

Abstract
Data-independent acquisition (DIA) approaches have awaken in the last years for there producible and precise quantification of thousands of proteins in complex samples. This information is stored in digital protein maps (DIA-maps) that can be interrogated in a targeted way by using ad hoc or publically-available peptide spectral libraries generated on the same sample species as for the generation of the DIA maps. The restricted availability of certain difficult-to-obtain human tissues (i.e. brain) together with the caveats of using spectral libraries generated under variable experimental conditions limits the potential of DIA. Therefore, DIA workflows would benefit from high-quality and extended spectral libraries that could be generated without the need of using valuable samples for library production. We describe here two new targeted approaches, using either classical data-dependent (DDA) repositories (not specifically built for DIA) or ad hoc mouse spectral libraries, which enable the profiling of human brain DIA dataset. The comparison of our results to both the most extended publically-available human spectral library and to a state-of-the-art untargeted method supports the use of these new strategies to improve future DIA profiling efforts.
Keywords: Data-independent acquisition, Spectral libraries
The changes in the proteome underlying the regulation of biological processes is of major interest in biomedical research and relies on the ability of proteomic methods to generate quantitative, reproducible and comprehensive data sets. To date, DDA mass spectrometry methods have been extensively used. In DDA, a subset of peptide precursors is selected from a survey scan (MS1) for subsequent fragmentation and acquisition of a fragment-ion spectrum (MS2). Their identification together with the quantitative information results in the discovery of differences in protein expression. However, the selection of peptide precursors is stochastic favoring the collection of high-abundant/intense peptides. As a result, roughly 27% of all detectable peptides can be identified in a single run [1, 2], and 30% of the selected can vary between replicates[3]. As an alternative approach, pioneer work [4-7] showed the potential of DIA methods where all peptide ions (in a given m/z range and above the detection limit of the LC-MS instrument) of a complex sample are fragmented independently of their intensity, theoretically enabling the identification and quantification of all peptide precursors. However, DIA-MS2 spectra stored in the form of DIA-maps are composed by fragmented ions belonging to different peptide precursors. This chimeric nature of the MS2 spectra is a bottleneck for identification using classical database search strategies.
The most widely used analytical solution of DIA methods is the use of reference spectral libraries for the targeted extraction of quantitative information of the peptides included in these libraries [8] using tools such as Spectronaut, Open SWATH, Skyline or Peak View. While this type of targeted approaches reaches excellent reproducibility [9], the analysis is restricted to a subset of the comprehensive peptide information stored in DIA-maps because reference spectral libraries are generated by DDA proteomics on the same sample species. Public repositories containing species-specific spectral libraries (i.e. Peptide Atlas) aim to solve this bottleneck, however the limited availability of certain tissue samples together with the caveats of using spectral libraries generated under variable experimental conditions [10, 11] prevents DIA to achieve its full potential.
Based on the correspondence between the peptide fragmentation pattern and the peptide sequence [12], we sought to improve the targeted analysis of human DIA-maps by developing two novel and complementary strategies. On the one hand, we used classical DDA repositories as means of human reference spectral libraries. On the other hand, we generated ad hoc spectral libraries using mouse tissues, a widely available and closely related animal model. Our results demonstrate our approaches having an improved performance in analyzing human DIA-maps when compared to the most extended pan-human spectral library [13] and to a recent untargeted approach [14].
First, we isolated regions of spinal cord (SC) and prefrontal cortex (PFC) from human and mouse (Fig. 1A). Our selection was guided by the important roles of these tissues in human pathophysiology and their low availability compared to other frequently used human matrices (e.g. plasma). Following, we constructed extended mouse and human ad hoc libraries (Ml and Hl, respectively) using a combined strategy. In order to reach the most comprehensive coverage possible of the expressed proteome we use multiple injections and long chromatography gradients, as well as three tissue fractions for each of the tissues: whole cell, membrane- and cytosol-enriched (Supporting Information Fig. 1 and Fig. 2). Finally, we created a mouse-derived human library (MdHl) by selecting only peptides displaying 100% identity with their human counterparts as annotated in Uniprot (of the 43,956 mouse peptides 26,953 overlap with human peptides).
Figure 1.
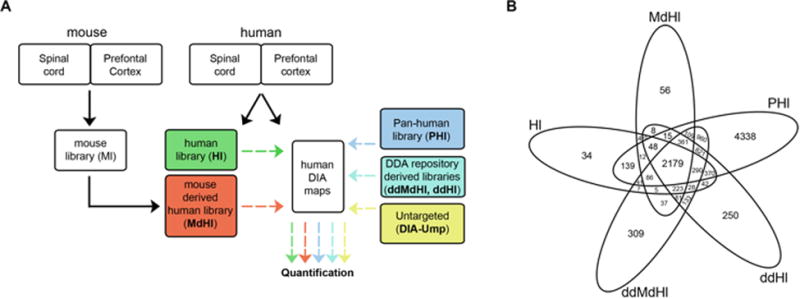
Experimental Workflow. (A) Adhoc and DDA-repository derived (dd) human- (H) and mouse-derived (Md) peptide spectral libraries (Hl, MdHl, ddHl and ddMdHl, respectively) were used for the quantification of DIA maps (human DIA maps) obtained from human PFC and SC. Their performance was compared to the pan-human peptide spectral library (PHl) and to DIA-Umpire (DIA-Ump). (B) Venn diagram showing the number of proteins in each of the five libraries.
As an additional strategy, we aimed to construct high-quality spectral libraries using the currently most extended datasets from classical DDA repositories of mouse and human cortex [15-17]. This relies on normalized retention times and on the ability to predict accurate retention times (RT) for peptides detected in different experimental conditions. For that, we applied a high-precision iRT algorithm [10] to all peptides from the mentioned datasets that resulted in two DDA-repository (dd) spectral libraries: a dd mouse-derived human library (ddMdHl) and a dd human library (ddHl).
Comparison of the four generated libraries with the currently most-extended pan-human spectral library (PHl) [13] revealed a significant amount of newly incorporated proteins(Fig. 1B and Supporting Information Table 1). A closer look revealed that 223 proteins of the Hl are not present in the PHl but are included in the three other libraries of which a significant amount are involved in physiological functions assumed by PFC and SC (e.g. transmission of nerve impulse; Supporting Information Fig. 3).
Next, we evaluated the ability of the generated libraries to profile differences in protein expression in human tissues. To this end, we obtained DIA-maps from three biological replicates of whole cell lysates of human SC and PFC (Fig. 1). Our results indicate that Hl profiled the highest number of unique peptides and proteins in both tissues and in all of the three biological replicates (Fig. 2, Supporting Information Table 2 and Supporting Information Fig. 4A-C), including also the highest number of peptides representing each protein (Supporting Information Fig. 4D). Importantly, ddHl, ddMdHl and MdHl out performe dPHl (114% more peptides for ddHl in PFC; Fig. 2 and Supporting Information Table 2) with comparable coefficients of variation (Supporting Information Fig. 5). The improvements shown here with inter-species resources (mouse) differ conceptually from previously published DDA database search strategies [18] as our approach does not rely on modifying the size of the database search space or on modifying the DIA map search space in order to influence the probability of positive peptide identification.
Figure 2.
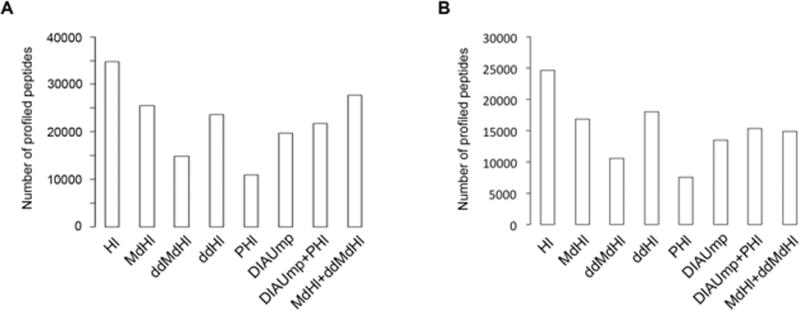
Performance of each library in human PFC and SC. (A) Number of peptides profiled by each of the spectral libraries in the human PFCDIA dataset and (B) in the human SC DIA dataset.
Our data partially confirm but also demonstrate the potential of our strategy to overcome well-known limitations when aiming to use spectral data obtained in different experimental conditions (i.e. different mass spectrometers which tend to produce different charge states of a given peptide). A closer look at the results show that the MdHl (generated using the same Q Exactive mass spectrometer as for the DIA maps) was able to extract complete quantitative information for 893 proteins more than the PHl (generated using a Triple TOF 5600+ mass spectrometer) in human PFC (Supporting Information Table 2), although 506 of those proteins were also represented in the PHl. A deeper analysis revealed that 56% of these proteins were represented by different peptides in the PHl spectral library, whereas in the remaining 44% different charge states, retention time and fragment ion selections were apparent. These differences go in line with the reported differences in spectral transferability between different instruments [19]. On the other hand we believe that the tissue similarity plays an important role: ddHl that is derived from the analysis of human frontal cortex (Supporting Information Table 1) is able to extract information of 32 more proteins than MdHl (Supporting Information Table 2) although the latter library was produced using exactly the same instrumentation as for the human DIA datasets. Further, ddHl extracted 925 proteins more than PHl (Supporting Information Table 2) that did not include any brain tissue for its generation [13]. In summary, our results show that although ad-hoc in-house spectral libraries from the same species as the DIA datasets and obtained using the same instrumentation continue as the most efficient choice, there is a big potential of applying high-precision iRT algorithms [10] to extended datasets obtained from classical DDA repositories (not specifically built for DIA).
Following, we evaluated whether the peptide assays included in all these libraries harbor not only proteotypic but also quantitative properties [20]. As shown in Figure 3A, the differences in abundance detected by ddHl, in comparison to Hlshowed remarkable consistency - 525 proteins were commonly identified with ddHl and Hl to be differentially expressed, including important synaptic proteins (Fig. 3B). Comparable results were obtained for ddMdHl and MdHl (Supporting Information Fig. 6).
Figure 3.
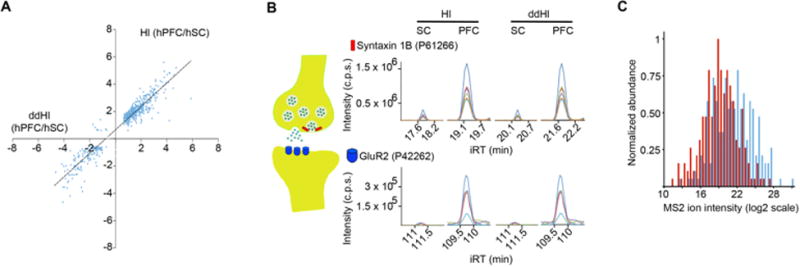
Quantification properties of each of the libraries. (A) Ratio of expression (hPFC/hSC) for 525 proteins calculated based on Hl and ddHl. Slope of regression 0.86 with a R2=0.91. (B) Examples of expression differences between the two human tissues for the presynaptic protein Syntaxin 1B and the postsynaptic protein GluR2 upon use of Hl and ddHl. (C) Distribution of the 204 fragment ion intensities identified uniquely by DIA-Umpire (red bars), or 204 randomly selected fragments shared with at least one of the five spectral libraries (blue bars) from the human PFC DIA dataset analysis.
Recent computational efforts have been developed to analyze DIA datasets without the need for spectral libraries [14, 21, 22]–referred to as untargeted approaches. Our data shows that library-based targeted extraction using Hl, MdHl and ddHl out performed the untargeted strategy of DIA mpire in terms of number of extracted peptides and proteins (Fig. 2, Supporting Information Table 2, Supporting Information Fig. 4 and Fig. 5). An alternative for the analysis of our human DIA datasets without relying in the use of ad hoc Hl or MdHl would be to use DIA-Umpire to generate a complementary library and combine it with the identifications when using the publically-available PHl. Our results indicate that the identifications reached by ddHl or MdHl alone or MdHl grouped with ddMdHl still outperformed a combination of DIA-Umpire and PHl (Fig. 2, Supporting Information Table 2 and Supporting Information Fig. 4).
The detection improvements over DIA-Umpire were comparable to or even bigger than the ones reached by latest untargeted solutions when similar conditions were compared: While MdHl alone and in combination with ddMdHl showed an improvement of 29% and 40%, respectively (Supporting Information Table 2), MSSPLIT-DIA [21] showed an approximate 25% improvement using a spectral library generated from the same sample. On the other hand, Group-DIA [22] did not show any improvement in peptide identifications when 3 DIA runs were considered (a similar number of runs as analyzed to construct the DIA maps in our study).
Interestingly, 204 of the peptides detected by DIA-Umpire were not present in any of the five other libraries. Further analysis showed that the intensities of the fragment ions exclusively profiled by DIA-Umpire were significantly lower in comparison to the ones commonly detected with the other five libraries (two sample t-test, two-tailed, P< 0.0001; similarity of the variance was confirmed using F test, P= 0.13; Fig. 3C). These results suggest that future integrative strategies would benefit from the combination of targeted and untargeted approaches in order to increase the analysis-depth of DIA-datasets.
DIA methods are in a prime position for proteomic-based biomarker discovery efforts. Several strategies are under development towards the goal to improve the amount of reliable peptide identifications from DIA maps, including computational [14, 21, 22] and technological efforts [23]. The novel strategies presented here add new possibilities to these efforts that can be easily implemented in future studies, unlocking extended classical DDA repositories [24] for comprehensive profiling of human samples by DIA.
Experimental Procedures
Human and mouse tissue
Native, unfixed, snap frozen CNS tissue of prefrontal cortex (PFC) and spinal cord (SC) of three non-neurological patients was obtained from the archives of the Institute of Neuropathology, University Medical Center, Göttingen (UMG), Germany. Post mortem tissue sampled for diagnostic purposes was used for the present study in conformity with the rules and regulations of the ethics committee of the UMG, reference number 6/5/16. Similar regions from three male 12-14 weeks old wild type C57Bl/6J mice were obtained. No randomization or blinding was used in this study. All mouse experiments are approved by the IACUC of the Max Planck Institute of Experimental Medicine (MPIEM).
Sample preparation for mass spectrometry acquisition
The frozen tissue was homogenized with help of a glass/Teflon homogenizer in 4% SDS lysis buffer (4% SDS in 100mM Tris, 10mM DTT, 5% glycerol, complete protease inhibitor cocktail (Roche), pH 7.5 and by shearing with a 25G needle. The homogenate was incubated for 10 minutes at 70°C, followed by centrifugation at 10,000×g for 5 minutes for removal of cell debris at room temperature (note: all centrifugation steps in this study were done at room temperature, except otherwise mentioned). The supernatant equals the whole cell lysate. Following, acetone precipitation of the proteins was done by addition of 5× volume pre-cooled acetone (Roth) and incubation for 2 hours at -20°C. The precipitated proteins were centrifuged at 14,000×g for 30 minutes, washed with ice-cold 80% Ethanol (Applied Chem) and centrifuged again at 14,000×g for 30 minutes. The air-dried proteins were resuspended under constant agitation in 2% SDS lysis buffer. For generation of the membrane and cytosolic fractions, the whole cell lysate was subjected to ultracentrifugation at 100,000×g, 18°C for 1 hour (TLA 100.3 rotor, Beckman Coulter). The pellet was dissolved in 4% SDS lysis buffer. The supernatant (cytosol) and dissolved pellet (membrane fraction) were subjected to acetone precipitation as described above.
The samples in 2% SDS were prepared for mass spectrometric acquisition using the FASP protocol with 30k Vivacon 500 spinfilters [25] (Sartorius). The peptides were desalted using C18 Macro Spin columns from The Nest Group according to manufacturer's instructions. After drying, the peptides were resuspended in 1% ACN and 0.1% formic acid. The Biognosys' HRM Calibration Kit, was added to all of the samples according to manufacturer's instructions (required for the DIA analysis using Biognosys' Spectronaut). All the three biological replicates for each of the four tissues (two in human and two in mouse) were processed in parallel.
Mass spectrometric acquisition
2 μg of the samples was analyzed on a self-made analytical column (75 μm × 30 cm) packed with 3 μm Magic C18AQ medium (Bruker) at 50°C, using an Easy-nLC connected to a Q Exactive mass spectrometer (Thermo Scientific). The peptides were separated by a 2 hours segmented gradient from 0 to 9 % ACN in 3 minutes, next to 28% ACN in 102 minutes, to 36% ACN in 12 minutes and to 44% in 3 minutes, with 0.1% formic acid at 300 nl/minute. Then a linear increase followed to 90% ACN in 2 minutes and 90% ACN was run for 8 minutes. For DDA acquisition, the “fast” method from Kelstrup [26] was used with the alteration described in Bruderer et al.[27]. In order to produce the most extended spectral libraries possible we have analyzed 10 independent samples for each of the tissues.
The HRM DIA method was performed using one technical replicate for each of the three biological replicates, and as described in Bruderer et al. with the following modification: the survey scan was recorded at 70,000 resolution in order to reach superior data quality through the introduction of MS1 based scores. The raw mass spectrometric DIA data and the spectral libraries were stored at the public repository Peptide Atlas (http://www.peptideatlas.org, No. PASS00782, the username is PASS00782 and the password is NN4585e).
MaxQuant data analysis
The DDA spectra were processed with the Max Quant Version 1.5.1.2 analysis software [28]. The spectra were searched by the Andromeda search engine [29]. The minimal peptide length was set to 6. Search criteria included carbamid methylation of cysteine as a fixed modification, oxidation of methionine and acetyl (protein N-terminus) as variable modifications. The mass tolerance for the precursor was 4.5 ppm and for the fragment ions was 20 ppm. The human DDA files were searched against a human UniProt KB/Swiss-Protfasta database (state 11.12.2014, 20,199 entries), the contaminants of MaxQuant and the Biognosys iRT peptide sequences [30] (11 entries). The murine DDA files were searched against a mouse UniProt KB/Swiss-Protfasta database (state 11.12.2014, 16,697 entries), the contaminants of Max Quant and the iRT peptide sequences.
Spectral library generation
To generate the spectral libraries, DDA spectra were analyzed as described above and a spectral library was generated using the spectral library generation functionality of Spectronaut (details can be found in the Supporting Information).
Spectronaut analysis
The DIA data were analyzed with Spectronaut 8, a mass spectrometer vendor independent software from Biognosys following default settings (details can be found in the Supporting Information).
DIA-Umpire analysis
Additionally, the DIA data was analyzed using the DIA-Umpire workflow as described in the DIA-Umpire manuscript[14] (details can be found in the Supporting Information).
Supplementary Material
Significance of the study.
A lot of interest has been generated around DIA methods applied to discovery-based proteomics, as they promise to overcome the well-known stochastic nature of classical DDA schemes. However, new analytical solutions are needed in order to increase the analyzed landscape of the DIA maps.
In this study we present a targeted analytical strategy to increase the quantitative information recovered from human DIA maps by using either the extended sources stored in classical DDA repositories or ad hoc mouse spectral libraries.
To the best of our knowledge this is the first demonstration of the possibility to unlock years of DDA research for the analysis of DIA datasets, which we expect will have an important impact in the growing field of DIA proteomics.
Acknowledgments
This work was supported by grants of the Deutsche For schungsgemeinschaft (DFG; GO 2481/3-1to D.G.V.,SCHM 2533/2-1to M.S.), a MPI PhD fellowship (to J.S.), the US National Institutes of Health grants NIH R01-GM-094231 (to A.I.N), the Research Program(For schungsförderungs programm) of the Faculty of Medicine, Georg-August-University, Göttingen and the Hertie Foundation (Gemeinnützige Hertie-Stiftung)(to C.S. and A.B).
Footnotes
Author Contributions: D.G.V. conceived and supervised the project. D.G.V., R.B. and C.S. designed experiments. J.S., M.S., A.B.F. performed tissue extraction and preparation. R.B. and L.R. acquired and analyzed mass spectrometry data. C.C.T. and A.I.N. performed the DIA-Umpire analysis. D.G.V. wrote the manuscript with input from all authors.
Competing Financial Interests: The authors R.B. and L.R. are employees of Biognosys AG (Switzerland). Spectronaut is a trademark of Biognosys AG. DGV received a research award from Biognosys AG that was not used for the current study. The rest of the authors declare no competing financial interests.
References
- 1.Michalski A, Cox J, Mann M. More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. Journal of proteome research. 2011;10:1785–1793. doi: 10.1021/pr101060v. [DOI] [PubMed] [Google Scholar]
- 2.Scheltema RA, Hauschild JP, Lange O, Hornburg D, et al. The Q Exactive HF, a Benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field Orbitrap analyzer. Molecular & cellular proteomics: MCP. 2014;13:3698–3708. doi: 10.1074/mcp.M114.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu H, Sadygov RG, Yates JR., 3rd Optimized fast and sensitive acquisition methods for shotgun proteomics on a quadrupole orbitrap mass spectrometer. Anal Chem. 2004;76:4193–4201. [Google Scholar]
- 4.Venable JD, Dong MQ, Wohlschlegel J, Dillin A, Yates JR. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nature methods. 2004;1:39–45. doi: 10.1038/nmeth705. [DOI] [PubMed] [Google Scholar]
- 5.Silva JC, Gorenstein MV, Li GZ, Vissers JP, Geromanos SJ. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Molecular & cellular proteomics: MCP. 2006;5:144–156. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- 6.Panchaud A, Scherl A, Shaffer SA, von Haller PD, et al. Precursor acquisition independent from ion count: how to dive deeper into the proteomics ocean. Anal Chem. 2009;81:6481–6488. doi: 10.1021/ac900888s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martins-de-Souza D, Faca VM, Gozzo FC. DIA is not a new mass spectrometry acquisition method. Proteomics. 2017 doi: 10.1002/pmic.201700017. [DOI] [PubMed] [Google Scholar]
- 8.Schubert OT, Gillet LC, Collins BC, Navarro P, et al. Building high-quality assay libraries for targeted analysis of SWATH MS data. Nature protocols. 2015;10:426–441. doi: 10.1038/nprot.2015.015. [DOI] [PubMed] [Google Scholar]
- 9.Selevsek N, Chang CY, Gillet LC, Navarro P, et al. Reproducible and consistent quantification of the Saccharomyces cerevisiae proteome by SWATH-mass spectrometry. Molecular & cellular proteomics: MCP. 2015;14:739–749. doi: 10.1074/mcp.M113.035550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bruderer R, Bernhardt OM, Gandhi T, Reiter L. High-precision iRT prediction in the targeted analysis of data-independent acquisition and its impact on identification and quantitation. Proteomics. 2016 doi: 10.1002/pmic.201500488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu JX, Song X, Pascovici D, Zaw T, et al. SWATH Mass Spectrometry Performance Using Extended Peptide MS/MS Assay Libraries. Molecular & cellular proteomics: MCP. 2016;15:2501–2514. doi: 10.1074/mcp.M115.055558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Z. Prediction of low-energy collision-induced dissociation spectra of peptides. Anal Chem. 2004;76:3908–3922. doi: 10.1021/ac049951b. [DOI] [PubMed] [Google Scholar]
- 13.Rosenberger G, Koh CC, Guo T, Rost HL, et al. A repository of assays to quantify 10,000 human proteins by SWATH-MS. Scientific data. 2014;1:140031. doi: 10.1038/sdata.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsou CC, Avtonomov D, Larsen B, Tucholska M, et al. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nature methods. 2015;12:258–264. doi: 10.1038/nmeth.3255. 257 p following 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma K, Schmitt S, Bergner CG, Tyanova S, et al. Cell type- and brain region-resolved mouse brain proteome. Nat Neurosci. 2015;18:1819–1831. doi: 10.1038/nn.4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilhelm M, Schlegl J, Hahne H, Moghaddas Gholami A, et al. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509:582–587. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- 17.Kim MS, Pinto SM, Getnet D, Nirujogi RS, et al. A draft map of the human proteome. Nature. 2014;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frank AM, Monroe ME, Shah AR, Carver JJ, et al. Spectral archives: extending spectral libraries to analyze both identified and unidentified spectra. Nature methods. 2011;8:587–591. doi: 10.1038/nmeth.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toprak UH, Gillet LC, Maiolica A, Navarro P, et al. Conserved peptide fragmentation as a benchmarking tool for mass spectrometers and a discriminating feature for targeted proteomics. Molecular & cellular proteomics: MCP. 2014;13:2056–2071. doi: 10.1074/mcp.O113.036475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Worboys JD, Sinclair J, Yuan Y, Jorgensen C. Systematic evaluation of quantotypic peptides for targeted analysis of the human kinome. Nature methods. 2014;11:1041–1044. doi: 10.1038/nmeth.3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J, Tucholska M, Knight JD, Lambert JP, et al. MSPLIT-DIA: sensitive peptide identification for data-independent acquisition. Nature methods. 2015 doi: 10.1038/nmeth.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Zhong CQ, Xu X, Cai S, et al. Group-DIA: analyzing multiple data-independent acquisition mass spectrometry data files. Nature methods. 2015 doi: 10.1038/nmeth.3593. [DOI] [PubMed] [Google Scholar]
- 23.Egertson JD, Kuehn A, Merrihew GE, Bateman NW, et al. Multiplexed MS/MS for improved data-independent acquisition. Nature methods. 2013;10:744–746. doi: 10.1038/nmeth.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Omenn GS, Lane L, Lundberg EK, Beavis RC, et al. Metrics for the Human Proteome Project 2015: Progress on the Human Proteome and Guidelines for High-Confidence Protein Identification. Journal of proteome research. 2015;14:3452–3460. doi: 10.1021/acs.jproteome.5b00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wisniewski JR, Zielinska DF, Mann M. Comparison of ultrafiltration units for proteomic and N-glycoproteomic analysis by the filter-aided sample preparation method. Anal Biochem. 2011;410:307–309. doi: 10.1016/j.ab.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 26.Kelstrup CD, Young C, Lavallee R, Nielsen ML, Olsen JV. Optimized fast and sensitive acquisition methods for shotgun proteomics on a quadrupole orbitrap mass spectrometer. Journal of proteome research. 2012;11:3487–3497. doi: 10.1021/pr3000249. [DOI] [PubMed] [Google Scholar]
- 27.Bruderer R, Bernhardt OM, Gandhi T, Miladinovic SM, et al. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen treated 3D liver microtissues. Molecular & cellular proteomics: MCP. 2015 doi: 10.1074/mcp.M114.044305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature biotechnology. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 29.Cox J, Neuhauser N, Michalski A, Scheltema RA, et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. Journal of proteome research. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 30.Escher C, Reiter L, MacLean B, Ossola R, et al. Using iRT, a normalized retention time for more targeted measurement of peptides. Proteomics. 2012;12:1111–1121. doi: 10.1002/pmic.201100463. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.