

Abstract
We present three linkage-disequilibrium (LD)-based recombination maps generated using whole-genome sequence data from 10 Nigerian chimpanzees, 13 bonobos, and 15 western gorillas, collected as part of the Great Ape Genome Project (Prado-Martinez J, et al. 2013. Great ape genetic diversity and population history. Nature 499:471–475). We also identified species-specific recombination hotspots in each group using a modified LDhot framework, which greatly improves statistical power to detect hotspots at varying strengths. We show that fewer hotspots are shared among chimpanzee subspecies than within human populations, further narrowing the time scale of complete hotspot turnover. Further, using species-specific PRDM9 sequences to predict potential binding sites (PBS), we show higher predicted PRDM9 binding in recombination hotspots as compared to matched cold spot regions in multiple great ape species, including at least one chimpanzee subspecies. We found that correlations between broad-scale recombination rates decline more rapidly than nucleotide divergence between species. We also compared the skew of recombination rates at centromeres and telomeres between species and show a skew from chromosome means extending as far as 10–15 Mb from chromosome ends. Further, we examined broad-scale recombination rate changes near a translocation in gorillas and found minimal differences as compared to other great ape species perhaps because the coordinates relative to the chromosome ends were unaffected. Finally, on the basis of multiple linear regression analysis, we found that various correlates of recombination rate persist throughout the African great apes including repeats, diversity, and divergence. Our study is the first to analyze within- and between-species genome-wide recombination rate variation in several close relatives.
Keywords: recombination, PRDM9, hotspots, primates
Introduction
The increasing availability of genetic maps from a variety of taxa has become a valuable resource for the scientific community for phasing (Browning SR and Browning BL 2011), QTL analysis (Altshuler et al. 2008), and most recently to aid in de novo genome assembly (Hahn et al. 2014; Kawakami et al. 2014). Despite the obvious utility of having genetic maps for these and other scientific applications, obtaining accurate estimates of genome-wide recombination rates can be both challenging and expensive. While the most straightforward approach is to directly observe meiotic events in genetic crosses or pedigrees, this requires large numbers of individuals observed over multiple generations and dense genetic markers. An alternative approach is to estimate recombination rates indirectly based on patterns of linkage disequilibrium (LD) between adjacent sites. This approach gives the benefits of finer-scale precision and requires a smaller sample size but still needs a dense set of markers. LD-based recombination maps have several limitations, including sensitivity to population structure, lack of sex-specific recombination rate estimates, and generation of historical recombination rate estimates rather than contemporary ones (Stumpf and McVean 2003; Clark et al. 2010). Nonetheless, recombination rates obtained via these two major approaches have been found to give fairly similar estimates when compared at broader size scales (Clark et al. 2010).
Both of these approaches have been used to estimate recombination rates in various human populations and our closest relative, the chimpanzee. The first genome-wide recombination map in humans was a pedigree-based map of a European population (Broman et al. 1998). Since then, other pedigree-based maps of human populations have been generated for Europeans (Kong et al. 2002; Coop et al. 2008) and Asians (Bleazard et al. 2013). Genome-wide LD-based recombination maps have also been generated using HapMap genotype data and include European (CEU), African (YRI), and Asian (CHB+JPT) population rate estimates (Myers et al. 2005). Recently, the 1000-genomes project constructed LD-based recombination maps from low-coverage whole-genome sequence data for the same populations (Altshuler et al. 2010). Another approach to fine-mapping recombination events in the genome has used local ancestry methods to build recombination maps for African-Americans (Hinch et al. 2011; Wegmann et al. 2011). Finally, in 2012, the first nonhuman primate fine-scale recombination map was published using ten unrelated whole-genome sequences of western chimpanzees (Auton et al. 2012).
Several studies have noted the importance of scale when comparing results between studies and when comparing recombination rates within and between species (Stevison and Noor 2010; Auton et al. 2012; Chan et al. 2012). Comparison of recombination rates between close relatives has shown that recombination rates have rapid turnover on the scale of recombination hotspots (1–2 kb) but are correlated between species when examined at intervals of approximately 1 Mb (Serre et al. 2005; Duret and Arndt 2008; Laayouni et al. 2011). However, most previous between-species comparisons have focused on either very closely related taxa or distant relatives (Smukowski and Noor 2011). Nonetheless, differences in the conservation of recombination rates at various scales suggest different mechanisms control broad and fine-scale patterns of recombination rates across the genome. While recombination rates are free to evolve in different directions as species diverge, meiotic recombination is a tightly regulated cellular process and thus broad-scale rates may be limited both mechanistically and evolutionarily in how much they can change (Brooks 1988; Kauppi et al. 2004). Mechanistically, recombination is necessary to stabilize chromosomes during meiosis, but excessive recombination or errors in this pathway can lead to aneuploidy, birth defects, disease, and/or various cancers (Hassold and Hunt 2001; Petronczki et al. 2003; Coop and Przeworski 2007). Evolutionarily, recombination helps to shuffle beneficial alleles onto common genetic backgrounds, facilitating the efficacy of natural selection (Crow 1994). However, too much recombination can break down these associations (Crow 1988).
One possible explanation for the difference in conservation of recombination rates at various scales is that the mechanisms controlling the distribution of recombination hotspots leads to rapid turnover of fine-scale recombination rates. In E. coli, hotspot determination is localized to χ sites (Smith 2012), whereas in mammals, such as humans and mice, it has been shown that the transcription factor PRDM9 binds to hotspots and recruits additional recombination machinery (Baudat et al. 2010; Cole et al. 2014). Despite recent efforts to comprehensively sequence PRDM9 across various taxa (Myers et al. 2010; Berg et al. 2011; Auton et al. 2012; Schwartz et al. 2014), the universal role of this protein in recruiting recombination machinery remains unclear. For example, in dogs, the PRDM9 protein sequence is truncated, and recombination hotspots are localized based on functional elements in the genome (Auton et al. 2013). In chimpanzees, there has thus far been no evidence that recombination rates are higher in regions with suspected PRDM9 binding (Auton et al. 2012). Unlike what has been shown in dogs, the chimpanzee PRDM9 protein is fully functional, and a recent survey of PRDM9 diversity in primates has shown pervasive diversifying selection for this protein throughout primates (Schwartz et al. 2014).
Despite the growing number of population-specific recombination maps in humans and the chimpanzee map (PanMap), there is not much information among primates for how recombination rate variation evolves. To fully understand the broad-scale evolution of recombination rates in great apes, more between-species comparisons are needed with different degrees of interspecific divergence and also more within species comparisons are needed that are not limited to human populations. Additionally, recombination rate estimates outside of these groups are imperative to assess PRDM9’s role in hotspot determination broadly across great apes. To address these fundamental questions regarding the time scale of recombination rate evolution, we present three new LD-based recombination maps for Nigerian chimpanzees, bonobos, and western gorillas collected as part of the Great Ape Genome Project (Prado-Martinez et al. 2013).
First, we compared patterns of fine-scale recombination rate variation within and between these groups, and existing recombination rate data in humans and chimpanzees. After identifying species-specific recombination hotspots from our population-scaled recombination rate estimates, we examined the amount of overlap between these localized regions within and between species to determine the time scale of hotspot turnover. We further sought to elucidate the role of PRDM9 in determining the location of recombination hotspots broadly across great apes. We used computational approaches to identify predicted DNA binding of the zinc fingers of each species-specific form of the protein PRDM9. While PRDM9 can have anywhere from 6 to 19 zinc fingers in primates (Schwartz et al. 2014) and all are experimentally shown to bind to DNA when expressed in E. coli (Billings et al. 2013), several of these zinc fingers seem to be less specific in their binding to DNA (Segurel 2013). Consistent with this result, previous studies examining the association between PRDM9 and recombination have focused on shorter submotifs within the full predicted binding sequence of PRDM9 that recur in population surveys of PRDM9 alleles (table 1) (Myers et al. 2005; Auton et al. 2012; Schwartz et al. 2014). For example, in humans, two major submotifs of PRDM9 are associated with binding to recombination hotspots. This includes a 13-bp submotif common to PRDM9 alleles found in European populations, and a 17-bp submotif more commonly associated with PRDM9 alleles found in African populations (Hinch et al. 2011), both matching the terminal zinc fingers in the full PRDM9 motif (table 1 and supplementary fig. S8, Supplementary Material online). For chimpanzees and bonobos, four submotif regions, including a recently described internal submotif, have been shown to recur in several alleles of PRDM9 across the Pan genus (Auton et al. 2012; Schwartz et al. 2014). For gorillas, an internal submotif was recently identified based on a smaller subset of PRDM9 alleles (Schwartz et al. 2014). None of these most recently described submotifs have been analyzed for their potential role in hotspot localization, mainly due to a lack of nonhuman primate recombination maps.
Table 1.
PRDM9 Submotif Summary.
| Submotif | Submotif Frequency in PRDM9 Alleles | Corresponding PRDM9 Allele | Source Population | Source Population Frequency |
|---|---|---|---|---|
| CCnCCnTnnCCnC | 34.5% (Berg et al. 2011) | Allele A | Human (CEU) | 84.6% (49.1% in YRI) (Schwartz et al. 2014) |
| CCnCnnTnnnCnTnnC | 34.5% (Berg et al. 2011) | Allele C | Human (YRI) | 13.4% (Schwartz et al. 2014) |
| AAnAAnCCC | 61.54% (Auton et al. 2012) | A1 (Auton et al. 2012) (Pan.p-1 [Schwartz et al. 2014]) | Bonobo | 62.5% (Schwartz et al. 2014) |
| CnnCCnAAnAA | 61.54% (Auton et al. 2012) | E1 (Auton et al. 2012) (Pan.t-3 [Schwartz et al. 2014]) | Eastern chimpanzee | 10.6% (Schwartz et al. 2014) |
| CnGnnAAnAnTT | 61.54% (Auton et al. 2012) | W6 (Auton et al. 2012) | Western chimpanzee | 25% (Auton et al. 2012) |
| AnTTnnAnTCnTCC | 66.7% (Schwartz et al. 2014) | Pt1 (Schwartz et al. 2014) | Pan troglydytes | 18.3% (Schwartz et al. 2014) |
| CCnAnnCCTC | 75.0% | Gg1 (Schwartz et al. 2014) | Gorilla | 42.9% |
| CTCnTCnTCnTC | 50.0% | Gg1 (see supplementary fig. S8, Supplementary Material online) | Gorilla | 42.9% |
Note.—Each submotif described is listed. Included information represents the PRDM9 allele from which each submotif was derived. Submotif frequency shows that for each, additional PRDM9 alleles contain a given submotif sequence. Additionally, the source population and the frequency are given. For values collected elsewhere, citations are included.
Next, we sought to compare the distribution of recombination rate across the genomes of great apes. Previously, it has been shown that nearly 80% of recombination events occur in < 20% of the physical sequence of the genome, occurring mostly in recombination hotspots (McVean et al. 2004). Further, recombination is more strongly biased toward hotspots in European recombination maps but less so in African or chimpanzee recombination maps. For this analysis, we adopted the use of the Gini coefficient, which has been used in economics to compare the distribution of wealth among countries, and was recently applied to analysis of the cumulative distribution of recombination in C. elegans (Dorfman 1979; Kaur and Rockman 2014). By directly comparing the area under the curve of these cumulative distribution functions, this approach allows for easier comparison among taxa (Kaur and Rockman 2014).
We further sought to compare broad-scale patterns of recombination rate divergence using this comparative recombination rate data set. This analysis included a comparison at various scales of the rate at which recombination and nucleotide sequences diverge between species to understand the relative constraints on each. We also examined how large-scale chromosomal differences impact recombination rates and the skew in recombination rates typically present in telomeric and centromeric regions.
Finally, because recombination rates have been shown to correlate with genetic features such as polymorphism, divergence, GC-content, repeat content, and specific sequence motifs (Jensen-Seaman et al. 2004; Coop and Przeworski 2007; Stevison and Noor 2010), we sought to determine the amount of variation in recombination rate that can be explained by various genetic features. The finding that recombination rate variation across the genome correlates with a variety of genetic features has led many to attempt to determine which evolutionary forces drive these associations. For example, many studies have found that recombination-mediated linked selection drives the ubiquitous correlation observed between recombination rate and nucleotide polymorphism in many taxa (Begun and Aquadro 1992; McGaugh et al. 2012; Webster and Hurst 2012). Alternatively, a biased repair process increasing the probability of transmission of GC-alleles, known as GC-biased gene conversion (gBGC), has been shown to explain the correlation between GC-content and recombination in most cases (Marais et al. 2001; Birdsell 2002; Marais 2003; Duret and Galtier 2009; Galtier et al. 2009), but see (Hey and Kliman 2002; Kliman and Hey 2003). For this analysis, we comprehensively represented the various genetic feature data available in primates and sought to normalize the explained variation by employing a multiple linear regression framework. In addition to the findings we present here, we anticipate the resources present here will be useful both for imputing and phasing future genotype data collected and in uncovering unique patterns of selection and demography in these species (McManus et al. 2015).
Results
We used whole-genome sequences (mean coverage 26.34×) from 10 Pan troglodytes ellioti, 13 Pan paniscus, and 15 Gorilla gorilla gorilla (supplementary table S1, Supplementary Material online) individuals to construct population-scaled recombination maps for each species, using a similar approach to that employed for the western chimpanzee map (Auton et al. 2012). The final maps were constructed from 4.2, 8.5, and 7.8 million single-nucleotide polymorphisms (SNPs) for bonobo, Nigerian chimpanzee, and western gorilla, respectively, as compared to 5.3 and 1.6 million sites used in the western chimpanzee and HapMap projects, respectively. The genome-wide population-scaled average recombination rates were 0.641, 0.8, and 0.944 ρ/kb in bonobo, Nigerian chimpanzee, and western gorilla, respectively. For a robust comparison between our maps and existing human and western chimpanzee maps, we identified blocks for each nonhuman genome that were syntenic with human (supplementary fig. S2–S4, Supplementary Material online). We later binned these syntenic blocks to 1 Mb (supplementary table S2 and fig. S5, Supplementary Material online), 500 kb, and 100 kb for downstream analysis. Additionally, figure 1 shows a plot of recombination rates across the genomes of the great apes compared here. To identify recombination hotspot locations, we implemented a version of LDhot that follows the approach of Myers et al. (2005). Briefly, for a 20-kb region, we used a likelihood ratio test to determine whether the central 2 kb had an elevated population-scaled recombination rate relative to the surrounding sequence (see supplementary fig. S6, Supplementary Material online, for comparison to other methods). Using this approach, we identified 10,704, 8,037, and 22,012 hotspots in bonobo, Nigerian chimpanzee, and western gorilla, respectively.
Fig. 1.

Broad-scale comparisons of recombination rates across great apes. Genome-wide plot of recombination rate estimates for Europe and African human populations from HapMap, western chimpanzees from PanMap, and the three maps generated here, grouped within humans (N = 2), chimpanzees and bonobos (N = 3), and gorillas (N = 1) to highlight both within- and between-species differences. Alternating chromosomes are plotted in different colors to emphasize boundaries.
Fine-Scale Comparisons
Hotspot Overlap within and between Species
We used two complementary approaches for exploring the degree of overlap in hotspot locations across populations. First, we examined the recombination rate at the syntenic locations of called hotspots (fig. 2). That is, if a hotspot is called in one population, we examined whether the estimated recombination rate at the syntenic region was elevated in closely related taxa. We call this “hotspot rate correlations” below. Using the publicly available hotspots from HapMap (Myers et al. 2005) and western chimpanzee (Auton et al. 2012), we found substantial hotspot rate correlation between human populations but little (or no) evidence of elevated rates at hotspot orthologs in other comparisons. It is worth noting that figure 2A inflates the hotspot rate correlation between human populations because the HapMap hotspots are a composite of population-specific hotspots from European, African, and Asian populations. Therefore, we compared our population-specific hotspot results for chimpanzees (fig. 2B and C) to the population-specific hotspot plots of European and African human populations (see supplementary fig. S6 in Auton et al. [2012]). Still, the degree of rate correlation in human hotspots is much higher than in figure 2B and C. One potential reason for a lack of shared hotspot rates between Nigerian and western chimpanzees could be that our method for identifying hotspots was slightly different than the methods used for both HapMap and western chimpanzee. We estimated hotspots in western chimpanzee using our method but did not see any qualitative difference in the degree of hotspot correlation with other populations (supplementary fig. S7, Supplementary Material online). Further, we found a similar lack of hotspot rate correlation in bonobo–chimpanzee comparisons (fig. 2D) and all comparisons involving gorilla (fig. 2E).
Fig. 2.
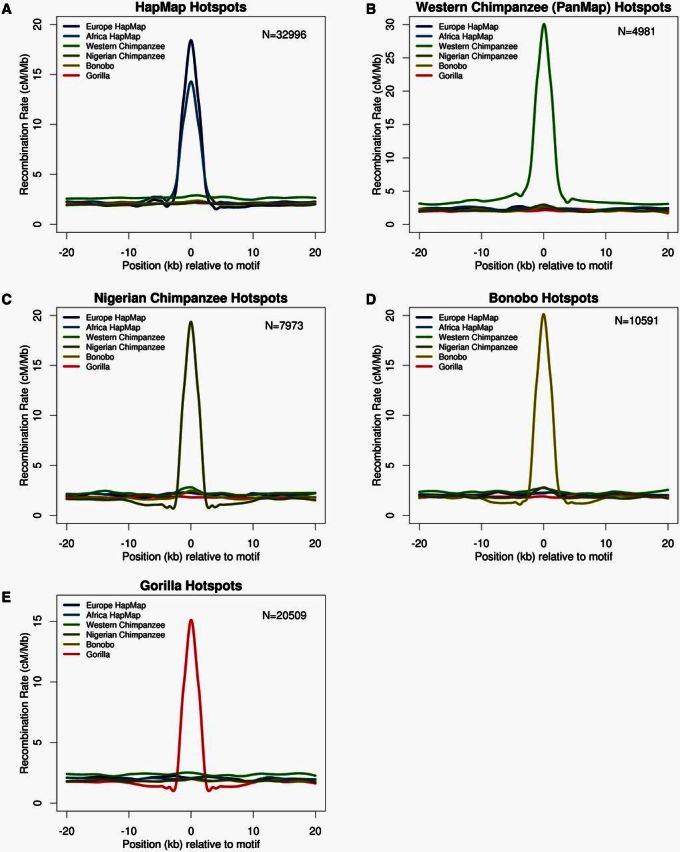
Hotspot rate correlation analysis. Degree of hotspot sharing and recombination rates for all maps in species-specific hotspots. Recombination rates for all maps 20 kb upstream and downstream of (A) human hotspots from HapMap, (B) western chimpanzee hotspot centers from PanMap, (C) Nigerian chimpanzee hotspot centers, (D) bonobo hotspot centers, and (E) western gorilla hotspot centers. Rates are shown in cM/Mb, and numbers of hotspots correspond to the number that mapped to hg18 genome.
In parallel, we also performed an analysis comparing the number of LDhot-inferred hotspots that overlap with each other (called here “hotspot overlaps” with the number of overlaps expected under a null model of random hotspot locations within syntenic blocks) (see Materials and Methods). For all comparisons between populations, we found an excess of overlapping hotspots over the null expectations (table 2). This observation can be explained in part due to the fact that recombination hotspot locations correlate with genomic features such as GC content (see below) that do not vary much between the closely related species examined in this study. The degree of hotspot overlap increased with decreasing divergence time between populations, ranging from 78 to 93% excess over null expectations in intra-chimpanzee comparisons to a 9–27% excess in comparisons involving gorilla. We hypothesize that the latter range reflects the baseline inflation due to genomic factors other than shared common descent of a recombination hotspot in the orthologous location in the gorilla–human common ancestor. If so, we note that there is a significant increase in hotspot overlap between chimpanzees and bonobos (36–47% excess), despite very little evidence for increased recombination rates in one species at the orthologs of hotspots identified in the other. It is also worth noting that the range of values for increased overlap of hotspots in the human–chimpanzee comparisons (9.6–18.1) are similar to the 8% estimate from a similar hotspot overlap analysis (Ptak et al. 2005). We also find that the observed percent overlap (table 2, below diagonal) is higher for comparisons with more hotspots (human and gorilla, samples sizes in fig. 2).
Table 2.
Hotspot Overlap Analysis.
| Panmap | Panmap (new) | Nigerian Chimpanzee | Bonobo | HapMap | Gorilla | |
|---|---|---|---|---|---|---|
| Panmap | — | 2167 | 77.7 | 46.9 | 11.3 | 27 |
| Panmap (new) | 35.6 | — | 92.9 | 35.9 | 18.1 | 22.8 |
| Nigerian chimpanzee | 2.15 | 3.3 | — | 39.9 | 9.6 | 12.3 |
| Bonobo | 2.33 | 2.87 | 2.59 | — | 13.2 | 9.2 |
| HapMap | 6.89 | 8.14 | 6.89 | 7.87 | — | 14.2 |
| Gorilla | 3.24 | 3.79 | 3.1 | 3.55 | 9.79 | — |
Note.—Above the diagonal values represent percent increase in hotspot overlap over null expectations in comparisons between populations. Below the diagonal values represent observed percent shared hotspots (compare to fig. 2). “Panmap” refers to hotspots called in P. troglodytes verus in Auton et al. (2012), while “Panmap (new)” refers to hotspots called from the same data using our method.
Localization of PRDM9 Predicted Binding Sites (PBS) to Hotspot Regions
Similar to the approach used in a recent mouse study (Brunschwig et al. 2012), we investigated the extent to which various PRDM9 submotifs (table 1) are represented in population-specific hotspots by calculating position weight matrices (PWMs) identified based on species-specific protein sequences of PRDM9 (table 1 and supplementary fig. S8, Supplementary Material online). We then compared the proportion of hotspots versus matched coldspots with a PBS (table 3). We also summed the total PBS count across all hotspots and coldspots over all submotifs and found significant enrichment of both the proportion of hotspots with a PBS and total motif count in the hotspots as compared to coldspots for all species (table 3), except when using our newly generated hotspots for western chimpanzee. Both human submotifs were significantly associated with higher PBS counts and proportions in hotspots versus coldspots, though the submotif derived from Allele C was marginally significant in CEU hotspots. Using the western chimpanzee hotspots from Auton et al. (2012), we found that the submotifs derived from PRDM9 alleles Pt1, W6, and to a lesser extent A1 were significantly associated with more PBS counts in hotspots versus coldspots. However, for our newly generated set of hotspots in the western chimpanzee, only the Pt1-derived submotif remained significantly enriched in hotspots. In the Nigerian chimpanzee, the submotifs derived from W6, E1, and Pt1 were all found significantly more often in hotspots than in coldspots. The lack of association between hotspots and the A1-derived submotif in Nigerian chimpanzees suggests that allele carrying this submotif may not be segregating in P. t. ellioti, but only direct sequencing of PRDM9 alleles in the relevant samples could address this question definitively. Bonobo showed a weak association with the submotifs derived from PRDM9 alleles A1, E1, and Pt1 but not W6, which is not surprising as this is most likely a derived allele of PRDM9 being only found in western chimpanzees. Finally, gorillas showed a strong association with both Gg1-derived submotifs (see table 3 and Materials and Methods).
Table 3.
PRDM9 Results Summary.
| Recombination Data | Proportion of Regions with a PBS |
Binomial Test P Value | Total Motif Count |
Binomial Test P Value | Hotspot Source | |||
|---|---|---|---|---|---|---|---|---|
| Submotif | Hotspots | Cold Spots | Hotspots | Cold Spots | ||||
| HapMap CEU | 0.50 | 0.45 | 2.70E-16 | 41,984 | 39,168 | 4.95E-23 | HapMap | |
| Allele A | 0.38 | 0.33 | 2.23E-23 | 27,808 | 25,390 | 1.06E-25 | ||
| Allele C | 0.29 | 0.27 | 1.29E-06 | 14,176 | 13,778 | 0.02 | ||
| HapMap YRI | 0.49 | 0.44 | 7.71E-22 | 40,732 | 37,687 | 1.58E-27 | HapMap | |
| Allele A | 0.38 | 0.32 | 9.04E-29 | 26,901 | 24,346 | 1.57E-29 | ||
| Allele C | 0.28 | 0.26 | 7.77E-07 | 13,831 | 13,341 | 3.01E-03 | ||
| Western chimpanzee | 0.64 | 0.60 | 0.02 | 5,776 | 5,480 | 0.01 | PanMap | |
| Western | 0.28 | 0.27 | 0.26 | 1,615 | 1,502 | 0.04 | ||
| A1 | 0.21 | 0.19 | 0.04 | 1,143 | 1,042 | 0.03 | ||
| E1 | 0.26 | 0.26 | 0.93 | 1,427 | 1,499 | 0.19 | ||
| Pt1 | 0.28 | 0.25 | 8.04E-04 | 1,591 | 1,437 | 0.01 | ||
| Western chimpanzee | 0.60 | 0.59 | 0.62 | 12,599 | 12,410 | 0.23 | This study | |
| Western | 0.27 | 0.28 | 0.26 | 3,393 | 3,534 | 0.09 | ||
| A1 | 0.21 | 0.21 | 0.92 | 2,490 | 2,496 | 0.94 | ||
| E1 | 0.25 | 0.26 | 0.13 | 3,163 | 3,241 | 0.34 | ||
| Pt1 | 0.28 | 0.25 | 1.61E-03 | 3,553 | 3,139 | 4.41E-07 | ||
| Nigerian chimpanzee | 0.63 | 0.58 | 2.55E-04 | 9,316 | 8,520 | 2.62E-09 | This study | |
| Western | 0.29 | 0.26 | 3.85E-05 | 2,654 | 2,356 | 2.70E-05 | ||
| A1 | 0.21 | 0.20 | 0.29 | 1,807 | 1,745 | 0.31 | ||
| E1 | 0.27 | 0.25 | 0.01 | 2,413 | 2,214 | 3.60E-03 | ||
| Pt1 | 0.27 | 0.25 | 0.01 | 2,442 | 2,205 | 5.35E-04 | ||
| Bonobo | 0.61 | 0.59 | 0.05 | 14,081 | 13,369 | 1.77E-05 | This study | |
| Western | 0.29 | 0.28 | 0.08 | 3,968 | 3,859 | 0.22 | ||
| A1 | 0.23 | 0.21 | 0.01 | 2,928 | 2,739 | 0.01 | ||
| E1 | 0.27 | 0.25 | 0.04 | 3,556 | 3,357 | 0.02 | ||
| Pt1 | 0.27 | 0.26 | 0.04 | 3,629 | 3,414 | 0.01 | ||
| Gorilla | 0.32 | 0.30 | 1.39E-05 | 10,384 | 9,421 | 8.08E-12 | This study | |
| Gg1-1 | 0.23 | 0.21 | 1.94E-03 | 6,429 | 6,080 | 1.86E-03 | ||
| Gg1-2 | 0.15 | 0.13 | 2.49E-06 | 3,955 | 3,341 | 6.93E-13 | ||
Note.—Comparison between identified hotspots and matched cold spot regions based on proximity, size, and GC-content. For each set of hotspots, the proportion of hotspots versus cold spots that contain a predicted binding sequence (PBS) based on species-specific PWM of the PRDM9 sequence. This is contrasted on the right with the total predicted binding motifs present in the combined sets of hotspots versus cold spots. Results for each relevant submotif and the combined results are presented for each genetic map with the source of the hotspots listed.
For each submotif of PRDM9, we further analyzed the PBS count relative to hotspot strength (supplementary fig. S9, Supplementary Material online) in hotspots versus coldspot regions. We posited that if PRDM9 activity is indicated by PBS count, then the difference in PBS count between hotspot and coldspot regions should be most pronounced in the strongest hotspots, where PRDM9 activity is likely to be high. On the basis of partitioning of hotspots by relative recombination intensity, we found that for the submotifs that are significant in table 3, there is evidence of more binding sites in hotspots relative to coldspots for increasing hotspot strength. The main deviation from table 3 is that the submotif derived from the putatively ancestral PRDM9 allele, A1, not significant overall in chimpanzee, has higher hotspot motif counts in stronger hotspots, suggesting some historical signature of binding remains at least for the strongest hotspots.
In addition to examining hotspot intensity and predicted PRDM9 binding activity, we examined the distribution of predicted binding sites across both hotspot and cold spot regions (supplementary fig. S10, Supplementary Material online). Based on a recent study in humans, there is an expectation that PRDM9 binds near the center of recombination hotspots (Pratto et al. 2014). To test if our data also supported this pattern, we compared the distribution of predicted binding locations of PRDM9 hits relative to the center of either hotspots or cold spots, limiting our analysis to hotspots less than 5 kb. Humans, bonobos, and gorilla exhibit a significant difference in the overall distribution of PBS hits in hotspots compared to cold spots based on a Wilcoxon test (supplementary fig. S10, Supplementary Material online), though these results are most striking for humans and not significant for chimpanzees.
To determine if the observed high GC content of the PRDM9 motifs drives the difference in predicted binding of hotspots and coldspots, we also partitioned the hotspot/coldspot regions based on their respective GC content and re-examined the distribution of PBS data (supplementary fig. S11, Supplementary Material online). If GC content drives the signal, then we would expect to observe a significant difference between hotspots and coldspots only in the highest GC bin. Our results indicate that PBS counts persist across all GC bins and for most submotifs, indicating that GC-content is not driving the results in table 3. There are three exceptions to these results: 1) the Pt1-derived submotif, significant in both western chimpanzee hotspot data sets, shows a possible GC-content signal based on PanMap hotspots (supplementary fig. S11C, Supplementary Material online), though this result is the opposite for the hotspots generated in this study (supplementary fig. S11D, Supplementary Material online), 2) for Nigerian chimpanzee, the lowest GC bin supports a GC signal, though the middle quartiles show a larger difference than either extreme GC bin, consistent with the overall results (supplementary fig. S11E, Supplementary Material online), and 3) for bonobo, the E1-derived submotif has a stronger signal with increasing %GC, suggesting the result in table 3 for this submotif could be partly driven by GC-content (supplementary fig. S11F, Supplementary Material online).
Genome-Wide Predictions of PRDM9 Binding to Compare to Previous Work in Chimpanzees
For a more direct comparison with the western chimpanzee analysis performed in Auton et al. (2012), we also performed a genome scan for each submotif and summed the results over all submotifs for each group. We then compared the results with a corresponding null motif (supplementary fig. S12, Supplementary Material online). The genome-wide scan did not reveal any significant association between recombination rate and the submotifs in Nigerian chimpanzee and bonobo, similar to previous results in western chimpanzees. Conversely, we saw higher recombination rate at both human submotifs in YRI, the submotif derived from Allele A in CEU, and the second submotif derived from Gg-1 in gorillas (table 1). We further split the genome-wide data based on whether each 1 kb region had 0, 1, or 2+ predicted binding sites and further split these into GC quantiles as was done for the hotspot/coldspot analysis (supplementary fig. S13, Supplementary Material online). For simplicity, we summed these results across all submotifs. We found that the recombination rate of both human and gorilla is higher in regions with higher PBS counts and that this difference is consistent across GC bins. When we compared these results to the null motifs, we found that the difference between PBS count categories was much less pronounced. In contrast, both the real and null motif result in the Pan species were similar and neither showed a marked increase in recombination rate with increased binding sites in any GC bins, consistent with the full genome-wide search results.
Distribution of Recombination Rate across Genome
Another way to compare the recombination rates within and between great apes is to look at the distribution of recombination rates across the genome. Similar to previous studies, we found a biased recombination rate distribution whereby the majority of recombination (∼75%) occurs in a small fraction of the physical genome (∼20%). From the corresponding Gini coefficient using the area under the curve of the Lorenz curve (fig. 3A), we found that the European human population has the strongest hotspot usage across the genome, similar to previous studies (fig. 3B). We confirm the differences between the CEU (Gini = 0.771), YRI (Gini = 0.688), and chimpanzee (Gini = 0.677) maps previously published. For the new maps, we calculate a Gini coefficient that lies within the values of the extremes of these previously published maps. Specifically, we calculate a Gini coefficient of 0.704 for gorilla, 0.713 for bonobo, and 0.726 for Nigerian chimpanzee. Using this statistic, we are able to show that the extent of recombination rate variation across the genome is quite similar across great apes, with more variation between human populations than across these diverse species. The values from the recently published survey of Gini coefficients across various species were included as a reference point in figure 3B (Kaur and Rockman 2014). It is also worth noting that the values in the Kaur and Rockman (2014) study were from direct measures of recombination rather than indirect methods used here. To illustrate this point, the human reference point included in figure 3B is from the Kong et al. (2010) study and is much higher than the CEU estimate here presumably from similar populations. This comparison suggests that Gini coefficients from population-scaled recombination rate estimates are likely underestimates of the values obtained for a direct pedigree-based method. This difference could reflect methodological differences in rate estimation, recent changes in the human recombination rate, or subpopulation differences in recombination rate. Finally, it is worth noting that the Gini coefficient estimated from LD data is sensitive to differences in Ne (Auton et al. 2013) and the slight variation between these taxa may mostly represent variation in effective population sizes rather than differences in the distribution of recombination rate across the genome.
Fig. 3.

Genome-wide distribution of recombination rates. Cumulative distribution or Lorenz curve of recombination rate plotted as proportion of recombination versus sequence for each recombination map (A). The diagonal represents a uniform distribution. Gini coefficients for each population map, and for comparison, other taxa reported in Kaur and Rockman (2014), including a human estimate from Kong et al. (2002) (B).
Broad-Scale Comparisons
As discussed in the introduction, most between-species comparisons of recombination rates have focused on either evolutionarily very close relatives or quite distant relatives. Because our data set represents a large swath of evolutionary distance and includes multiple within- and between-species pairs, we wanted to compare the rate of nucleotide divergence to recombination rate divergence across great apes. We first binned the genome into 1 Mb, 500 kb, or 100 kb syntenic blocks, then calculated the Spearman rank correlation coefficient between all pairwise recombination rates. We then compared the correlation coefficients to the amount of nucleotide sequence divergence between each pair (fig. 4 and supplementary fig. S15A and B, Supplementary Material online). Using these data, we see closely related pairs of populations display a rapid decline in recombination rate correlation with increasing sequence divergence. Additionally, when we replace the YRI-CEU comparison with the quality control comparison of 10YRI-10CEU (fig. 4, gray dot) representing smaller samples sizes similar to the maps generated here, we still see a steep decline in recombination rate comparisons for the Pan species relative to nucleotide changes. In contrast, comparisons using species pairs with higher nucleotide divergence have relatively similar recombination rate correlations, regardless of which human comparison is used. Further, the correlation at decreasing bin sizes (supplementary fig. S15, Supplementary Material online) suggests that sequence divergence explains less of the variance in recombination rates at finer scales, as has been shown previously (Auton et al. 2012).
Fig. 4.

Recombination rate versus nucleotide divergence. Spearman rank correlation coefficient between all recombination maps at 1 Mb (y axis) versus the pairwise nucleotide divergence between pairs (x axis) with various comparisons labeled.
Another interesting broad-scale recombination pattern is the skew in recombination rates at the ends and near the center of chromosomes. We quantified the extent of this skew across species, controlling for differences in recombination rate in each chromosome (supplementary fig. S16, Supplementary Material online). While centromeric regions recovered to the mean of the chromosome within 5 Mb of the centromere, the skew at telomeres was more pronounced and continued for nearly 15 Mb from the chromosome end. We further looked at large-scale chromosomal changes across great apes, including the chromosome 2 fusion in humans and the chromosome 5/17 translocation in gorillas. We found that other nonhuman primates also have high recombination rates across the junction of chromosomes 2a/2b supporting its historical telomeric origin. However, bonobos have lower recombination rates across this region similar to what has been seen in humans (supplementary fig. S17A, Supplementary Material online), though this is most likely due to reduced sequencing coverage in this area for bonobos leading to less accuracy for recombination rate estimates. We further found that the translocation event in gorillas did not influence broad-scale recombination rates, likely because it did not involve centromeric or telomeric regions (supplementary fig. S17B and C, Supplementary Material online).
Multiple Linear Regression Analysis
Using a multiple linear regression framework, we evaluated the correlates of various genetic features with both the rate of recombination, as well as the increase in recombination rate relative to the human–chimpanzee ancestor of Munch et al. (2014) (fig. 5). Briefly, this study used an HMM to reconstruct approximately 1 million ancestral crossover events between humans and chimpanzees. Note that we are reporting standardized (beta) coefficients, so the x-axis in figure 5 represents the relative importance of each of these factors in predicting recombination rate and change in rate. Further, the power of using so many different taxa that share a common ancestor is that it now becomes possible to disentangle results that would be ambiguous with only one genetic map. As correlations between recombination rates and our independent variables, such as GC-content, diversity, and divergence, may have a complicated, possibly interacting/nonlinear relationship in genic versus nongenic contexts, we excluded genic, as well as phylogenetically conserved bases from phastCons elements (see Materials and Methods) to simplify the interpretation of the results. To further disentangle substitution patterns from the quasi-selective effects of gBGC, we looked solely at transversions that were strong to strong (G↔C) or weak to weak (A↔T) for our divergence and diversity statistics, while we looked at the change in GC-content in substitutions for assessing equilibrium GC-content as per Duret and Arndt (2008). It is worth noting that the error bars at fine scales are much smaller owing to the larger number of intervals at smaller size scales. Likewise, there is likely more power to detect differences in the coefficients at finer scales, though several factors show larger coefficients at larger scales, for example, diversity.
Fig. 5.
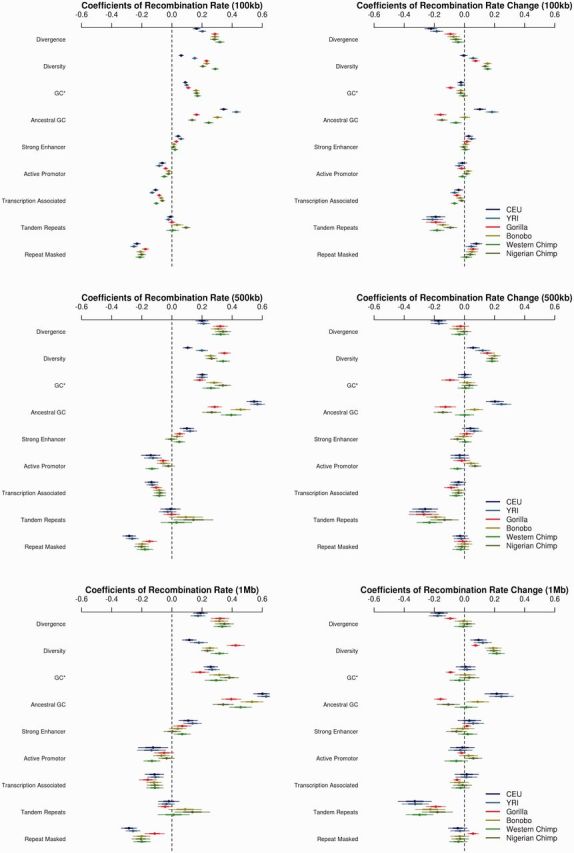
Genetic correlates of recombination rate. The predictors of recombination rate (left) and the change in recombination rate since the human–chimpanzee ancestor (right) at varying physical scales (top to bottom). Within each pane, multiple linear regression coefficient estimates (with standard errors, SEs) are shown for each of the independent variables across taxa. Larger coefficients reflect larger effects, while smaller SEs correspond to larger correlations.
Both diversity and divergence show significant positive correlations with recombination rate across all taxa and all size scales (fig. 5A–C). Further, diversity as a predictor of recombination rate variation becomes more pronounced at larger size scales (supplementary fig. S14A, Supplementary Material online). While the diversity correlation is consistent with previous work, the divergence correlation is a bit more puzzling. Typically, the correlation between divergence and recombination is used to disentangle the effects of mutagenic recombination or patterns of linked selection (Begun and Aquadro 1992). There is evidence that recombination has a mutagenic effect (Pratto et al. 2014; Arbeithuber et al. 2015), though these associations largely involve CpG mutations, and in the case of Arbeithuber et al. (2015), were only seen in transitions. Further, gBGC, associated with higher recombination, may also impact estimates of divergence by influencing the fixation probability of alleles. However, as we only used S↔S and W↔W transversion mutations, neither of these two processes should influence our coefficient estimates. Further, as linked selection does not influence substitution rates, save in the ancestral species (Birky and Walsh 1988), which is the same ancestor in this analysis across taxa, this correlation cannot be attributed solely to linked selection in the ancestor of humans and orangs because the human confidence intervals do not overlap the confidence intervals of the other apes. Similar findings were obtained from the use of the African individuals sequenced in Prado-Martinez et al. (2013), suggesting this is not an artifact of different sequencing technologies. Further, divergence is positively correlated with recombination rates, while it is weakly associated with a deceleration in recombination rate relative to the human-chimpanzee ancestor, especially at smaller scales (100 kb) (fig. 5A vs. 5D).
Consistent with previous studies, the rate of recombination is positively correlated with both ancestral GC-content (Kong et al. 2002; Jensen-Seaman et al. 2004) and an estimate of the equilibrium GC-content (GC*, see Materials and Methods) (Duret and Arndt 2008; Munch et al. 2014). Further, the magnitude of the coefficients for ancestral GC-content are largely nonoverlapping, with ancestral GC consistently a stronger predictor than GC* in all groups except Nigerian chimpanzee. Note that the ρ values inferred from LDhat were converted into z-scores, so this ordering does not reflect mere tautologies but rather may reflect an increased association of ancestral GC-content with local recombination rate. Additionally, the difference between ancestral GC and GC* is most apparent in humans, which may reflect some demographic change in the human-specific lineage. The relative strength of these associations is consistent across all scales (supplementary fig. S14B, Supplementary Material online).
The ENCODE annotations (strong enhancer, active promoter, and transcription associated) provide some of the smallest effect sizes of all annotations. Similar to previous works (Kong et al. 2002; Jensen-Seaman et al. 2004), recombination is negatively correlated with genic activity, as measured by transcription and active promoters. Enhancers show an occasional significant positive correlation with recombination rate, which may just reflect recombination being low in gene-rich regions (see supplementary material, Supplementary Material online).
Discussion
By extending the number of whole-genome fine-scale genetic maps, we present a major advance in understanding recombination rate evolution across great apes. First, our results show that few hotspots are shared between chimpanzees and bonobos, suggesting that complete hotspot turnover takes on the order of millions of years. Although our results show nearly complete hotspot turnover in the Pan species examined here, our hotspot overlap analysis (table 2) suggests higher sharing in Pan species than expected by chance. Second, we have shown that PRDM9 binding likely determines the locations of recombination hotspots across great apes not just in humans as had been shown previously. While we report significant enrichment of putative PRDM9 binding in recombination hotspot regions as compared with coldspot regions, we did not observe a significant association between PRDM9 binding broadly across the genome and local increases in recombination rates. Below we discuss some of the reasons for the incongruence between the genome-wide results and the hotspot results. Further, we have analyzed broad-scale patterns of recombination rate evolution and showed that the divergence in recombination rates between species occurs rapidly relative to the divergence of nucleotide sequences and perhaps more closely tracks population divergence times. We showed that in gorilla, a translocation between chromosomes 5 and 17 does not display any broad-scale recombination rate differences when compared to other species studied here. Finally, we showed that a subset of genetic features explains the majority of the variation in recombination rate and that the amount of variation explained depends in part on species-specific patterns of natural selection and demography.
Within- and between-Species Comparisons of Recombination Rate Evolution
Access to multiple within- and between-species comparisons across great apes allowed us to perform a comprehensive comparison of recombination rate variation at fine scales. The two chimpanzee subspecies diverged approximately 400–600 ka (Bowden et al. 2012), whereas the two human populations diverged approximately 70–80 ka. We find that most hotspots are shared between human populations but that very few hotspots are shared between chimpanzee subspecies, indicating a very rapid change in the hotspot landscape over a short evolutionary time period. The lower coverage of individuals (mean 9.45× coverage) and the high error rate (34–41%) at the fine scale (<10 kb) reported in the western chimpanzee study (Auton et al. 2012) indicates that individual SNP calls may not be accurate and direct comparisons with western chimpanzee may not be appropriate. Though we find similar results (of very few shared hotspots) in comparisons between bonobos and chimpanzees (fig. 2D and table 2), suggesting that near-complete hotspot turnover happens within 1–2 My.
It is worth noting that a recent study examining the recombination rates of chimpanzees at human hotspots was able to identify approximately 30 overlapping regions between chimpanzees and human on chromosome 21, where both YRI and western chimpanzees had high inferred recombination rates (Wang and Rannala 2014). It is unclear whether the extent of hotspot overlap found in the study was more than what is expected by chance, especially if one controls for GC content. Further work will be needed to answer this question. Nonetheless, our results of a quicker turnover are consistent with a recent study suggesting very little overlap of recombination hotspots between modern humans and Denisovans, as inferred from a disruption in equilibrium GC-content indicative of past or present recombination activity in primates (Lesecque et al. 2014). They further propose that the rapid turnover of hotspots presents a solution to the hotspot paradox, whereby the self-destructive nature of hotspots drives selection for new PRDM9 alleles. While they did not directly estimate recombination rates in Denisovans, they were able to quantify the expected lifespan of a recombination hotspot in humans to be approximately 3 My based on degeneration of the human PRDM9 submotif sequences in the Densiovan genome. Our results suggest that hotspot turnover in chimpanzees may occur more rapidly, due in part to the higher polymorphism at PRDM9. Most likely, the recent bottleneck in modern humans slowed this hotspot turnover process by reducing the diversity at PRDM9, which seems to occur more rapidly along the chimpanzee branch than the human branch. This presents an interesting example of how demographic history can impact the time scale of recombination rate evolution.
PRDM9 Predicted to Bind to Great Ape Recombination Hotspots
As listed in table 3, we find strong evidence that PRDM9 likely binds to recombination hotspot regions more frequently than to coldspot regions broadly across great apes. In fact for all other groups of great apes, we find at least one submotif of PRDM9 enriched across hotspot regions as compared to coldspot regions. Because the signal in western chimpanzees is mainly reflected in the newly identified internal submotif, it is not surprising that the earlier study in this group failed to identify an important role for PRDM9 in recombination rate association. Indeed the western-chimpanzee-specific submotif of PRDM9 is most strongly associated with predicted binding in Nigerian chimpanzee recombination hotspots. This is possibly due to shared PRDM9 alleles between these two subspecies. However, the Nigerian chimpanzees have not been previously included in surveys of PRDM9 diversity. Further, the putatively ancestral A1-derived motif does not seem to be active in this group despite its activity in bonobos, supporting the rapid turnover of hotspot landscapes observed between these groups. Additionally, the PRDM9 submotif which is putatively ancestral across chimpanzee subspecies seems to be active in the outgroup of bonobo, suggesting it has been active since prior to the bonobo-chimpanzee split. By breaking hotspots into groups with potential binding of various submotifs of PRDM9, we can break down the hotspot landscape. This supports recent evidence in humans that LD-based recombination rate estimates represent a composite landscape with distinct landscapes superimposed to yield a population average (Pratto et al. 2014).
While this composite landscape of recombination activity may help explain the lack of association in the previous chimpanzee recombination study, another source of complication was the approach for identifying an association. By searching across the whole genome as opposed to focused hotspot regions as we did here, the earlier study was more prone to difficulties of computational predictions of PRDM9 binding (see supplementary material, Supplementary Material online, for details). Nonetheless, to compare our results to those of Auton et al. (2012), we further examined rate differences associated with PBSs along the genome irrespective of local recombination rate. We compared recombination rates in regions with a PBS based on the species-specific PRDM9 PWM versus a null version generated by shuffling the original PWM. We found higher recombination rates near PRDM9 PBSs versus the null PBS for gorilla, similar to humans (supplementary fig. S12, Supplementary Material online), and irrespective of GC content (supplementary fig. S13, Supplementary Material online) but not for Pan species. We attribute this to the loss of sensitivity of a genome-wide search in the Pan group due to the high diversity at the PRDM9 locus in the Pan genus (Schwartz et al. 2014). The higher allelic diversity at PRDM9, especially in Pan species, likely contributes to population rate estimates based on patterns of LD being a composite of multiple distinct hotspot landscapes (Pratto et al. 2014). Further, LD-based maps are less likely to reflect recent changes in the recombination landscape and the rapid turnover of hotspots in this group may also render a genome-wide approach difficult in uncovering a true association between specific PRDM9 submotifs and recombination rates. In human and gorilla, both the age of the alleles and the frequency in the population sampled (table 1) likely yield higher power to detect such a signal, which may also explain the reduced signal for both the submotif derived from Human Allele C and the gorilla submotif Gg1-1. Like previous studies, a lack of a signal in our genome-wide results can be explained by a variety of computational challenges.
However, our overall results provide strong evidence that PRDM9 helps localize recombination hotspots in great apes. Further, we showed that the difference in PBS count between hotspot and cold spot regions is more pronounced as both recombination hotspots of increasing strength and as regions closer to the center of the hotspot region are considered. We also showed that GC content does not drive this pattern, which persists even across regions with the lowest GC-content. Therefore, our study represents the first evidence that PRDM9 may be more ubiquitous in determining recombination rate activity in primates. Future work should both focus on direct recombination initiation maps from individuals homozygous for specific PRDM9 alleles as was done recently in humans (Pratto et al. 2014) and should also work to generate Chip-Seq data for PRDM9 during meiosis to gain a better understanding of the binding locations of this protein in vivo (Segurel 2013).
Distribution of Recombination Rate across the Genome
We found that variation in the great ape recombination maps presented here are similar to other vertebrate taxa which have functional PRDM9 (Schwartz et al. 2014). These results are consistent with previous work reporting a dominant PRDM9 allele for determining hotspot locations across the genome in European populations, driving the bias towards hotspot usage (Altshuler et al. 2010). Further, higher allelic diversity and levels of within population heterozygosity of PRDM9 likely contribute to a more even distribution of recombination rates across the genome, with distinct hotspot landscapes averaged over the longer population history of chimpanzee, bonobo, and gorilla (Berg et al. 2011; Schwartz et al. 2014). However, because the amount of variation in Gini coefficients across great apes is rather small, it would be difficult to distinguish between the impact of PRDM9 variation and variation in effective population size (Ne) (Auton et al. 2013). Further, the previous application of the Gini coefficient to recombination rate data excluded LD-based maps due to the potential biases caused by natural selection and gene conversion. Nonetheless, while not converted to Gini coefficients, previous studies have made inferences from the cumulative distribution of recombination rates across the genome from LD-based maps that seem to agree with differences in PRDM9 diversity (Frazer et al. 2007; Altshuler et al. 2010; Auton et al. 2012).
Broad-Scale Recombination Rate Changes Occur More Rapidly than Nucleotide Divergence
Because fine-scale recombination rates change rapidly within species, focusing on broad-scale recombination rate changes between species allowed us to identify changes that occurred over longer evolutionary time frames. These results suggest that the amount of change in recombination rate over time plateaus after a few millions years with gorilla versus human comparisons largely overlapping chimpanzee versus human ones (fig. 4 and supplementary fig. S15, Supplementary Material online).
We further analyzed the skew of recombination rates at chromosome ends across great apes and found a stronger skew at telomeric regions than centromeric regions. Previous studies account for this skew by removing approximately 5–10 Mb nearby centromeres and telomeres (Serre et al. 2005), which while sufficient for centromeres but may not adequately account for the telomeric skew in great apes. We also plotted recombination rates across the chromosome 5/17 translocation present in gorillas and found that unlike the chromosome 2 fusion event, this does not seem to disrupt the broad-scale recombination landscape (supplementary fig. S17B and C, Supplementary Material online). This is perhaps due to the fact that while the chromosome 2 fusion in humans leads to broad-scale rate differences, this change can be explained by the conversion from telomeric to centromeric regions. However, the translocation in gorillas does not involve any chromosome end regions nor does it alter the relative distance to the chromosome ends for either chromosome.
Regression Analysis
We used a multiple linear regression analysis to evaluate the relationships between recombination rate and rate change with various genetic features. Unlike simple correlations, our approach allows us to determine the relative importance of each feature to the overall variation in each of these variables. We find the strongest positive predictors of recombination rate (fig. 5A–C) are diversity, divergence, and ancestral GC content across all scales analyzed here. We showed that the equilibrium GC content (GC*) increases in importance with increasing scale. We also showed that repeat masked regions and to a lesser extent genic annotations are negatively associated with recombination rate. We further examined recombination rate change in relation to the human-chimpanzee ancestor. Both divergence and repeats are associated with a decelerated recombination rate, while diversity is associated with an accelerated recombination rate.
A relationship between the local Ne (measured as π/divergence to the human-orang ancestor) and recombination is not surprising, as it is consistent with the predictions of background selection (Hudson and Kaplan 1995; Nordborg et al. 1996). As bases under direct forms of selection (genes and conserved elements) are not included in this analysis, and ENCODE annotations are partitioned into separate coefficients, if directional selection is invoked as an explanation for this relationship, then it is due to the action of selection at linked sites. As linkage to selected bases should be more pronounced in areas of low recombination, the coefficient differences we see across taxa may reflect differences in differences in the distribution of fitness effects across apes. Alternative explanations include the possible inclusion of unannotated bases under directional selection (Asthana et al. 2007), which would also explain why diversity is correlated with an accelerated recombination rate. However, if recombination rates are accelerating, then levels of background selection would become reduced, which may also explain this relationship. The strong positive correlation with divergence is surprising, as we constrained ourselves to only looking at W->W and S->S transversions, which should be invariant to both gCBC and the documented mutagenic effects of recombination found in Arbeithuber et al. (2015). An explanation for this strong positive association despite having removed most functionally annotated sites is that functional density (regions subject to selection) is nonrandomly distributed and perhaps is higher in low recombining regions (Cutter and Payseur 2013), where the bases in question are unannotated, or alternatively, that recombination-associated processes influence the probability of substitution at strong to strong and weak to weak transversion sites in a previously undocumented fashion.
Overall, these results suggest that despite rapid turnover in local recombination rates, correlations between specific genetic features and recombination rates are consistent across great apes, but the degree is contingent on the sample size or the total depth of the coalescent history for a given population as well as population-specific factors such as the strength of selection or unique demographic processes in these taxa.
Materials and Methods
Samples, Sequencing,and SNP Calling
Samples for the fine-scale recombination maps presented here were collected and described in the Great Ape Genome Diversity Consortium (Prado-Martinez et al. 2013). From the 88 samples described, 38 were used here to estimate genome-wide recombination rates for three populations of three major species: 13 bonobos without known geographical origin (Pan paniscus); 10 chimpanzees from Nigeria (Pan troglodytes ellioti); and 15 western gorillas from Cameroon and Congo (Gorilla gorilla gorilla). This subset of individuals is described in supplementary table S1, Supplementary Material online.
A detailed description of sequencing, reference mapping, and SNP/variant calling can be found in Prado-Martinez et al. (2013). Briefly, samples were sequenced on an Illumina sequencing platform (HiSeq 2000) with data production at four different sequencing centers, sequence reads were mapped to both the human reference genome (hg18) and each species-specific reference (PanTro 2.1.4, Ensembl release 65 for Pan and gorGor3, Ensembl release 62 for Gorilla). SNP calling was performed using the Genome Analysis Toolkit (GATK) software (DePristo et al. 2011). Final coverage for the individuals used here is included in supplementary table S1, Supplementary Material online.
Recombination Rate Estimation
The processes of data filtering and rate estimation were carefully matched to be similar to those used in the recent PanMap project (Auton et al. 2012). Using both the human-based mapping and the species-specific mapping of the reads for each species, the data were filtered using a combination of VCFtools (Danecek et al. 2011) and custom scripts (Stevison 2015). See supplementary methods, Supplementary Material online, for details on this filtering process. To maintain comparable inherited segments of the genome, regions that were syntenic between each nonhuman primate and humans were defined as described in supplementary methods, Supplementary Material online. See supplementary figure S1, Supplementary Material online, for the distribution of block sizes for each species. See supplementary figures S2–S4, Supplementary Material online, for plots of each set of coordinates as mapped in the human reference versus the nonhuman reference genome, highlighting large-scale differences from human in orientation for each species.
Computational phasing and imputation to infer bases at missing sites was performed on each syntenic block using the software fastPHASE v1.2 (Scheet and Stephens 2006). Then, for improved phasing accuracy, the variants were re-phased using PHASE v2.1 (Stephens and Donnelly 2003) as described in Auton et al. (2012). See supplementary methods, Supplementary Material online, for additional details. An additional filter based on minor allele frequency was performed afterward (cutoff = 0.05). Filtered syntenic blocks were split into 4,000 SNP blocks with 100 SNP overlap and converted to input for the software LDhat v2.1 (Fearnhead and Donnelly 2001; International HapMap 2005). LDhat was run for 60 million iterations with a block penalty of 5, sampling every 40,000 steps (Auton et al. 2012).
Comparisons to Existing Maps
To get comparable recombination rate data for the published human and western chimpanzee maps, source data were downloaded and converted as described in supplementary methods, Supplementary Material online. Further, our map estimates were converted from ρ/kb to cM/Mb following the approach of McVean et al. (2004), which yielded Ne estimates of 13,428, 16,781, and 19,785 for bonobos, Nigerian chimpanzees, and gorillas, respectively. These Ne values yielded the same adjusted rate estimate of approximately 1.193 cM/Mb for bonobo, Nigerian chimpanzee, and gorillas (see supplementary methods, Supplementary Material online, for details).
Together with western chimpanzee, the block boundaries for regions that are syntenic to human were intersected across the four nonhuman maps and the two human maps as described in supplementary methods, Supplementary Material online. These “multi-syntenic” blocks were generated to give average rate estimates in bins of 1 Mb, 500 kb, and 100 kb. Supplementary figure S5, Supplementary Material online, shows a boxplot of the mean values across all six maps using the 1 Mb binned data set and the full 1 Mb binned data set can be found in supplementary table S2, Supplementary Material online.
Hotspot Determination and Sharing between Populations
LDhot uses a composite-likelihood framework based on the work of Hudson(2001) and McVean et al. (2002). The Auton et al. (2012) implementation tests every 2 kb region (with a 1 kb increment) as a potential hotspot by analyzing the 200-kb region centered around the region of interest. Auton et al. (2014) used a smaller window size (100 kb) but the same basic approach for identifying candidate regions. Our new approach here is to use a 20-kb window size to yield greatly improved power to detect less intense recombination hotspots. A detailed comparison of our method and the two former approaches can be found in supplementary methods, Supplementary Material online.
To identify the degree of overlap between hotspots (labeled “hotspot overlap” above), we started with 442 syntenic blocks that were >1 Mb in length. We trimmed 10 kb off each end of these blocks. Then, we randomly permuted the start sites of each hotspot, keeping it in the same syntenic block it started in and keeping the hotspot lengths unchanged. We did this 1,000 separate times and tabulated the average number of permuted hotspots whose boundaries overlapped each other (by at least 1 bp). We also tabulated the excess of observed hotspot overlaps compared with the expected number (i.e., the observed number of hotspot overlaps divided by the average number of simulated hotspot overlaps for a pair of populations) (table 2).
Comparisons between Existing and Newly Identified Hotspots
To compare recombination rates at hotspots identified here and in previous studies, source data were downloaded and converted as described in supplementary methods, Supplementary Material online. Next, the coordinates for each full recombination map were rescaled to reflect the relative location ±20 kb to the center of each set of hotspots. Then, a loess smoothing was applied to the rate estimates using the rescaled coordinates across all hotspots in each map (fig. 2). Finally, the same method used to identify hotspots for the three recombination maps generated in this study was applied to the phased haplotype data from PanMap. This resulted in a set of 9,993 hotspots in western chimpanzee (as compared to 5,038 from the original set of western chimpanzee hotspots). Supplementary figure S7, Supplementary Material online, shows the plot of this new set of western chimpanzee hotspots with rates from all six compared maps (similar to fig. 2B).
Examining the Relationship between PRDM9 Binding Motifs and Hotspots
Previous work has shown higher recombination rates in PRDM9 predicted binding sites in humans but not in western chimpanzees. To further investigate the importance of PRDM9 in localizing population-specific hotspots, we downloaded the protein sequences of PRDM9 from previous studies (Berg et al. 2011; Auton et al. 2012; Schwartz et al. 2014). We then predicted binding motifs for the zinc-fingers of the protein sequence using http://zf.princeton.edu/ (last accessed October 29, 2014) and the polynomial SVM model as described in Persikov et al. (2009). This analysis included eight submotifs (table 1) with different combinations searched across six recombination maps (including the newly defined set of hotspots for western chimpanzee). See supplementary methods, Supplementary Material online, for submotif descriptions and sources.
To examine the prevalence of PRDM9 in explaining hotspot distribution, we computationally identified matched coldspots across the genome as described in supplementary methods, Supplementary Material online. We then extracted the fasta sequence for both the hotspots and coldspots from the masked version of the species-specific reference genome. Next, we used the software “fimo” to identify PBSs within the fasta sequence of the hotspots and coldspots (Grant et al. 2011). In table 3, we report the results for individual submotifs for each map and set of hotspots used here and the analysis summed across all submotifs. In addition to total number of predicted binding regions for both hotspots and coldspots, we summed up the number of nonzero regions for each to yield a proportion of regions with any suspected PRDM9 binding. To explain why our results were different from those of Auton et al. (2012), we also performed a genome scan for each submotif. In supplementary figure S11, Supplementary Material online, we plot the results of our genome-wide survey of recombination rate enrichment at PRDM9 submotif predicted binding regions as compared to a null motif (see supplementary methods, Supplementary Material online).
Genomic Distribution of Recombination Rates
To get cumulative distributions of recombination rates across each recombination map, the absolute physical and genetic distance for each interval was calculated, sorted relative to genetic distance, and summed to 1 for both physical and genetic distance values across the full data set (Stevison 2015). From these data, we plotted the Lorenz curve and Gini coefficient for all six maps (fig. 3).
Broad-Scale Comparisons
Pairwise nucleotide divergence between each population was taken from supplementary table S5.2 of Prado-Martinez et al. (2013). To compare each map, the 1 Mb rate estimates from the multisyntenic regions as defined between all six maps were fitted to a regression in R and the Pearson correlation coefficient between each pairwise comparison was computed.
Using the 1 Mb binned multisynteny data set, we examined variation in the skew of recombination typically observed at chromosome ends (Serre et al. 2005). We then plotted the skew at telomeres (supplementary fig. S15A, Supplementary Material online) and centromeres (supplementary fig. S15B, Supplementary Material online) in 1 Mb bins for the first and last 25 Mb relative to the chromosome ends for all six comparative maps. In addition to skews in recombination due to chromosomal location, we examined how large-scale changes in chromosomal structure impacted recombination rates in great apes. We examined the chromosome 2 junction across great apes and the translocation of human chromosomes 5 and 17 in gorillas.
Multiple Linear Regression Analysis
From the UCSC genome browser, we downloaded three classes of repeats, Repeating Elements (v. 3.2.7), Simple Tandem Repeats, Microsatellites, two classes of functional elements, exons from the CCDS project and phastCons elements from the 28-way placental mammals alignments, and three ENCODE annotations pertaining to gene activity—Transcription-associated, Active Promotor, and Strong Enhancer—from the Genome Segments track (from GM12878, combined Segway + ChromHMM).
We calculated nucleotide diversity (π), divergence, ancestral GC content, and GC* using the ancestral-sequence inferred for the common ancestor of humans and orangutans using the same methodology as described in Prado-Martinez et al. (2013). GC-flux was defined as the number of AT to GC substitutions divided by the number of GC to AT substitutions, and GC* was defined as GC-flux/(1+GCflux) as per Munch et al. (2014). Further details can be found in supplementary methods, Supplementary Material online.
Supplementary Material
Acknowledgments
This work was supported by the National Human Genome Research Institute of the National Institutes of Health under award number R01_HG005226 to M.F.H. and J.D.W. and National Institute of General Medical Sciences of the National Institutes of Health under Award Number F32GM101744 to L.S.S. A.E.W. was supported by National Science Foundation Graduate Research Fellowship Grant DGE-1143953. Computations for this study were performed on the QB3 cluster at UCSF. The authors thank Molly Przeworski and Laure Segurel for early access to gorilla PRDM9 sequences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- Altshuler D, Daly MJ, Lander ES. 2008. Genetic mapping in human disease. Science 322:881–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler D, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Collins FS, De la Vega FM, Donnelly P, Egholm M, et al. 2010. A map of human genome variation from population-scale sequencing. Nature 467:1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbeithuber B, Betancourt AJ, Ebner T, Tiemann-Boege I. 2015. Crossovers are associated with mutation and biased gene conversion at recombination hotspots. Proc Natl Acad Sci U S A. 112:2109–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asthana S, Noble WS, Kryukov G, Grant CE, Sunyaev S, Stamatoyannopoulos JA. 2007. Widely distributed noncoding purifying selection in the human genome. Proc Natl Acad Sci U S A. 104:12410–12415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, Fledel-Alon A, Pfeifer S, Venn O, Segurel L, Street T, Leffler EM, Bowden R, Aneas I, Broxholme J, et al. 2012. A fine-scale chimpanzee genetic map from population sequencing. Science 336:193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, Rui Li Y, Kidd J, Oliveira K, Nadel J, Holloway JK, Hayward JJ, Cohen PE, Greally JM, Wang J, et al. 2013. Genetic recombination is targeted towards gene promoter regions in dogs. PLoS Genet. 9:e1003984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, Myers S, McVean G. 2014. Identifying recombination hotspots using population genetic data. arXiv 1403.4264. Available from: http://arxiv.org/abs/1403.4264.
- Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, Coop G, de Massy B. 2010. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327:836–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun DJ, Aquadro CF. 1992. Levels of naturally-occurring DNA polymorphism correlate with recombination rates in Drosophila melanogaster. Nature 356:519–520. [DOI] [PubMed] [Google Scholar]
- Berg IL, Neumann R, Sarbajna S, Odenthal-Hesse L, Butler NJ, Jeffreys AJ. 2011. Variants of the protein PRDM9 differentially regulate a set of human meiotic recombination hotspots highly active in African populations. Proc Natl Acad Sci U S A. 108:12378–12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billings T, Parvanov ED, Baker CL, Walker M, Paigen K, Petkov PM. 2013. DNA binding specificities of the long zinc-finger recombination protein PRDM9. Genome Biol. 14:R35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birdsell JA. 2002. Integrating genomics, bioinformatics, and classical genetics to study the effects of recombination on genome evolution. Mol Biol Evol. 19:1181–1197. [DOI] [PubMed] [Google Scholar]
- Birky CW, Jr, Walsh JB. 1988. Effects of linkage on rates of molecular evolution. Proc Natl Acad Sci U S A. 85:6414–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleazard T, Ju YS, Sung J, Seo JS. 2013. Fine-scale mapping of meiotic recombination in Asians. BMC Genet. 14:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden R, MacFie TS, Myers S, Hellenthal G, Nerrienet E, Bontrop RE, Freeman C, Donnelly P, Mundy NI. 2012. Genomic tools for evolution and conservation in the chimpanzee: Pan troglodytes ellioti is a genetically distinct population. PLoS Genet. 8:e1002504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman KW, Murray JC, Sheffield VC, White RL, Weber JL. 1998. Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am J Hum Genet. 63:861–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks LD. 1988. The evolution of recombination rates In: Michod RE, Levin BR, editors. The evolution of sex: an examination of current ideas. Sunderland (MA): Sinauer Associates; 87–105. [Google Scholar]
- Browning SR, Browning BL. 2011. Haplotype phasing: existing methods and new developments. Nat Rev Genet. 12:703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunschwig H, Levi L, Ben-David E, Williams RW, Yakir B, Shifman S. 2012. Fine-scale maps of recombination rates and hotspots in the mouse genome. Genetics 191:757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AH, Jenkins PA, Song YS. 2012. Genome-wide fine-scale recombination rate variation in Drosophila melanogaster. PLoS Genet. 8:e1003090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, Wang X, Matise T. 2010. Contrasting methods of quantifying fine structure of human recombination. Annu Rev Genomics Hum Genet. 11:45–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole F, Baudat F, Grey C, Keeney S, de Massy B, Jasin M. 2014. Mouse tetrad analysis provides insights into recombination mechanisms and hotspot evolutionary dynamics. Nat Genet. 46:1072–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coop G, Przeworski M. 2007. An evolutionary view of human recombination. Nat Rev Genet. 8:23–34. [DOI] [PubMed] [Google Scholar]
- Coop G, Wen XQ, Ober C, Pritchard JK, Przeworski M. 2008. High-resolution mapping of crossovers reveals extensive variation in fine-scale recombination patterns among humans. Science 319:1395–1398. [DOI] [PubMed] [Google Scholar]
- Crow JF. 1988. The importance of recombination In: Michod RE, Levin BR, editors. The evolution of sex: an examination of current ideas. Sunderland (MA): Sinauer Associates; p. 56–73. [Google Scholar]
- Crow JF. 1994. Advantages of sexual reproduction. Dev Genet. 15:205–213. [DOI] [PubMed] [Google Scholar]
- Cutter AD, Payseur BA. 2013. Genomic signatures of selection at linked sites: unifying the disparity among species. Nat Rev Genet. 14:262–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. 2011. The variant call format and VCFtools. Bioinformatics 27:2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorfman R. 1979. Formula for the Gini coefficient. Rev Econ Stat. 61:146–149. [Google Scholar]
- Duret L, Arndt PF. 2008. The impact of recombination on nucleotide substitutions in the human genome. PLoS Genet. 4(5):e1000071. doi:10.1371/journal.pgen.1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Galtier N. 2009. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet. 10:285–311. [DOI] [PubMed] [Google Scholar]
- Fearnhead P, Donnelly P. 2001. Estimating recombination rates from population genetic data. Genetics 159:1299–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, et al. 2007. A second generation human haplotype map of over 3.1 million SNPs. Nature 449:851–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtier N, Duret L, Glemin S, Ranwez V. 2009. GC-biased gene conversion promotes the fixation of deleterious amino acid changes in primates. Trends Genet. 25:1–5. [DOI] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, Noble WS. 2011. FIMO: scanning for occurrences of a given motif. Bioinformatics 27:1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn MW, Zhang SV, Moyle LC. 2014. Sequencing, assembling, and correcting draft genomes using recombinant populations. G3 4:669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassold T, Hunt P. 2001. To err (meiotically) is human: the genesis of human aneuploidy. Nat Rev Genet. 2:280–291. [DOI] [PubMed] [Google Scholar]
- Hey J, Kliman RM. 2002. Interactions between natural selection, recombination and gene density in the genes of Drosophila. Genetics 160:595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinch AG, Tandon A, Patterson N, Song YL, Rohland N, Palmer CD, Chen GK, Wang K, Buxbaum SG, Akylbekova EL, et al. 2011. The landscape of recombination in African Americans. Nature 476:170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR. 2001. Two-locus sampling distributions and their application. Genetics 159:1805–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson RR, Kaplan NL. 1995. Deleterious background selection with recombination. Genetics 141:1605–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap C. 2005. A haplotype map of the human genome. Nature 437:1299–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen-Seaman MI, Furey TS, Payseur BA, Lu YT, Roskin KM, Chen CF, Thomas MA, Haussler D, Jacob HJ. 2004. Comparative recombination rates in the rat, mouse, and human genomes. Genome Res. 14:528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppi L, Jeffreys AJ, Keeney S. 2004. Where the crossovers are: recombination distributions in mammals. Nat Rev Genet. 5:413–424. [DOI] [PubMed] [Google Scholar]
- Kaur T, Rockman MV. 2014. Crossover heterogeneity in the absence of hotspots in Caenorhabditis elegans. Genetics 196:137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami T, Smeds L, Backstrom N, Husby A, Qvarnstrom A, Mugal CF, Olason P, Ellegren H. 2014. A high-density linkage map enables a second-generation collared flycatcher genome assembly and reveals the patterns of avian recombination rate variation and chromosomal evolution. Mol Ecol. 23: 4035–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliman RM, Hey J. 2003. Hill-Robertson interference in Drosophila melanogaster: reply to Marais, Mouchiroud and Duret. Genet Res. 81:89–90. [DOI] [PubMed] [Google Scholar]
- Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B. 2002. A high-resolution recombination map of the human genome. Nat Genet. 31:241–247. [DOI] [PubMed] [Google Scholar]
- Kong A, Thorleifsson G, Gudbjartsson DF, Masson G, Sigurdsson A, Jonasdottir A, Walters GB, Jonasdottir A, Gylfason A, Kristinsson KT, et al. 2010. Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467:1099–1103. [DOI] [PubMed] [Google Scholar]
- Laayouni H, Montanucci L, Sikora M, Mele M, Dall'Olio GM, Lorente-Galdos B, McGee KM, Graffelman J, Awadalla P, Bosch E, et al. 2011. Similarity in recombination rate estimates highly correlates with genetic differentiation in humans. PLoS One 6:e17913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesecque Y, Glemin S, Lartillot N, Mouchiroud D, Duret L. 2014. The red queen model of recombination hotspots evolution in the light of archaic and modern human genomes. PLoS Genet. 10:e1004790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais G. 2003. Biased gene conversion: implications for genome and sex evolution. Trends Genet. 19:330–338. [DOI] [PubMed] [Google Scholar]
- Marais G, Mouchiroud D, Duret L. 2001. Does recombination improve selection on codon usage? Lessons from nematode and fly complete genomes. Proc Natl Acad Sci U S A. 98:5688–5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaugh SE, Heil CSS, Manzano-Winkler B, Loewe L, Goldstein S, Himmel TL, Noor MAF. 2012. Recombination modulates how selection affects linked sites in Drosophila. PLoS Biol. 10:e1001422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus KF Kelley JL Song S Veeramah KR Woerner AE Stevison LS Ryder OA Ape Genome Project G, Kidd JM Wall JD et al. . 2015. Inference of gorilla demographic and selective history from whole-genome sequence data. Mol Biol Evol. 32:600–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean G, Awadalla P, Fearnhead P. 2002. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 160:1231–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean G, Myers SR, Hunt S, Deloukas P, Bentley DR, Donnelly P. 2004. The fine-scale structure of recombination rate variation in the human genome. Science 304:581–584. [DOI] [PubMed] [Google Scholar]
- Munch K, Mailund T, Dutheil JY, Schierup MH. 2014. A fine-scale recombination map of the human-chimpanzee ancestor reveals faster change in humans than in chimpanzees and a strong impact of GC-biased gene conversion. Genome Res. 24:467–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. 2005. A fine-scale map of recombination rates and hotspots across the human genome. Science 310:321–324. [DOI] [PubMed] [Google Scholar]
- Myers S, Bowden R, Tumian A, Bontrop RE, Freeman C, MacFie TS, McVean G, Donnelly P. 2010. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science 327:876–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg M, Charlesworth B, Charlesworth D. 1996. The effect of recombination on background selection. Genet Res. 67:159–174. [DOI] [PubMed] [Google Scholar]
- Persikov AV, Osada R, Singh M. 2009. Predicting DNA recognition by Cys2His2 zinc finger proteins. Bioinformatics 25:22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronczki M, Siomos MF, Nasmyth K. 2003. Un menage a quatre: the molecular biology of chromosome segregation in meiosis. Cell 112:423–440. [DOI] [PubMed] [Google Scholar]
- Prado-Martinez J, Sudmant PH, Kidd JM, Li H, Kelley JL, Lorente-Galdos B, Veeramah KR, Woerner AE, O'Connor TD, Santpere G, et al. 2013. Great ape genetic diversity and population history. Nature 499:471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratto F, Brick K, Khil P, Smagulova F, Petukhova GV, Camerini-Otero RD. 2014. DNA recombination. Recombination initiation maps of individual human genomes. Science 346:1256442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptak SE, Hinds DA, Koehler K, Nickel B, Patil N, Ballinger DG, Przeworski M, Frazer KA, Paabo S. 2005. Fine-scale recombination patterns differ between chimpanzees and humans. Nat Genet. 37:429–434. [DOI] [PubMed] [Google Scholar]
- Scheet P, Stephens M. 2006. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am J Hum Genet. 78:629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JJ, Roach DJ, Thomas JH, Shendure J. 2014. Primate evolution of the recombination regulator PRDM9. Nat Commun. 5:4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segurel L. 2013. The complex binding of PRDM9. Genome Biol. 14:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serre D, Nadon R, Hudson TJ. 2005. Large-scale recombination rate patterns are conserved among human populations. Genome Res. 15:1547–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GR. 2012. How RecBCD enzyme and Chi promote DNA break repair and recombination: a molecular biologist's view. Microbiol Mol Biol Rev. 76:217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smukowski CS, Noor MA. 2011. Recombination rate variation in closely related species. Heredity 107:496–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Donnelly P. 2003. A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 73:1162–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevison L. 2015. Great ape recombination respository. Available from: http://github.com/lstevison/great-ape-recombination/.
- Stevison LS, Noor MAF. 2010. Genetic and evolutionary correlates of fine-scale recombination rate variation in Drosophila persimilis. J Mol Evol. 71:332–345. [DOI] [PubMed] [Google Scholar]
- Stumpf MPH, McVean GAT. 2003. Estimating recombination rates from population-genetic data. Nat Rev Genet. 4:959–968. [DOI] [PubMed] [Google Scholar]
- Wang Y, Rannala B. 2014. Bayesian inference of shared recombination hotspots between humans and chimpanzees. Genetics 198:1621–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MT, Hurst LD. 2012. Direct and indirect consequences of meiotic recombination: implications for genome evolution. Trends Genet. 28:101–109. [DOI] [PubMed] [Google Scholar]
- Wegmann D, Kessner DE, Veeramah KR, Mathias RA, Nicolae DL, Yanek LR, Sun YV, Torgerson DG, Rafaels N, Mosley T, et al. 2011. Recombination rates in admixed individuals identified by ancestry-based inference. Nat Genet. 43:847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.