

Abstract
Summary
We present a user-friendly, cloud-based, data analysis pipeline for the deconvolution of pooled screening data. This tool, CRISPRcloud, serves a dual purpose of extracting, clustering and analyzing raw next generation sequencing files derived from pooled screening experiments while at the same time presenting them in a user-friendly way on a secure web-based platform. Moreover, CRISPRcloud serves as a useful web-based analysis pipeline for reanalysis of pooled CRISPR screening datasets. Taken together, the framework described in this study is expected to accelerate development of web-based bioinformatics tool for handling all studies which include next generation sequencing data.
Availability and implementation
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Genetic screening allows for the unbiased interrogation of a genome to ask targeted questions (Miles et al., 2016). While originally centered around random mutagenesis or arrayed RNAi (Mohr et al., 2014; Simon et al., 2015), a growing amount of studies are moving towards large-scaled, pooled approaches (Gilbert et al., 2014; Schlabach et al., 2008; Shalem et al., 2014; Wang et al., 2014). The advent of CRISPR has heralded a dramatic increase in the number of pooled screens (Shalem et al., 2015). For instance, in the past year, the number of datasets for CRISPR screens in Gene Expression Omnibus have more than tripled from the previous year (from 39 datasets in 2015 to 121 datasets in 2016). In these, a collection of modifiers (e.g. sgRNAs) are introduced to a population (e.g. in cell culture or into an organism). Following phenotypic (e.g. enriching for surviving cells following a toxic insult) or enrichment-based (e.g. Fluorescence activated cell sorting (FACS) of a given fluorophore) screening, genomic DNA is extracted and enrichment of each modifier is assessed using next generation sequencing (NGS). Once a daunting and expensive approach to generate hypotheses in an unbiased fashion, pooled screening approaches are now more feasible and accessible than ever.
Nevertheless, the bioinformatics hurdle of data deconvolution following sequencing remains a roadblock for investigators with little-to-no computational knowledge. Previous tools have been generated to analyze high-throughput screening datasets but, like most bioinformatics tools, these did not always provide a good user experience (Table 1). There are several reasons to explain these shortcomings. First, these tools often require a self-installation step to use and often require the combination of several packages to work adequately (Li et al., 2015; Winter et al., 2015, 2017). Second, raw sequence data handling is not provided and therefore users must independently manipulate the data (leading to undue errors) by uploading their data to platforms such as RIGER and ATARI (Luo et al., 2008; Shao et al., 2013). Third, most available tools are specialized for dropout studies such that enrichment-based studies (or any other study with more complex data involving 3 or more groups) do not fit in the analysis pipeline. Lastly, the inflexibility in statistical analysis output in these programs may lead to improper conclusions.
Table 1.
A comparison of CRISPRcloud with existing methods for the multifunctionality
| Feature | RIGER | ATARIS | MAGeCK-VISPR | caRpools | CRISPRAnalyzeR | CRISPRcloud |
|---|---|---|---|---|---|---|
| Web interface | ✓ | ✓ | ✓ | ✓ | ||
| Cloud service | ✓ | |||||
| FASTQ file handling | ✓ | ✓ | ✓ | ✓ | ||
| GUI | ✓ | ✓ | ✓ | |||
| Multiple group studies | ✓ | ✓ | ✓ | ✓ | ||
| Paired data analysis | ✓ | |||||
| Quality control | ✓ | ✓ | ✓ | ✓ | ||
| Reference | Luo et al. (2008) | Shao et al. (2013) | Li et al. (2015) | Winter et al. (2015) | Winter et al. (2017) |
Note: The ‘✓’ symbol indicates the corresponded function is supported in the tools.
To overcome these issues, we generated a web-based analysis pipeline for pooled screens: CRISPRcloud. CRISPRcloud is a user-friendly cloud based platform that lends itself to customization per the screens at hand.
In this system, the user can upload raw sequencing files confidentially, can extract valuable screening information through customizable statistical analysis and can output various end-point results in a personalized manner.
2 Materials and methods
We developed CRISPRcloud and have started sharing this web-service to the public (Fig. 1a). CRISPRcloud provides a user-friendly graphical interface (Fig. 1b) and enables those performing the screens who are not necessarily well versed in bioinformatics to mine their own data.
Fig. 1.
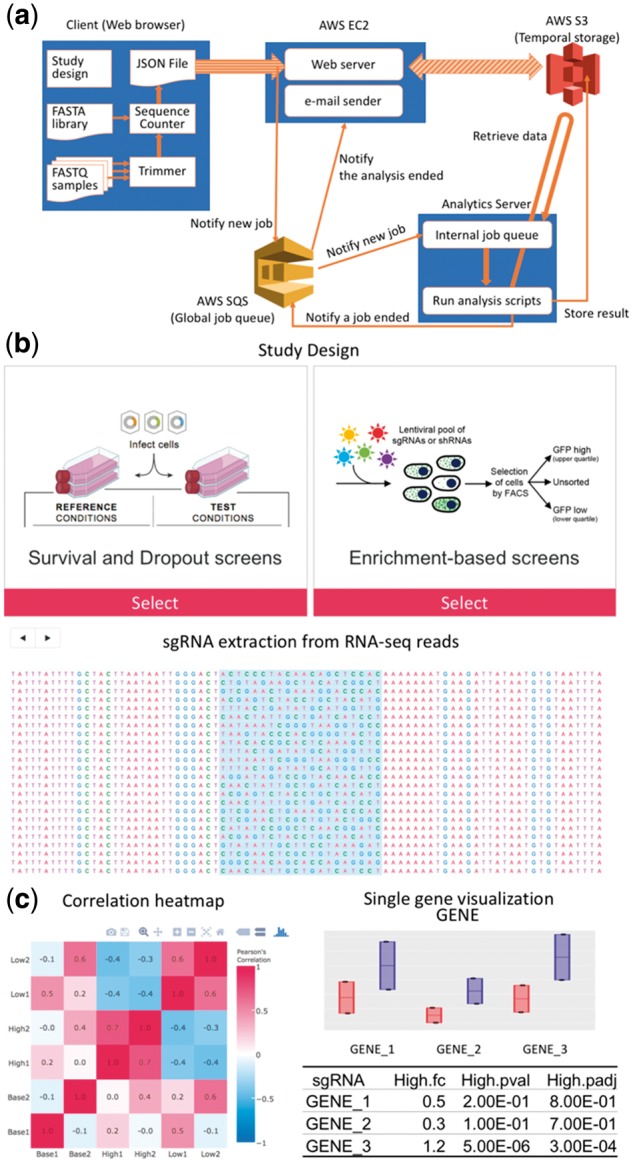
(a) A detailed illustration of the pipeline of CRISPRcloud. (b) Screenshots of GUI to input user data in the CRISPRcloud website. (c) An example of the visualized report
To generate this platform, we had to overcome several major roadblocks for web-based tools on CRISPR screening. For one, the file size of NGS datasets from pooled screening is large (∼10 GB per sample), it is impractical to transfer such large amount of data through the Internet. The client-side web technology like JavaScript and HTML5 have rapidly developed in the recent year, allowing high-performance computation on large files through modern web browsers. These program languages enabled us to perform the initial steps of data analysis at the client-side allowing for rapid and secure data minimization. In practice, CRISPRcloud reduces several gigabyte-size files into a single megabyte-size file. The only data received by function ‘GETS’ at the server side is the single-guided RNA (sgRNA) read counts data, the study design information and the user e-mail address only to notify when the analysis finishes. When it ends successfully, the server erases the user email data and counts data. It also removes the result reports after a week. This step drastically increases the response time and data transfer load.
To address the bottleneck on computing power associated with a centralized server solution, CRISPRcloud provides a scalable service through the infrastructure provided by Amazon Elastic Compute Cloud (EC2) (https://aws.amazon.com/ec2/), Amazon Simple Storage Service (S3) (https://aws.amazon.com/s3/) and Amazon Simple Queue Service (SQS) (https://aws.amazon.com/sqs/) at a cost-effective manner.
For the statistical analysis, CRISPRcloud supports two different types of experiment design (dropout and enrichment based experiment) and three choices on hypothesis testing methods for each sgRNA (a Student’s t-test with log-transformation of counts (Wu et al., 2010), DESeq2 (Love et al., 2014) and the inverted beta-binomial test – a test used for paired datasets (Pham and Jimenez, 2012)). Once the selected hypothesis tests are finished, CRISPRcloud provides various gene scoring metrics to ease selection of hit genes in the screens (See Supplementary Document for details). Users are provided with a rich set of functions to visualize, query, analyze, export and share their data via an address-encrypted link (Fig. 1c). Moreover, users can easily mine further information on their interested genes from the external databases, such as CRISPRtools (predicting sgRNA efficiency for in vivo mouse experiment), GenomeCRISPR (searching other public CRISPR screens) and MARRVEL (searching multiple genetic variants and disease databases simultaneously), for the next step of the pooled screens (Peterson et al., 2017; Rauscher et al., 2017; Wang et al., 2017). CRISPRcloud can finish the pre-processing within a couple of hours (takes minutes for the small coverage dataset and about an hour for the large dataset on MacBook Pro with 3.3 GHz Intel Core i7 and 16 GB memory, see Supplementary Table S1 for detailed information of the running time) and take an additional few minutes to have a result of the statistical analysis. Therefore, this tool can pass the result to user rapidly; even user run CRISPRcloud on a standard laptop.
3 Discussion and conclusion
The decreasing costs and time-frames associated with pooled genome-wide screening approaches is incentivizing an increasing number of investigators. Nevertheless, the sequencing-to-data gap remains discouraging. We developed CRISPRcloud as a solution to this issue. The multifunctionality of CRISPRcloud is clear with current technologies such as shRNA and CRISPR but can easily be extended to any technology where short sequence barcoding is used (e.g. ORF screens and CRISPRi/a) (Gilbert et al., 2014; Horlbeck et al., 2016; Konermann et al., 2015). Moreover, this user-friendly system allows for re-analysis of published datasets at user-defined statistical analysis parameters. The analysis results can be shared through an encrypted link to collaborators.
Taken together, this study highlights a web-based platform upon which investigators can securely deposit raw sequencing files, extract the valuable data and perform statistical analyses, generate and prioritize hit lists and export datasets for downstream validation. This cloud-based novel system enables screen-based biological analysis for any interested user.
Funding
This work has been supported by National Institute of General Medical Sciences R01-GM120033, National Science Foundation - Division of Mathematical Sciences DMS-1263932, Cancer Prevention Research Institute of Texas RP170387, Houston Endowment (Z.L.), Huffington Foundation, Howard Hughes Medical Institute (H.Y.Z.), and Canadian Institutes of Health Research Fellowship 201210MFE-290072-173743 (M.W.C.R.).
Conflict of Interest: none declared.
Supplementary Material
References
- Gilbert L.A. et al. (2014) Genome-scale CRISPR-mediated control of gene repression and activation. Cell, 159, 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horlbeck M.A. et al. (2016) Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife, 5, e19760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S. et al. (2015) Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature, 517, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. et al. (2015) Quality control, modeling, and visualization of CRISPR screens with MAGeCK-VISPR. Genome Biol., 16, 281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I. et al. (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol., 15, 550.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo B. et al. (2008) Highly parallel identification of essential genes in cancer cells. Proc. Natl. Acad. Sci., 105, 20380–20385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles L.A. et al. (2016) Design, execution, and analysis of pooled in vitro CRISPR/Cas9 screens. FEBS J., 283, 3170–3180. [DOI] [PubMed] [Google Scholar]
- Mohr S.E. et al. (2014) RNAi screening comes of age: improved techniques and complementary approaches. Nat. Rev. Mol. Cell Biol., 15, 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson K.A. et al. (2017) CRISPRtools: a flexible computational platform for performing CRISPR/Cas9 experiments in the mouse. Mamm. Genome, doi: 10.1007/s00335-017-9681-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham T.V., Jimenez C.R. (2012) An accurate paired sample test for count data. Bioinformatics, 28, i596–i602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauscher B. et al. (2017) GenomeCRISPR - a database for high-throughput CRISPR/Cas9 screens. Nucleic Acids Res., 45, D679–D686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlabach M.R. et al. (2008) Cancer proliferation gene discovery through functional genomics. Science, 319, 620–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O. et al. (2014) Genome-scale CRISPR-Cas9 knockout screening in human cells. Science, 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O. et al. (2015) High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet., 16, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao D.D. et al. (2013) ATARiS: computational quantification of gene suppression phenotypes from multisample RNAi screens. Genome Res., 23, 665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon M.M. et al. (2015) Current strategies for mutation detection in phenotype-driven screens utilising next generation sequencing. Mamm. Genome, 26, 486–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. et al. (2017) MARRVEL: Integration of Human and Model Organism Genetic Resources to Facilitate Functional Annotation of the Human Genome. Am. J. Hum. Genet., 100, 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T. et al. (2014) Genetic screens in human cells using the CRISPR-Cas9 system. Science, 343, 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter J. et al. (2015) CaRpools: An R package for exploratory data analysis and documentation of pooled CRISPR/Cas9 screens. Bioinformatics, 32, 632–634. [DOI] [PubMed] [Google Scholar]
- Winter J. et al. (2017) CRISPRAnalyzeR: Interactive analysis, annotation and documentation of pooled CRISPR screens. bioRxiv, 109967. [Google Scholar]
- Wu Z.J. et al. (2010) Gene expression profiling of human breast tissue samples using SAGE-Seq. Genome Res., 20, 1730–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.