

Abstract
Motivation
Glycans play a central role in many essential biological processes. Glycan Reader was originally developed to simplify the reading of Protein Data Bank (PDB) files containing glycans through the automatic detection and annotation of sugars and glycosidic linkages between sugar units and to proteins, all based on atomic coordinates and connectivity information. Carbohydrates can have various chemical modifications at different positions, making their chemical space much diverse. Unfortunately, current PDB files do not provide exact annotations for most carbohydrate derivatives and more than 50% of PDB glycan chains have at least one carbohydrate derivative that could not be correctly recognized by the original Glycan Reader.
Results
Glycan Reader has been improved and now identifies most sugar types and chemical modifications (including various glycolipids) in the PDB, and both PDB and PDBx/mmCIF formats are supported. CHARMM-GUI Glycan Reader is updated to generate the simulation system and input of various glycoconjugates with most sugar types and chemical modifications. It also offers a new functionality to edit the glycan structures through addition/deletion/modification of glycosylation types, sugar types, chemical modifications, glycosidic linkages, and anomeric states. The simulation system and input files can be used for CHARMM, NAMD, GROMACS, AMBER, GENESIS, LAMMPS, Desmond, OpenMM, and CHARMM/OpenMM. Glycan Fragment Database in GlycanStructure.Org is also updated to provide an intuitive glycan sequence search tool for complex glycan structures with various chemical modifications in the PDB.
Availability and implementation
http://www.charmm-gui.org/input/glycan and http://www.glycanstructure.org.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Carbohydrates are one of the four essential classes of macromolecules in all living organisms, and their sequence and structure are diverse due to a multitude of ways to link individual sugar units and introduce various chemical modifications to each sugar unit (Varki et al., 2015). Carbohydrate moieties, referred to as glycans, can be covalently attached to proteins (glycoproteins) and lipids (glycolipids), or exist as diffusible ligands. Glycosylation is the most common post-translational modification of protein, and more than 50% of all eukaryotic proteins are thought to be glycosylated (Apweiler et al., 1999). Thus, it is not surprising that glycans play critical roles in a vast array of biological processes by altering protein structure, dynamics, and stability, and thus affect not only enzymatic activity and in vivo circulation half-life of protein pharmaceuticals, but also protein-protein and protein-lipid interactions through specific protein-glycan recognitions (Lee et al., 2015; Pinho and Reis, 2015; Rudd et al., 2004). In this context, knowledge of glycans’ three-dimensional (3D) structures is imperative to better understand their biological roles.
The Protein Data Bank (PDB) (Berman et al., 2000) includes various carbohydrate moieties in the form of pure polysaccharides or glycoconjugates. Among the 125 461 PDB entries as of December 2016, 10 712 (8%) contain carbohydrate structures. However, it is estimated that 30% of carbohydrate structures in the PDB contain at least one error such as wrong assignment of anomeric configuration, lack of the atoms, and wrong atom connectivity (Joosten and Lütteke, 2016; Lütteke and von der Lieth, 2004). These errors make it difficult to precisely recognize and annotate the carbohydrates. To facilitate the recognition of carbohydrates, several web services, such as GLYCOSCIENCES.de (Lütteke et al., 2006) and Glyco3D (Perez et al., 2015) have been developed to provide convenient ways to automatically annotate carbohydrates in PDB files. For instance, pdb-care (Lütteke and von der Lieth, 2004) in GLYCOSCIENCES.de detects carbohydrate structures from the PDB format file, reports the carbohydrate structural errors, and displays LINUCS (Bohne-Lang et al., 2001), a nomenclature that is a linear notation for carbohydrate structures. Glyco3D (Perez et al., 2015) provides the database of manually curated 3D structures of monosaccharide, oligosaccharide, polysaccharide, glycosaminoglycan-binding proteins, lectin, and monoclonal antibodies from the PDB. In addition, carbohydrate 3D models can be generated using Carbohydrate Builder and Glycoprotein Builder in GLYCAM web server (http://www.glycam.org), SWEET-II (Bohne et al., 1999) and GlyProt (Bohne-Lang and von der Lieth, 2005) in GLYCOSCIENCE.de, POLYS (Engelsen et al., 2014), and CarbBuilder (Kuttel et al., 2016). However, these tools offer rather limited options for building glycan structures in solution or membrane environments and generating related structure files for biomolecular simulations. CHARMM-GUI Glycan Reader (Jo et al., 2008; Jo et al., 2011) (http://www.charmm-gui.org/input/glycan) greatly simplifies the building of simulation systems for the PDB entries that contain glycoprotein and polysaccharides by automatic detection and annotation of carbohydrates, recognition of glycosidic linkages to proteins, correction of structural errors, and generation of inputs for biomolecule simulations (Lee et al., 2016) by combining with other functional modules in CHARMM-GUI such as PDB Reader & Manipulator (Jo et al., 2014), Quick MD Simulator, and Membrane Builder (Jo et al., 2009; Wu et al., 2014).
Biological function of proteins and nucleic acids can be modulated by chemical modifications. For instance, phosphorylation at a specific site on a protein causes major conformational changes, leading to activation or deactivation of a protein function (Chaffey, 2003). Similarly, chemical modifications of glycans such as methylation, acylation, phosphorylation, and sulfation can modulate their biological functions (Yu and Chen, 2007). For example, sulfation patterns on heparan sulfate chains in the proteoglycans determine the binding affinity to the various ligands such as fibroblast growth factors (FGFs) (Muthana et al., 2012). O-acetylated sialic acid has been proposed to be a diagnostic marker for cancer and plays significant roles in the regulation of ganglioside-mediated apoptosis (Mandal et al., 2015). To understand the atomistic detail of biological roles of glycans, molecular dynamics (MD) simulations have been used (Jo et al., 2016; Qi et al., 2016). For example, Re et al. employed replica-exchange MD simulations to study N-glycan structures depending on their modification such as core fucosylation and addition of bisecting N-acetyl glucosamine that are known to change the affinity of protein–glycan interactions (Re et al., 2012). However, few tools of building carbohydrate structures are able to handle chemical modifications.
This work reports Glycan Reader that is improved to provide automatic detection of most sugar types and chemical modifications including glycolipids in the PDB. In addition, CHARMM-GUI Glycan Reader is updated to generate the simulation systems and inputs of various glycoconjugates with most sugar types and chemical modifications. It also offers a new functionality to edit the glycan structures through addition/deletion/modification of glycosylation types, sugar types, chemical modifications, glycosidic linkages, and anomeric states. Glycan Fragment Database (GFDB) (Jo and Im, 2013) in GlycanStructure.Org (http://www.glycanstructure.org/fragment-db) is also updated to provide an intuitive glycan sequence search tool for complex glycan structures with various chemical modifications in the PDB. The specific algorithms and the implementations of Glycan Reader in CHARMM-GUI and GlycanStructure.Org are described in detail in the next section. We also report the PDB glycan statistics and MD simulation results of FGF monomer in complex with a heparin analogue as a case study, which is followed by a brief summary.
2 New features and improvements in Glycan Reader
2.1 Reading of PDBx/mmCIF format
As PDBx/mmCIF (PDB exchange/macromolecular Crystallographic Information File) (Bourne et al., 1997) became the standard PDB archive format since 2014, in addition to conventional PDB format, a new functionality to handle PDBx/mmCIF format has been incorporated into Glycan Reader. PDBx/mmCIF supports large structures having more than 62 chains and 99,999 atoms, which could not be handled by the PDB format. In addition, the PDBx/mmCIF format is more flexible and extensible in that one can add new data items to address ever-increasing size and complexity of deposited structures (Sen et al., 2014). In the case of the PDB format, Glycan Reader uses HETATM and CONECT record to build molecular topologies (Jo et al., 2011). Similarly, the first step of reading a PDBx/mmCIF file is to read HETATM record in the _atom_site category. Then, Glycan Reader builds the connectivity of HETATM using the inter-atom connectivity (from _struct_conn category) and the intra-atom connectivity [from _chem_comp_atom and _chem_comp_bond categories in the chemical component dictionary (http://www.wwpdb.org/data/ccd)].
2.2 Recognition of various sugar types and chemical modifications
Glycan Reader is improved to recognize more sugar types and detect various chemical modifications. 46 sugar types are newly added for a total of 78 monosaccharides (Supplementary Table S1), supporting most common sugar types referred in the third edition of “Essential of Glycobiology” (Varki et al., 2015). In addition, Glycan Reader can now detect 25 chemical modification types (including six lipid acyl chain types; Supplementary Table S2).
The assignment protocol of carbohydrate residue types follows the original Glycan Reader work (Jo et al., 2011). Briefly, the workflow consists of the following four steps: (1) detecting the potential carbohydrate monomers, (2) identifying anomeric carbons in each monomer, (3) determining carbohydrate name, and (4) building the linkages between sugars and to proteins. To detect the potential carbohydrate monomers and identify anomeric carbons in each monomer, Glycan Reader constructs a graph, composed of the nodes (i.e., atom types) and edges (i.e., bonds between atoms), using the molecular topologies in a given PDB file. Five- or six-membered rings that are composed of four or five carbon and one oxygen atoms are recognized as a potential carbohydrate structure after the detection of all cycles in the connected component of the graph by cycle basis algorithm (Paton, 1969). The other chemical moieties that are not part of potential carbohydrate structures are assigned as potential chemical modifications if they are linked to the potential carbohydrate structure via glycosidic bonds (–O–R, –N–R) and not attached to other potential carbohydrate structures. For each potential carbohydrate structure, the anomeric carbon is determined by checking if one of the carbon atoms connected to the ring oxygen has oxygen, nitrogen, or sulfur atom attached to it. If there are no such carbon atoms due to the lack of the electron density or structural errors, a carbon atom that is connected to the ring oxygen and has no attached exocyclic carbon atom is assigned as an anomeric carbon. If a potential carbohydrate structure has a six-membered ring, their backbone carbon atoms are used for VF2 subgraph isomorphism algorithm (Cordella et al., 2004) to find matching template graphs of sialic acid, octulosonic acid, heptose, and hexose (Fig. 1). For a five-membered ring, the template graphs of hexofuranose, hex-2-ulofuranose, and pentofuranose are used (Supplementary Fig. S1). Once their backbone atoms are identified, Glycan Reader measures the improper angles of the chiral centers to which hydroxyl groups are attached and finds additional chemical moieties to the backbone atoms to determine the sugar types.
Fig. 1.

Pyranose templates and available chemical modification sites. Six-membered rings that are composed of five carbon and one oxygen atoms are compared with the above templates through the VF2 isomorphism to identify the sugar types and assign the backbone atoms. R is the available position of chemical modifications
The common carbohydrate derivatives, such as N-acetyl glucosamine and glucuronic acid, are determined by including the N-acetyl or carboxyl group in their template graph (Fig. 1). In the case of sialic acid, a sugar type is determined among Neu5Ac, Neu5Gc, Neu, and Kdn based on the moiety in C5 atom. Deoxy sugars like rhamnose are identified when the recognized sugar has no attached hydroxyl groups to the relevant backbone carbon positions. Pentopyranose like xylose is detected when there is no C6 atom.
After the sugar-type assignment step, Glycan Reader inspects whether the remaining chemical moieties correspond to a certain lipid type as part of a glycolipid. Glycan Reader recognizes lipid moieties associated with Gram-negative bacterial lipid A (LIPA1, LIPA2), phosphatidylinositol diacylglycerols (PIDAG), diacylglycerols (DAG), ceramides (CER), 2,3-di-phytanylglycerols (DPHG), acyl chains (detergent, DET), and S-linked acyl chains (SDET). X-ray structures of glycolipids in the PDB may not have all acyl chains resolved due to their high flexibility, so identification of glycolipid types are determined by the presence of the lipid head group (with minimum acyl chain information) attached to the carbohydrate ring. Figure 2 shows the template structures used to identify each glycolipid type using the VF2 subgraph isomorphism algorithm. Glycan Reader uses specific minimal acyl chains (represented as red color) to determine a glycolipid type. Following the order in Figure 2, the lipid templates are examined by subgraphs of a query molecule and a matched lipid type is selected if one of the subgraphs is isomorphic, meaning that all matched nodes have the same atom types and are a superset of a given template. For modeling purpose, various CHARMM lipid patches in terms of acyl chain types and lengths are provided through Glycan Reader in CHARMM-GUI (see below). For example, seven patches for CER are currently available: CER160 (18:1/16:0), CER180 (18:1/18:0), CER181 (18:1/18:1), CER200 (18:1/20:0), CER220 (18:1/22:0), CER240 (18:1/24:0), and CER241 (18:1/24:1). After glycolipid detection, the remaining exocyclic chemical moieties are assigned to other chemical modifications.
Fig. 2.
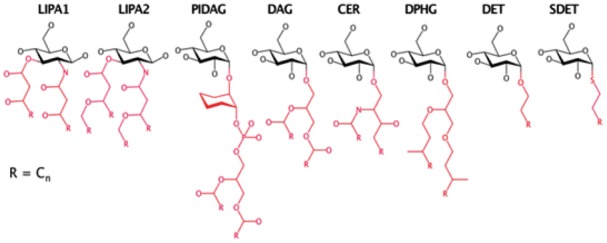
Glycolipid templates. The minimal substructures representing different acyl chain types are colored in red (Color version of this figure is available at Bioinformatics online.)
Glycan Reader has 19 template structures of common chemical modifications that appear in more than 10 PDB entries (Supplementary Table S2). They are represented as graph templates used for VF2 graph matching to recognize the corresponding chemical modifications. We have also generated the corresponding CHARMM patch residues if they did not exist in the current CHARMM carbohydrate force field (Guvench et al., 2011). Note that atom names in the recognized sugars as well as the detected chemical modification groups are renamed based on those in the corresponding CHARMM residues and patches to read these coordinates in CHARMM-GUI.
The linkage building protocol is the same as before, but Glycan Reader can now recognize more linkage types (Supplementary Table S3). The newly available glycosidic linkages are 1-1, 1–2, 1–3, 1–4, and 1–6 S-linked linkages; 1–6 and 1–7 linkages between pyranose and heptopyranose; 2–8 linkage between octulosonic acids; 2–6 linkage between pyranose and sialic acids; 2–8 and 2–9 linkages between sialic acids; 1–2 and 1–3 linkages between pyranose and furanose; and 1–2 and 1–3 between furanoses. The force field parameters of these new sugar residues and patches were transferred by analogy based on the standard CHARMM force field.
2.3 Improvements in CHARMM-GUI
CHARMM-GUI is a web-based graphical user interface (GUI) that can be used to prepare complex biomolecular systems for molecular simulations. CHARMM-GUI generates input files for a number of widely used programs (Lee et al., 2016), such as CHARMM (Brooks et al., 2009), NAMD (Phillips et al., 2005), GROMACS (Abraham et al., 2015), AMBER (Case et al., 2005), GENESIS (Jung et al., 2015), LAMMPS (Plimpton, 1995), Desmond (Bowers et al., 2006), OpenMM (Eastman et al., 2013), and CHARMM/OpenMM (Arthur and Brooks, 2016) to facilitate the usage of common and advanced simulation techniques. As part of its input generation module, CHARMM-GUI provides a GUI for Glycan Reader that has been updated and newly designed with the aforementioned new features, which are elaborated below. Note that, while it exists as a separate module in CHARMM-GUI, Glycan Reader is also linked to other functional modules as part of PDB Reader & Manipulator, allowing users to easily generate molecular simulation systems with carbohydrates or glycoproteins, and visualize the electrostatic potential on glycoprotein surfaces (Jo et al., 2008; Jo et al., 2016).
When carbohydrates are included in a user specified PDB entry or uploaded structure file in PDB or PDBx/mmCIF format, the glycan primary sequences are displayed in the CASPER sequence representation (Lundborg and Widmalm, 2011) on the PDB manipulation page. When the user clicks the edit button for a specific glycan chain, a new window pops up, displaying the sequence and symbolic representations of the selected glycan chain (Fig. 3). Symbolic glycan representations follow the SNFG (Symbol Nomenclature for Glycans) system (Varki et al., 2015). The user can add, remove, or modify sugar types and anomeric states (α and β), glycosidic linkages, glycosylation types, and chemical modifications. If the sugars are detected as part of a glycolipid, appropriate lipid patch is provided, and the user can edit the lipid lengths. The coordinates of lipid tails are first transferred from the ones in the PDB structure, and the missing coordinates are generated based on the CHARMM internal coordinate (IC) information.
Fig. 3.
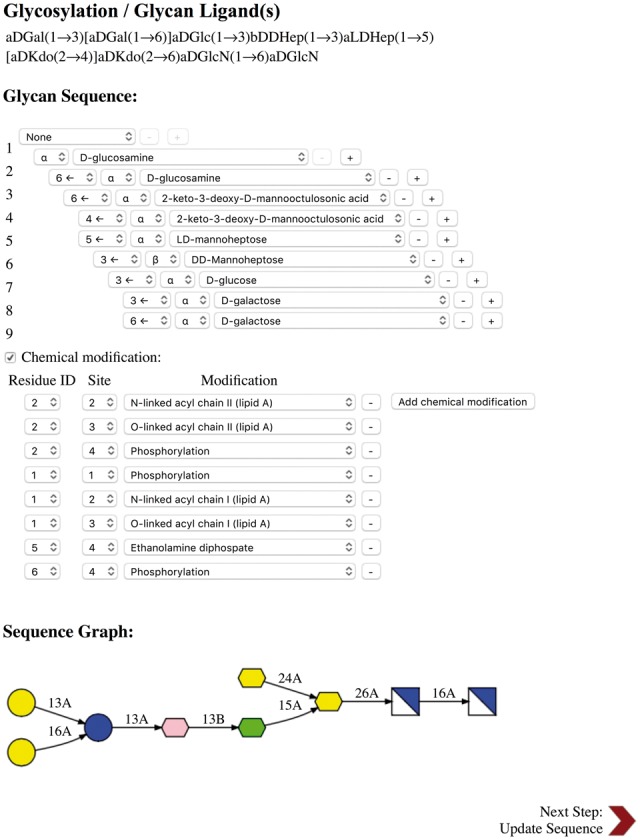
CHARMM-GUI Glycan Reader snapshot for a glycan chain in PDB:2FCP. When a PDB entry or uploaded PDB file contains glycan chains, their sequences are displayed in CASPER format under Glycosylation/Glycan Ligand(s) in the PDB manipulation section. When the edit button next to a specific CASPER sequence is clicked, Glycosylation/Glycan Ligand(s) is displayed in a new pop-up window, and one can view/edit glycosylation, sugar, linkage, and chemical modification types. All running steps of Glycan Reader are illustrated in Supplementary Figure S2
2.4 Improvements in GlycanStructure.Org
Glycan Reader (Fig. 4A) in GlycanStructure.Org (http://www.glycanstructure.org/glycanreader) has also been updated to provide users with glycan sequences in Glycan Reader sequence (GRS) format with symbolic representations of all natural sugars (Fig. 4B). GRS format is the internal format used in Glycan Reader for the primary glycan sequence, containing the glycan residue ID in the PDB, the sugar residue names and chemical modifications in the CHARMM force field, and a hierarchical linkage information between sugars. A chemical modification table lists sugar residues having chemical modifications with the carbon position, CHARMM patch name, chemical formula, and name of each chemical modification.
Fig. 4.

Snapshots from GlycanStructure.Org. (A) Glycan Reader in GlycanStructure.Org. (B) When a PDB entry is specified or a structure is uploaded in (A), detected glycan structure information is displayed. (C) Using the “Search GFDB” button in (B), the selected glycan chain information including the chemical modifications is transferred to GFDB (Glycan Fragment Database) to search the selected glycan structures in the PDB
GFDB in GlycanStructure.Org provides an intuitive glycan sequence search tool that allows users to search complex glycan structures in the PDB. After a glycan search is complete, GFDB allows users to visually examine the torsion angle (ϕ, ψ, and ω) distributions of selected glycosidic bonds and to obtain the representative glycan structures using a clustering analysis of the searched glycan structures. To incorporate the new features of Glycan Reader, GFDB has been improved to include most carbohydrates in the PDB and to submit a query glycan sequence with or without chemical modifications (Fig. 4C). The chemical modification option is provided below the glycan sequence input, so that users can search glycan sequences more specifically in terms of user-selected chemical modifications in each sugar. If the chemical modification is set to ‘any’, GFDB searches for a query glycan sequence without considering chemical modifications. However, if chemical modifications are specified, the GFDB search result is limited to the chemically modified glycans. Note that users can also use ‘Search GFDB’ in the result of Glycan Reader in GlycanStructure.Org (Fig. 4B). The updated numbers of glycan structures in GFDB are shown in Figure 5, which is further discussed in the next section.
Fig. 5.
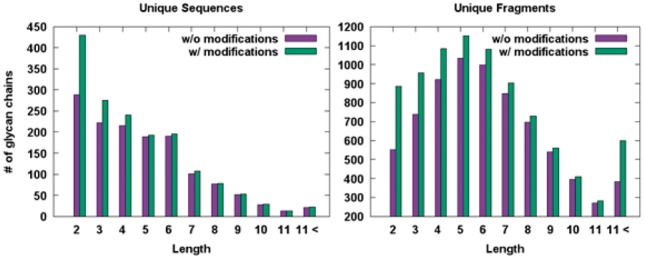
Histograms for the numbers of unique and fragment glycan sequences in terms of glycan lengths in GFDB
3 Results and discussion
3.1 PDB statistics
As of December 2016, the PDB contains 43 767 glycan chains detected by Glycan Reader. 10 731 out of the 125 424 PDB entries (8.56%) have at least one glycan chain. 10 573 out of the 10 731 PDB entries (98.53%) were solved by X-ray crystallography and the others by solution NMR, electron microscopy, fiber diffraction, powder diffraction, neutron diffraction, solid-state NMR, and theoretical models (Fig. 6A). Compared to the previous version, there are 5932 glycan chains and 1813 PDB entries that are newly identified, including complex glycolipids, chemical modifications, and rare cases of glycosidic linkages (e.g., S-glycosidic bond). Among 43 767 glycan chains, there are 24 544 N-linked glycan chains (56.08%), 1252 O-linked glycans (2.86%), 17 856 free glycan ligands (40.80%), and 115 glycan chains covalently linked to Asp and Glu residues (0.26%) (Fig. 6B). N-acetyl-d-glucosamine (GlcNAc) is the most abundant monosaccharide in the PDB (38 991; 47.37%), followed by d-glucose (14 144; 17.18%), d-mannose (13 839; 16.81%) (Fig. 6C).
Fig. 6.
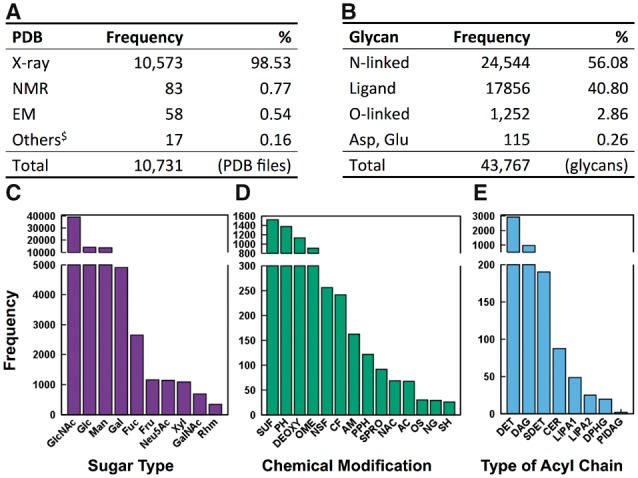
RCSB glycan statistics as of December, 2016. (A) Numbers of PDB entries that have at least one glycan chain in terms of experimental methods. $Others are fiber diffraction, powder diffraction, neutron diffraction, solid-state NMR, and theoretical models. (B) Numbers of glycan chains in the PDB in terms of their types. (C) Histogram for the numbers of monosaccharides in terms of sugar types. (D) Histogram for the numbers of chemical modifications. (E) Histogram for the numbers of glycans in terms of lipid acyl chain types
Figure 6D shows glycan chemical modifications with their number frequencies more than 20 in the PDB. Note that Figure 6D does not include the functional groups of sugar residue types such as N-acetylation at C2 of GlcNAc or carboxylate at C6 of GlcA. Sulfation, phosphorylation, and O-methylation are the most frequent modifications in the PDB. Sulfation is frequently found at C2 and C6 of sugar residues within glycosaminoglycans (with N-linked sulfate at C2); at C6 of glyco lipids such as glycosyl ceramide and sulfoquinovosyl diacylglycerol; and in several hexopyranoses like N-acetyl-d-glucosamine-6-sulfate and O3-sulfonyl galactose. Phosphorylation is frequently found at C1, C4, and C6 of hexopyranoses, C2 and C6 of fructofuranose, C1 and C5 of ribose, C7 of mannoheptose, and C8 of Kdo in lipopolysaccharides (LPS). O-methylation has been found at C2 of Neu5Ac and Kdo, and carboxylate at C5 of GlcA or IdoA, and general hexopyranoses.
There are a total of 1,948 undefined chemical modification groups detected in the 904 PDB entries. The number of unique undefined modifications is 403 (data not shown). The description and examples of the undefined modifications in the PDB are shown in Supplementary Table S4 for the modifications that appear in more than 10 PDB entries. These molecules are generally carbohydrate analogues in which a sugar is bound to another functional group (e.g., 4-nitrophenyl) via a glycosidic bond.
Note that detection of deoxylation is nontrivial because oxygen atoms often do not exist due to missing electron density or an error in the PDB structure (Jo et al., 2011). The most frequent case is the lack of the oxygen atom attached to anomeric carbon (Lütteke et al., 2004). In PDBx/mmCIF format, the ‘zero_occ_atoms’ category contains information about the missing atoms in residues by the authors of the PDB structures. We used this category to determine whether it is deoxylation or not. Nonetheless, the ‘zero_occ_atoms’ category does not cover all missing atom information, so it is still possible that sugars with missing oxygen atoms are assigned as deoxy sugars. CHARMM-GUI Glycan Reader allows users to edit a given glycan structure, so users can change each residue back to normal sugars with determination of α or β configuration.
Figure 6E shows the number frequencies of glycolipids in the PDB. Detergents (DET) are most prevalent in the PDB glycolipids. They usually consist of monosaccharide or disaccharide with octyl or dodecyl acyl chain. Their abundance is well understood because detergents are utilized to isolate the proteins from the cell membrane and to crystalize them (le Maire et al., 2000). Diacylglycerols (DAG) are found mostly in the photosystem like bacterial rhodopsin as forms of monogalactosyl diacylglycerol, digalactosyl diacylglycerol, and sulfoquinovosyl diacylglycerol. 2,3-di-phytanylglycerols (DPHG) have similar chemical compositions with DAG and are also found in the photosystems. Ceramides (CER) are found in glycosphingolipids (e.g., cerebrosides and gangliosides such as GM1). Lipid A (as part of LPS) anchors LPS in the outer membrane of the Gram-negative bacteria and possesses an archetypal structure of a β-(1→6)-linked d-GlcN disaccharide that is acylated with four to eight fatty acids of different lengths, and there exist complex chemical substitutions in lipid A from certain bacterial species (Kim et al., 2016). Phosphatidylinositol diacylglycerols (PIDAG) have the form of phosphatidyl-myoinositol mannosides (PIM), which influences the interaction of the immune system with M. tuberculosis. Mice that develop antibodies for PIM are better at sustaining or defeating TB infection (Mehta and Khuller, 1988; Zajonc et al., 2006).
As of December 2016, the PDB has 1,638 unique sequences and 8641 unique fragments with chemical modifications. Without considering chemical modifications, the number of unique sequences and fragments are 1396 and 7372 (Fig. 5). These unique structures are stored in GFDB for fast structure search, which can then be used to examine torsional angles between specific monosaccharides to characterize the glycan structures. These unique glycan and fragment structures with and without chemical modifications could be used to model glycan structures from their sequences using a comparative modeling approach (Lee et al., 2015).
Note that Glycan Reader corrects structural errors of glycan chains in the PDB (Jo et al., 2011). For instance, Glycan Reader builds the missing glycosidic linkages by examining the distance between the anomeric carbon and the exocyclic oxygen in the neighboring residue. If it is in close proximity (e.g. <2.5 Å), a glycosidic linkage is generated between the two residues. Among the 10 731 PDB entries that have at least one glycan chain, 624 (5.81%) PDB entries contain structural errors (Supplementary Table S5). There is no way to systemically measure the recognition accuracy of sugar types and glycolipid templates via Glycan Reader, because chemical component dictionary (CCD; http://www.wwpdb.org/data/ccd) also contains errors in glycan annotation. In particular, wrong annotation of anomeric configuration is the most abundant cases (Lütteke et al., 2004). However, without consideration of anomeric configuration, the sugar types annotated by Glycan Reader are highly identical to those in the CCD (Supplementary Data S2). Few cases of type mismatches between Glycan Reader and CCD arise from the isomeric configuration of carbohydrates (e.g., galactose and glucose). In the cases of glycolipids, they are usually composed of several chemical types from the CCD. When we inspected them manually, most glycolipid annotations by Glycan Reader are correct. DET (detergent acyl chain) has a few cases of type mismatches because the number of nodes in the DET template is relatively small compared to other glycolipid templates (Fig. 2). However, since many lipid acyl chains are not resolved in the electron density due to their high flexibility, having a smaller template is beneficial in detecting glycolipids with DET.
3.2 Case study: MD simulation of heparin analogue and FGF-1 protein complex
To illustrate the utility of Glycan Reader in CHARMM-GUI, we built and simulated the molecular systems of FGF-1 monomer complexed with a heparin sulfate analogue (hexasaccharide in PDB ID: 2ERM; Fig. 7A–C) using the default options in Quick MD Simulator. FGF-1 belongs to fibroblast growth factor (FGF) family and binds to heparin sulfate and FGFR receptor, which modulate roles of FGFs in regulating cell growth, survival, differentiation, and migration (Ornitz, 2000). We built 20 independent simulation systems using all 20 NMR models in PDB:2ERM and performed 100-ns NPT (constant number, pressure, and temperature) simulations for each system using NAMD (Phillips et al., 2005) and the CHARMM36 force field (Guvench et al., 2011; Huang and MacKerell, 2013) with the inputs provided by CHARMM-GUI. The root mean-square deviations (RMSDs) of FGF-1 monomer and the heparin analogue in the 20 trajectories indicate stable complex structures resulting from the favorable protein-carbohydrate interactions during the 100-ns MD simulations (Supplementary Fig. S3). The distributions of ϕ and ψ glycosidic angles from the 20 MD simulations are consistent with those from the initial 20 NMR models (Fig. 7D and E and Supplementary Figs S4 and S5).
Fig. 7.
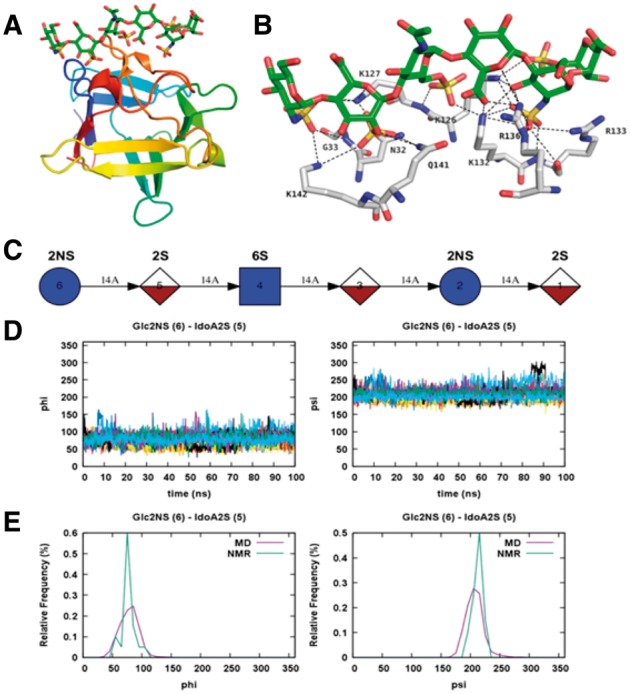
MD simulations of FGF-1 monomer with a heparin analogue. (A) FGF-1 monomer in complex with a heparin analogue in PDB ID: 2ERM. (B) The hydrogen bonds between the FGF-1 monomer and heparin analogue are shown by dotted lines. (C) Symbolic representation of the heparin analogue. (D) Time series of ϕ and ψ glycosidic angles between Glc2NS (residue 6 in C) and IdoA2S (residue 5 in C) obtained from the 100-ns MD simulations starting from 20 different initial NMR models. (E) Frequency distribution curves of ϕ and ψ angles obtained from the MD simulations in comparison with those calculated from the 20 NMR models
4 Summary
We have described the improved functional features in Glycan Reader, which are important in glycan modeling and simulation. They include (1) handling of both PDB and PDBx/mmCIF formats, (2) identification of most sugar types and chemical modifications including various glycolipids in the PDB, and (3) its implementation in CHARMM-GUI and GlycanStructure.Org. The introduction of such key features into Glycan Reader in CHARMM-GUI enables facile generation of the simulation systems of complex glycoconjugates with most sugar types and chemical modifications in the PDB. The update of GFDB in GlycanStructure.Org provides an intuitive glycan sequence search tool for complex glycan structures even with various chemical modifications in the PDB. These tools are expected to be useful in carrying out innovative and novel glycan modeling and simulation research to acquire insight into structures, dynamics, and underlying mechanisms of complex glycoconjugate systems.
Funding
The National Science Foundation DBI-1707207, the National Institutes of Health (GM087519 and GM103695), and XSEDE MCB070009.
Conflict of Interest: none declared.
Supplementary Material
References
- Abraham M.J. et al. (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 1–2, 19–25. [Google Scholar]
- Apweiler R. et al. (1999) On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta, 1473, 4–8. [DOI] [PubMed] [Google Scholar]
- Arthur E.J., Brooks C.L. 3rd (2016) Parallelization and improvements of the generalized born model with a simple sWitching function for modern graphics processors. J. Comput. Chem., 37, 927–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.M. et al. (2000) The Protein Data Bank. Nucleic Acids Res., 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohne A. et al. (1999) SWEET - WWW-based rapid 3D construction of oligo- and polysaccharides. Bioinformatics, 15, 767–768. [DOI] [PubMed] [Google Scholar]
- Bohne-Lang A. et al. (2001) LINUCS: linear notation for unique description of carbohydrate sequences. Carbohydr Res., 336, 1–11. [DOI] [PubMed] [Google Scholar]
- Bohne-Lang A., von der Lieth C.W. (2005) GlyProt: in silico glycosylation of proteins. Nucleic Acids Res., 33, (Web Server issue):W214–W219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne P.E. et al. (1997) Macromolecular crystallographic information file. Methods Enzymol., 277, 571–590. [DOI] [PubMed] [Google Scholar]
- Bowers K.J. et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the ACM/IEEE SC 2006 Conference 2006. p. 43.
- Brooks B.R. et al. (2009) CHARMM: the biomolecular simulation program. J. Comput. Chem., 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D.A. et al. (2005) The Amber biomolecular simulation programs. J. Comput. Chem., 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffey N., (2003) Molecular biology of the cell. Ann. Bot., 91, 401–401. [Google Scholar]
- Cordella L.P. et al. (2004) A (sub)graph isomorphism algorithm for matching large graphs. IEEE Trans Pattern Anal. Mach. Intell., 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Eastman P. et al. (2013) OpenMM 4: a reusable, extensible, hardware independent library for high performance molecular simulation. J. Chem. Theory Comput., 9, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelsen S.B. et al. (2014) POLYS 2.0: an open source software package for building three-dimensional structures of polysaccharides. Biopolymers, 101, 733–743. [DOI] [PubMed] [Google Scholar]
- Guvench O. et al. (2011) CHARMM additive all-atom force field for carbohydrate derivatives and its utility in polysaccharide and carbohydrate-protein modeling. J. Chem. Theory Comput., 7, 3162–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., MacKerell A.D. (2013) CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem., 34, 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S. et al. (2014) CHARMM-GUI PDB manipulator for advanced modeling and simulations of proteins containing nonstandard residues. Adv. Protein Chem. Struct. Biol., 96, 235–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S. et al. (2016) CHARMM-GUI 10 years for biomolecular modeling and simulation. J. Comput. Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S., Im W. (2013) Glycan fragment database: a database of PDB-based glycan 3D structures. Nucleic Acids Res., 41, (Database issue):D470–D474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S. et al. (2008) CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem., 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- Jo S. et al. (2009) CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys. J., 97, 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S. et al. (2016) Preferred conformations of N-glycan core pentasaccharide in solution and in glycoproteins. Glycobiology, 26, 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S. et al. (2011) Glycan Reader: automated sugar identification and simulation preparation for carbohydrates and glycoproteins. J. Comput. Chem., 32, 3135–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S. et al. (2008) PBEQ-Solver for online visualization of electrostatic potential of biomolecules. Nucleic Acids Res., 36, W270–W275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joosten R.P., Lütteke T. (2016) Carbohydrate 3D structure validation. Curr. Opin. Struct. Biol., 44, 9–17. [DOI] [PubMed] [Google Scholar]
- Jung J. et al. (2015) GENESIS: a hybrid-parallel and multi-scale molecular dynamics simulator with enhanced sampling algorithms for biomolecular and cellular simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci., 5, 310–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. et al. (2016) Bilayer properties of lipid A from various Gram-negative bacteria. Biophys. J., 111, 1750–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuttel M.M. et al. (2016) CarbBuilder: software for building molecular models of complex oligo- and polysaccharide structures. J. Comput. Chem., 37, 2098–2105. [DOI] [PubMed] [Google Scholar]
- Le Maire M. et al. (2000) Interaction of membrane proteins and lipids with solubilizing detergents. Biochim. Biophys. Acta, 1508, 86–111. [DOI] [PubMed] [Google Scholar]
- Lee H.S. et al. (2015) GS-align for glycan structure alignment and similarity measurement. Bioinformatics, 31, 2653–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.S. et al. (2015) Effects of N-glycosylation on protein conformation and dynamics: Protein Data Bank analysis and molecular dynamics simulation study. Sci. Rep., 5, 8926.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. et al. (2016) CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput., 12, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundborg M., Widmalm G. (2011) Structural analysis of glycans by NMR chemical shift prediction. Anal. Chem., 83, 1514–1517. [DOI] [PubMed] [Google Scholar]
- Lütteke T. et al. (2006) GLYCOSCIENCES.de: an Internet portal to support glycomics and glycobiology research. Glycobiology, 16, 71R–81R. [DOI] [PubMed] [Google Scholar]
- Lütteke T. et al. (2004) Data mining the protein data bank: automatic detection and assignment of carbohydrate structures. Carbohydr Res., 339, 1015–1020. [DOI] [PubMed] [Google Scholar]
- Lütteke T., von der Lieth C.W. (2004) pdb-care (PDB carbohydrate residue check): a program to support annotation of complex carbohydrate structures in PDB files. BMC Bioinform., 5, 69.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal C. et al. (2015) Functions and biosynthesis of O-acetylated sialic acids. Top Curr. Chem., 366, 1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta P.K., Khuller G.K. (1988) Protective immunity to experimental tuberculosis by mannophosphoinositides of mycobacteria. Med. Microbiol. Immunol., 177, 265–284. [DOI] [PubMed] [Google Scholar]
- Muthana S.M. et al. (2012) Modifications of glycans: biological significance and therapeutic opportunities. ACS Chem. Biol., 7, 31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz D.M. (2000) FGFs, heparan sulfate and FGFRs: complex interactions essential for development. Bioessays, 22, 108–112. [DOI] [PubMed] [Google Scholar]
- Paton K. (1969) An algorithm for finding a fundamental set of cycles of a graph. Commun. ACM, 12, 514–518. [Google Scholar]
- Perez S. et al. (2015) Glyco3D: a portal for structural glycosciences. Methods Mol. Biol., 1273, 241–258. [DOI] [PubMed] [Google Scholar]
- Phillips J.C. et al. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem., 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho S.S., Reis C.A. (2015) Glycosylation in cancer: mechanisms and clinical implications. Nat. Rev. Cancer., 15, 540–555. [DOI] [PubMed] [Google Scholar]
- Plimpton S. (1995) Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys., 117, 1–19. [Google Scholar]
- Qi Y. et al. (2016) Roles of glycans in interactions between gp120 and HIV broadly neutralizing antibodies. Glycobiology, 26, 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Re S. et al. (2012) Conformational flexibility of N-glycans in solution studied by REMD simulations. Biophys. Rev., 4, 179–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd P.M. et al. (2004) Sugar-mediated ligand-receptor interactions in the immune system. Trends Biotechnol., 22, 524–530. [DOI] [PubMed] [Google Scholar]
- Sen S. et al. (2014) Small molecule annotation for the Protein Data Bank. Database (Oxford), 2014, bau116.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A. et al. (2015) Symbol nomenclature for graphical representations of glycans. Glycobiology, 25, 1323–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A. et al. (2015) In: Varki A.et al. (ed.) Essentials of Glycobiology. Cold Spring Harbor (NY). [Google Scholar]
- Woods R.J. 2005–2017. GLYCAM Web; Complex Carbohydrate Research Center. University of Georgia, Athens, GA. [Google Scholar]
- Wu E.L. et al. (2014) CHARMM-GUI membrane builder toward realistic biological membrane simulations. J. Comput. Chem., 35, 1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H., Chen X. (2007) Carbohydrate post-glycosylational modifications. Org. Biomol. Chem., 5, 865–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajonc D.M. et al. (2006) Structural characterization of mycobacterial phosphatidylinositol mannoside binding to mouse CD1d. J. Immunol., 177, 4577–4583. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.