quality of the isolated RNA was evaluated using a Thermo Scientific NanoDrop 2000/2000C spectrophotometer at 260/280 nm. PrimeScript™ RT reagent Perfect Real Time Kit (TaKaRa Biotechnology, Dalian, China) was used to reverse-transcribe RNA into cDNA. The primers for detecting amelogenin, enamelin, Klk4 and Mmp20 are shown in Table 1. SYBR Premix Ex Taq™II (TaKaRa Biotechnology, Dalian, China) was used for real-time PCR. Amplification and detection were performed with an ABI 7500 real-time PCR instrument (Applied Biosystems, Foster, CA, USA) at the following conditions: 30 s hot start at 95 °C in the holding stage; 40 cycles of 15 s at 95 °C, 34 s at 60 °C in the cycling stage; and 15 s at 95 °C, 1 min at 60 °C, 30 s at 95 °C, and 15 s at 60 °C in the melt curve stage. The relative expression levels were calculated using the comparative threshold cycle (DDCT) method.
Table 1.
Primer sequences for real-time PCR analyses
| Gene name | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| Gapdh | CATGTTCCAGTATGACTCCACTC | GGCCTCACCCCATTTGATGT |
| Amelogenin | GATGGCTGCACCACCAAATC | CTGAAGGGTGTGACTCGGG |
| Enamelin | TGCAGAAATCCGACTTCTCCT | CATCTGGAATGGCATGGCA |
| Klk4 | CCGGATCATACAAGGCCAGG | TGCGGATGCACCAAGACTC |
| Mmp20 | ACACATTTCGACAATGCTGAGA | AAGTTGGGTACATCAGTGCTG |
MC3T3-E1 cells (1 × 105/ml) were treated in triplicate with the 50 or 100 nM GIBCO® STEMPRO in completed culture medium for several days. Total RNA was extracted from using TRIzol (Invitrogen) and reverse-transcribed into cDNA using the PrimeScript™ PT reagent kit (TaKaRa). Amplification of target genes were performed by real-time quantitative PCR using cDNA as template, the specific primers and the SYBR® PrimeScript® RT-PCR kit (Takara) on an ABI PRISM 7900 real-time PCR system (Applied Biosystems, Carlsbad, CA). PCR amplifications were performed in duplicate at 95 °C for 15 s and subjected to 40 cycles of 95 °C for 5 s, 60 °C for 30 s, 95 °C for 15 s, 60 °C for 15 s, and 95 °C for 15 s. The primers used were: ALP: 5-CCC TCT CCA AGA CAT ATA ACAC-3′ and 5′-TTG CCC TGA GTG GTG TTG-3′; osteocalcin (Ocn): 5′-GGA CCA TCT TTC TGC TCA CT 3′ and 5′-CGG AGT CTG TTC ACT ACC TTA T-3′; Dlx2: 5′-CCG CTC GAG AAG GAT GGA CAA GTC AGA C-3′ and 5′-ATA GTT TAG CGG CCG CCT GCG AAT AAT GAA CAG AG-3′; and β-actin: 5′-AAC AGT CCG CCT AGA AGC AC-3′ and 5′-CGT TGA CAT CCG TAA AGA CC-3′. The relative levels of miR-185-5p expression to control U6 snRNA were quantified using the TaqMan MicroRNA expression assay (Applied Biosystems). The relative levels of target gene mRNA transcripts to control β-actin were determined by 2-△△Ct.
ALP staining and activity
MC3T3-E1 cells were cultured overnight in six-well plates at 3 × 105 cells/well. Two days after transfection, the cells were treated with STEMPRO for 7 days, then ALP staining and activity assays were conduced. Subsequently, the cells were fixed with 95% ethanol (v/v) and then incubated with a substrate solution from an ALP staining kit (Beyotime® Institute of Biotechnology, Shanghai, China) in the dark, according to the manufacturer’s protocol. For ALP activity assays, after incubation, the treated cells were washed twice with PBS, and 200 µl of lysis buffer was added to the cell layer and kept on ice for 5 min. The cell lysate was sonicated for 1 min and centrifuged at 1000 × g at 4 °C for 10 min. ALP activity was assayed by a spectrophotometric method using a LabAssay™ ALP kit. The absorbance at 405 nm of each well was measured with a microplate reader according to the manufacturer’s instruction40.
Alizarin red staining
For detection of calcification during osteoblast differentiation, MC3T3-E1 cells with STEMPRO-treated after 21 days were washed twice with PBS and fixed with 500 µl of ice-cold 70% ethanol for 10 min. The fixed cells were stained with 500 µl of Alizarin red solution (Sigma) according to manufacturer’s instructions. To quantify matrix mineralization, alizarin red S-stained cultures were incubated in 100 mM cetylpyridinium chloride for 1 h at room temperature to solubilize calcium-bound alizarin red S, the absorbance of which was measured at 562 nm and normalized to the cells without any treatment.
Western blot analysis
Different groups of MC3T3-E1 cells (2 × 106/tube) were lysed in radioimmune precipitation assay lysis and extraction buffer (Thermo Fisher Scientific). Individual cell lysates (10 µg/lane) were separated by SDS-PAGE and transferred to an Immobilon-P polyvinylidene difluoride membrane (Millipore, Billerica, MA). After being blocked with SuperBlock T20 PBS blocking buffer (Thermo Fisher Scientific, Pittsburgh, PA), the membranes were incubated with rabbit monoclonal antibodies against Dlx2 (1:1000), rabbit polyclonal antibodies against ALP (1:1000), and goat polyclonal antibodies against Ocn (1:1000), respectively. The bound antibodies were detected with 1:10,000 diluted HRP-conjugated secondary antibodies and visualized using Pierce ECL Western blotting substrate (Thermo Fisher Scientific), followed by exposure to film and being digitally imaged.
Statistical analysis
Data are expressed as mean ± S.E. Data were analyzed by one-way or two-way analysis of variance. Multiple comparisons analysis between the groups was performed by using the Bonferroni post hoc test method. P < 0.05 was considered statistically significant. Statistical analyses were carried out using StatView 5.0 software (SAS Institute, Cary, NC) and GraphPad Prism 4.0 software.
Electronic supplementary material
Acknowledgements
This work was supported by National Nature Science Foundation of China (31571298 and 81570961).
Competing interests
The authors declare no conflicts of interest.
Footnotes
This article has been retracted. Please see the retraction notice for more detail: https://doi.org/10.1038/s41419-025-07762-2
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Huaiguang Chang, Yue Wang contributed equally to this study
Edited by E. Candi
Change history
06/17/2025
This article has been retracted. Please see the Retraction Notice for more detail: 10.1038/s41419-025-07762-2
Contributor Information
Hongshan Zhao, Email: hongshan@bjmu.edu.cn.
Hailan Feng, Email: kqfenghl@bjmu.edu.cn.
Supplementary information
The online version of this article (10.1038/s41419-017-0078-4) contains supplementary material.
References
- 1.Mundlos, S. Cleidocranial dysplasia: clinical and molecular genetics. J. Med. Genet.36, 177–182 (1999). [PMC free article] [PubMed] [Google Scholar]
- 2.Chen, S. et al. Altered gene expression in human cleidocranial dysplasia dental pulp cells. Arch. Oral Biol.50, 227–236 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Fukuta, Y. et al. Histological and analytical studies of a tooth in a patient with cleidocranial dysostosis. J. Oral Sci.43, 85–89 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Bae, S. C. et al. PEBP2 alpha B/mouse AML1 consists of multiple isoforms that possess differential transactivation potentials. Mol. Cell Biol.14, 3242–3252 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Otto, F., Kanegane, H. & Mundlos, S. Mutations in theRUNX2 gene in patients with cleidocranial dysplasia. Hum. Mutat.19, 209–216 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Thirunavukkarasu, K., Mahajan, M., McLarren, K. W., Stifani, S. & Karsenty, G. Two domains unique to osteoblast-specific transcription factor Osf2/Cbfa1 contribute to its transactivation function and its inability to heterodimerize with Cbfbeta. Mol. Cell Biol.18, 4197–4208 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Javed, A. et al. Groucho/TLE/R-esp proteins associate with the nuclear matrix and repress RUNX (CBF(alpha)/AML/PEBP2(alpha)) dependent activation of tissue-specific gene transcription. J. Cell. Sci.113, 2221–2231 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Zhou, G. et al. CBFA1 mutation analysis and functional correlation with phenotypic variability in cleidocranial dysplasia. Hum. Mol. Genet.8, 2311–2316 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Ryoo, H. M., Kang, H. Y., Lee, S. K., Lee, K. E. & Kim, J. W. RUNX2 mutations in cleidocranial dysplasia patients. Oral Dis.16, 55–60 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Wang, X.-P. & Fan, J. Molecular genetics of supernumerary tooth formation. Genesis49, 261–277 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erson, A. E. & Petty, E. M. MicroRNAs in development and disease. Clin. Genet.74, 296–306 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Hwang, H. W. & Mendell, J. T. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br. J. Cancer94, 776–780 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bergwitz, C. et al. Identification of novel CBFA1/RUNX2 mutations causing cleidocranial dysplasia. J. Inherit. Metab. Dis.24, 648–656 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Quack, I. V. B. et al. Mutation analysis of core binding factor A1 in patients with cleidocranial dysplasia. The American Journal of Human Genetics.65, 1268–1278(1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li, H. et al. Expression and function of Dlx genes in the osteoblast lineage. Dev. Biol.316, 458–470 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCollum, M. & Sharpe, P. T. Evolution and development of teeth. J. Anat.199, 153–159 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomas, B. L. et al. Role of Dlx-1 and Dlx-2 genes in patterning of the murine dentition. Development124, 4811–4818 (1997). [DOI] [PubMed] [Google Scholar]
- 18.D’Alessandro, G., Tagariello, T. & Piana, G. Cleidocranial dysplasia: etiology and stomatognathic and craniofacial abnormalities. Minerva Stomatol.59, 117–127 (2010). [PubMed] [Google Scholar]
- 19.Mundlos, S. Cleidocranial dysplasia: clinical and molecular genetics. J. Med. Genet.36, 177–182 (1999). [PMC free article] [PubMed] [Google Scholar]
- 20.Mundlos, S. et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell89, 773–779 (1997). [DOI] [PubMed] [Google Scholar]
- 21.Wang, S. et al. Cloning and characterization of subunits of the T-cell receptor and murine leukemia virus enhancer core-binding factor. Mol. Cell Biol.13, 3324–3339 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Otto, F., Lubbert, M. & Stock, M. Upstream and downstream targets of RUNX proteins. J. Cell. Biochem.89, 9–18 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Cohen, M. M.Jr. Biology of RUNX2 and cleidocranial dysplasia. J. Craniofac. Surg.24, 130–133 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Dhamija, S. & Krebsbach, P. H. Role of Cbfa1 in ameloblastin gene transcription. J. Biol. Chem.276, 35159–35164 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Kobayashi, I. et al. Type II/III Runx2/Cbfa1 is required for tooth germ development. Bone38, 836–844 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Flynt, A. S. & Lai, E. C. Biological principles of microRNA-mediated regulation: shared themes amid diversity. Nat. Rev. Genet.9, 831–842 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Obernosterer, G., Leuschner, P. J., Alenius, M&Martinez, J. Post-transcriptional regulation of microRNA expression. RNA12, 1161–1167 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neilson, J. R., Zheng, G. X., Burge, C. B. & Sharp, P. A. Dynamic regulation of miRNA expression in ordered stages of cellular development. Genes Dev.21, 578–589 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Venugopalan, S. R. et al. Hierarchical interactions of homeodomain and forkhead transcription factors in regulating odontogenic gene expression. J. Biol. Chem.286, 21372–21383 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lezot, F. et al. Biomineralization, life-time of odontogenic cells and differential expression of the two homeobox genes MSX-1 and DLX-2 in transgenic mice. J. Bone Miner. Res.15, 430–441 (2000). [DOI] [PubMed] [Google Scholar]
- 31.Lezot, F. et al. Epithelial Dlx-2 homeogene expression and cementogenesis. J. Histochem. Cytochem.48, 277–284 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Peng, L. et al. Comparative analysis of mesenchymal stem cells from bone marrow, cartilage, and adipose tissue. Stem Cells Dev.17(4), 761–773 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Thomas, B. L., Porteus, M. H., Rubenstein, J. L. & Sharpe, P. T. The spatial localization of Dlx-2 during tooth development. Connect. Tissue Res.32, 27–34 (1995). [DOI] [PubMed] [Google Scholar]
- 34.Thomas, B. L., Liu, J. K., Rubenstein, J. L. & Sharpe, P. T. Independent regulation of Dlx2 expression in the epithelium and mesenchyme of the first branchial arch. Development127, 217–224 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Lézot, F. et al. Physiological implications of DLX homeoproteins in enamel formation. J. Cell. Physiol.216, 688–697 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Liu, J. K., Ghattas, I., Liu, S., Chen, S. & Rubenstein, J. L. Dlx genes encode DNA-binding proteins that are expressed in an overlapping and sequential pattern during basal ganglia differentiation. Dev. Dyn.210, 498–512 (1997). [DOI] [PubMed] [Google Scholar]
- 37.Fincham, A. G., Moradian-Oldak, J. & Simmer, J. P. The structural biology of the developing dental enamel matrix. J. Struct. Biol.126, 270–299 (1999). [DOI] [PubMed] [Google Scholar]
- 38.Yang, T. et al. High amounts of fluoride induce apoptosis/cell death in matured ameloblast-like LS8 cells by downregulating Bcl-2. Arch. Oral Biol.58, 1165–1173 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Duan, X., Mao, Y., Wen, X., Yang, T. & Xue, Y. Excess fluoride interferes with chloride-channel-dependent endocytosis in ameloblasts. J. Dent. Res.90, 175–180 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Li, Z. et al. A microRNA signature for a BMP2-induced osteoblast lineage commitment program. Proc. Natl. Acad. Sci .USA105, 13906–13911 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.