

Abstract
Motivation
RNA editing generates post-transcriptional sequence alterations. Detection of RNA editing sites (RESs) typically requires the filtering of SNVs called from RNA-seq data using an SNP database, an obstacle that is difficult to overcome for most organisms.
Results
Here, we present a novel method named SPRINT that identifies RESs without the need to filter out SNPs. SPRINT also integrates the detection of hyper RESs from remapped reads, and has been fully automated to any RNA-seq data with reference genome sequence available. We have rigorously validated SPRINT’s effectiveness in detecting RESs using RNA-seq data of samples in which genes encoding RNA editing enzymes are knock down or over-expressed, and have also demonstrated its superiority over current methods. We have applied SPRINT to investigate RNA editing across tissues and species, and also in the development of mouse embryonic central nervous system. A web resource (http://sprint.tianlab.cn) of RESs identified by SPRINT has been constructed.
Availability and implementation
The software and related data are available at http://sprint.tianlab.cn.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
RNA editing generates post-transcriptional sequence alterations (Farajollahi and Maas, 2010; Maydanovych and Beal, 2006), primarily the modification of RNA nucleotides, including adenosine-to-inosine [A-to-I, detected as A-to-G (Ramaswami et al., 2012)] and cytosine-to-uracil (C-to-U, detected as C-to-T) editing (Farajollahi and Maas, 2010; Picardi et al., 2015). A-to-I editing accounts for over 95% of all editing events in most human tissues (Bahn et al., 2012; Ramaswami et al., 2012; Zhang and Xiao, 2015), and is a universal process in metazoan (Grice and Degnan, 2015). C-to-U editing is found in both animals and plants (Blanc and Davidson, 2003; Blanc et al., 2014; Shikanai, 2015). RNA editing can affect protein coding (Benne, 1996), alternative splicing (Rueter et al., 1999), microRNA binding (Borchert et al., 2009) and other biological processes (Blanc and Davidson, 2003; Zipeto et al., 2015), and may be a major contributor to transcriptome diversity (Fumagalli et al., 2015; Han et al., 2015; Paz-Yaacov et al., 2015). It has been shown to play an important role in brain development (Wahlstedt et al., 2009), and has also been linked to a number of human diseases (Slotkin and Nishikura, 2013; Zipeto et al., 2015), such as Alzheimer disease (Gaisler-Salomon et al., 2014) and Amyotrophic lateral sclerosis (Hideyama et al., 2012).
Detection of RNA Editing Sites (RESs) typically starts from mapping RNA-seq data to a reference genome and/or a transcriptome to first identify single nucleotide variants (SNVs) (Bahn et al., 2012; John et al., 2016; Li et al., 2011; Peng et al., 2012; Picardi and Pesole, 2013; Piechotta et al., 2017; Ramaswami et al., 2012; Sun et al., 2016; Wang et al., 2016; Zhang and Xiao, 2015), which is then followed by filtering out single nucleotide polymorphisms (SNPs). Since the matched DNA-seq data are usually not available for a given study, a complete SNP database is needed in order to filter out SNPs (Ramaswami et al., 2012). However, most organisms either do not have an SNP database or have only an incomplete SNP database, which has significantly hindered the study of RNA editing from a broad evolutionary point of view. In 2013, Ramaswami et al. proposed an approach to identify conserved RESs by using an incomplete SNP database and multiple individuals’ RNA-seq data (Ramaswami et al., 2013). However, this approach is limited in that it could detect only a small subset of RESs (Zhang and Xiao, 2015). Recently, Zhang et al. developed a method named GIREMI (Zhang and Xiao, 2015) that identifies RESs based on the mutual information (MI) of adjacent SNV in the same reads (or the same pair). Though this approach does not require a complete SNP database, it still needs prior knowledge of a significant fraction of SNPs to derive the MI cutoff. To our knowledge, there is currently no method that can detect RESs without the use of any SNP information.
Since A-to-I editing is catalyzed by adenosine deaminases acting on RNA (ADARs, including ADAR1, ADAR2 and ADAR3) (Nishikura, 2010) on double-stranded RNA (Ramaswami et al., 2012; Zipeto et al., 2015), while C-to-U editing is by apolipoprotein B (ApoB) mRNA editing enzyme-1 (APOBEC1) (Zipeto et al., 2015) on single-stranded RNA (Blanc et al., 2014; Zipeto et al., 2015), it is unlikely to observe both types of RESs within the same genomic region. On the other hand, it has been reported that A-to-I RESs tend to be clustered in the genome (Ramaswami et al., 2012). Given that the density of SNP in a genome is usually low and the distribution of different types of SNP (e.g. A-to-G or A-to-C) should be independent to each other, we reason that it may be possible to distinguish RESs from SNPs by investigating the distribution of SNV duplets [defined as two consecutive SNV with the same type of variation (e.g. both are A-to-G or A-to-C)]. Indeed, in this study we found RES-based and SNP-based SNV duplets have very distinctive distributions, and have therefore developed an SNP-free RNA editing IdeNtification Toolkit (SPRINT) to identify RESs by clustering SNV duplets. When detecting RESs, most methods only investigated those RNA reads mapped to the reference genome (Bahn et al., 2012; Ramaswami et al., 2012; Zhang and Xiao, 2015). However, Porath et al. analyzed unmapped reads by masking adenosine (A) sites with guanine (G), and found genomic regions with extensive A-to-I RESs, a phenomenon called hyper RNA editing (Porath et al., 2014). Here, SPRINT also integrated the detection of hyper RNA editing sites (hyper-RESs). We have conducted thorough validations to prove the efficacy of SPRINT in detecting RESs, and have also compared SPRINT with a number of existing methods. Finally, we have applied SPRINT to investigate RNA editing across tissues and species, and also in the development of mouse embryonic central nervous system.
2 Materials and methods
2.1 Reference genome and annotations
Reference genomes (human, hg19; chimpanzee, panTro3; mouse, mm9; C.elegans, ce10), gene category annotations (e.g. CDS, 5’-UTR, etc.) and repeats annotations of human, chimpanzee, mouse and C.elegans were downloaded from UCSC genome browser (http://genome.ucsc.edu). MacaM_Rhesus_Genome_v7 (Zimin et al., 2014) (http://www.unmc.edu/rhesusgenechip/index.htm) was used as the reference genome for rhesus, and RepeatMasker (Tempel, 2012) (http://www.repeatmasker.org) was used with the command option of ‘perl RepeatMasker –species macaca MacaM_Rhesus_Genome_v7.fa’ to produce repeat annotations in rhesus genome. RepeatMasker libraries were obtained from GIRI (Genetic Information Research Institute, http://www.girinst.org). Rhesus gene annotations, MacaM_Rhesus_Genome_Annotation_v7.6.8 (Zimin et al., 2014), were obtained from http://www.unmc.edu/rhesusgenechip/index.htm. Human dbSNP (version 141) was downloaded from UCSC Table Browser (http://genome.ucsc.edu/cgi-bin/hgTables). PhyloP (Pollard et al., 2010) conservation scores of human and mouse were downloaded from UCSC (http://hgdownload.soe.ucsc.edu/downloads.html). PhyloP (Pollard et al., 2010) conservation scores were based on individual nucleotides, and the corresponding file names for human and mouse were ‘hg19.100way.phyloP100way.bw’ and ‘chrN.phyloP30way.wigFix’ (‘N’ refers to the ID of chromosome), respectively. Human conservation score file was in BIGWIG format (binary format), which was converted to WIG file (text format) using ‘bigWigTowig’ (http://hgdownload.soe.ucsc.edu/downloads.html#utilities_downloads).
2.2 RNA-seq datasets
The RNA-seq data of GM12878 (lymphoblastoid cell line) were downloaded from the ENCODE project (http://genome.ucsc.edu/ENCODE/downloads.html). The RNA-seq data of glioma cell line U87MG (including both wild-type and ADAR1 knockdown) (Bahn et al., 2012), C.elegans (including both wild-type and adars knockdown) (Zhao et al., 2015), mouse liver (including wild-type, Apobec-1 knockdown and ad-Apobec-1) and intestine (including both wild-type and Apobec-1 knockdown) (Blanc et al., 2014), and the brain, heart, liver and testis of human, chimpanzee, rhesus and mouse (Ruiz-Orera et al., 2015), as well as mouse embryonic and adult tissues were all downloaded from NCBI SRA database (http://www.ncbi.nlm.nih.gov/gds/). The detailed information about the above RNA-seq datasets can be found in Supplementary Table S1.
2.3 Reads processing, reads mapping and SNV calling
Before mapping, the first 6 bases of each read were trimmed to avoid mapping errors caused by random-hexamer primers. Then, Burrows-Wheeler Aligner (Li and Durbin, 2009; Li and Homer, 2010) (BWA, version 0.7.12) was applied to map RNA-seq reads to the reference genome. Note that for all organism to be analyzed, BWA is the only program used in SPRINT for reads mapping. Paired-end reads were mapped separately with the command options of ‘bwa aln fastqfile’ and ‘bwa samse -n4’ as described by Ramaswami et al. (2012). The detailed mapping information was in SAM file (Li et al., 2009). Samtools (Li et al., 2009) (version 1.2) was used to convert SAM files to BAM files and to sort BAM files, and picard-tools (version 1.119, http://broadinstitute.github.io/picard/) was then used to remove PCR duplicates in the sorted BAM files with the command option of ‘MarkDuplicates.jar REMOVE_DUPLICATES = true’. Those reads with high mapping quality (≥20) were regarded as mapped reads. Low complexity reads were removed from unmapped reads (refer to Fig. 1a). Then, both unmapped reads and the reference genome were masked by replacing A with G (the reference genome was also masked by replacing T with C in order to retrieve RESs in antisense transcripts), as described by Porath et al. (2014). Remapping of masked reads to masked reference genome was conducted, and remapped reads were obtained by recovering remapped masked reads.
Fig. 1.
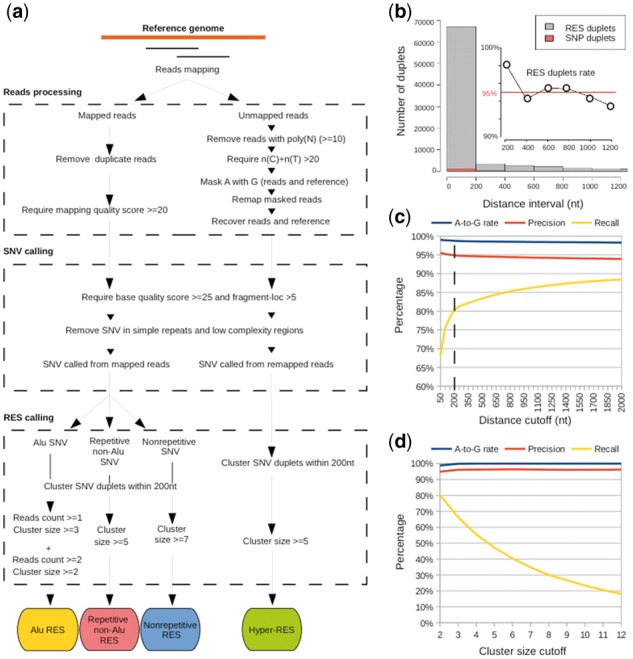
The workflow and the methodology of SPRINT. (a) The work flow of SPRINT. (b) The number of SNV duplets (two consecutive SNVs with the same type of variation) at different distance intervals. SNP and RES duplets refer to SNV duplets in which both SNVs are SNPs and RESs, respectively. The sub-figure plots the fraction of RES duplets among all SNV duplets (RES duplet rate) at a given distance interval. In the horizontal axis of sub-figure, ‘200’ means ‘0–200’; ‘400’ means ‘200–400’ etc. (c) The A-to-G rate, the precision and the recall of the RESs identified by SPRINT when the cluster size cutoff is set at 2 while the distance cutoff varies. (d) Similar to (c) except that the distance cutoff is fixed at 200 nt while the cluster size cutoff varies. The SNVs used in (b–d) are those SNVs called by SPRINT in Alu regions of GM12878 (cytosolic) with two or more read counts
For SNVs calling, we first identified all mismatches. Then, we followed Ramaswami et al. (2012) to use base quality scores and repeat annotation database to filter out those mismatches likely resulted from system errors (sequencing and/or mapping errors) (for details, refer to Fig. 1a). In case an organism does not have repeat annotation database available, we used RepeatMasker (Tempel, 2012) to obtain repeat annotations. Ramaswami et al. (2012) further used transcript annotations to filter out mismatches close to exon/intron junctions because mapping errors occur frequently near splicing sites. Considering that most organisms do not have complete transcript annotations, here we introduced a new measure called ‘fragment-loc’ that refers to the distance of a mismatch to the nearest end of the mapped fragment it belongs to (a read may be split into more than one fragment when mapped to the reference genome) (Supplementary Fig. S1a). Analyzing the mismatches in the mapped reads from GM12878 [ENCODE, cytosolic, poly (A)+], we found there was a sharp increase in the number of mismatches when fragment-loc is less than 5 (Supplementary Fig. S1b), i.e. close to the end of mapped fragments, and consequently used ‘fragment-loc’ of 5 as an additional filter to filter out mismatches. The remaining mismatches were then called SNVs.
2.4 Mouse gene expression analysis and gene set enrichment analysis
For mouse gene expression analysis, mouse Ensembl gene annotations (version 67, ftp://ftp.ensembl.org/pub/release-67/) were used to annotate reference genes. To calculate mouse gene expression level, Tophat2 (Kim et al., 2013) (version 2.1.0) was applied with default command options to map RNA-seq reads to the reference genome. Note that Tophat2 is only used for calculating the expression level of mouse genes. When detecting the RESs in mouse, we use BWA for reads mapping. Then, cufflinks (Trapnell et al., 2012) was used to calculate gene expression level with the command option of ‘cufflinks –u -G reference_genes.gtf’, and cuffmerge (Trapnell et al., 2012) was used to assemble transcriptomes with the command option of ‘cuffmerge –g reference_genes.gtf –s mm9.fa’. Finally, cuffdiff (Trapnell et al., 2012) was used to calculate the fold change of expression level with the command option of ‘cuffdiff –u merged.gtf’. DAVID (Huang da et al., 2009a,b) (https://david.ncifcrf.gov/) was used for gene set enrichment analysis on GO BP terms (GOTERM_BP_FAT), with ‘FDR’ as the method for multiple test correction, and the significance threshold was set at 0.05.
2.5 Competing tools
We compare SPRINT with the following four tools on detecting RNA editing. These four tools are JACUSA (Piechotta et al., 2017), GIREMI (Zhang and Xiao, 2015), RNAEditor (John et al., 2016) and REDItools (Picardi and Pesole, 2013). GIREMI is downloaded from https://github.com/zhqingit/giremi. RNAEditor is downloaded from https://github.com/djhn75/RNAEditor. The related tools and annotations for installing RNAEditor are downloaded following the instruction of RNAEditor’s documents. REDItools is downloaded from https://sourceforge.net/projects/reditools/. JACUSA is downloaded from https://github.com/dieterich-lab/JACUSA. RNAEditor is the only tool in these three that includes reads mapping, SNV calling and RES calling. After trimming the first six bases of each read, we run RNAEditor with the command-line option of ‘python RNAEditor.py –i read1.fastq read2.fastq –c configuration.txt’. Reads mapping is not included in GIREMI, REDItools (de novo) and JACUSA (RRD). Consequently, the BAM file produced by SPRINT is used for these tools. To call SNVs for GIREMI, we use samtools (version 1.4) and bcftools (version, 1.4) with the command option of ‘samtools mpileup –vf reference_genome.fa BAM_file | bcftools call –cv - > VCF_file’. After removing all homozygous sites with more than two types of alleles in the VCF_file, we annotate the VCF_file with Ensembl Genes (Version 75) and convert the VCF_file into the input_fIle of GIREMI. Then, GIREMI is used with the command option of ‘giremi –f reference_genome.fa –o output_file –l input_file BAM_file’ to call RESs. REDItools (de novo) is used with the command option of ‘python REDItoolDenovo.py –i BAM_file –f reference_genome.fa –o output_file –V 0.05 –l –t 20’. JACUSA (RRD) is used with the commond option of ‘java -jar JACUSA_v1.2.0.jar call-2 -T 1.56 -p 20 -s –r output_file BAM_file_CTRL BAM_file_KD’. Then, we filter the sites called by REDItools (de novo) and JACUSA (RRD) using dbSNP141 to obtain RESs.
3 Results
3.1 Development of SPRINT, an SNP-free RNA editing IdeNtification toolkit
SPRINT consists of three major steps: reads processing, SNVs calling and RESs calling (Fig S1a). Briefly speaking, reads from RNA-seq data are first processed into mapped reads (those with high mapping quality scores) and remapped reads (those unmapped reads that are remapped to the reference genome after masking A with G). Then, SNVs are called separately from mapped and remapped reads, which is then followed by the calling of RESs from SNVs. For convenience, the RESs identified from mapped and remapped reads are called regular-RESs and hyper-RESs, respectively. For details about reads processing and SNV calling, please refer to Figure 1a, Supplementary Figure S1a, b and Section 2. Below, we describe the RESs calling in SPRINT.
For SNVs called from mapped reads, we follow Ramaswami et al. (2012) to classify them into three categories—those in Alu (or Alu family) regions, and those in repetitive non-Alu and non-repetitive regions, before conducting RESs calling. To examine the quality of the called SNVs, we select the SNVs called by SPRINT in Alu regions from the mapped reads of GM12878 (cytosolic). Following Ramaswami et al. (2012), we filter those SNVs with two or more read counts using human dbSNP [version 141, a complete SNP database to GM12878 (Djebali et al., 2012)] to call RESs. We obtain 133 571 RESs with an A-to-G rate of 97.4% that is comparable to what Ramaswami et al. (2012) (95.8%, GM12878, cell) and Zhang and Xiao (2015) (99.7%, GM12878, cytosolic) reported for RESs in Alu regions, demonstrating that the SNVs called by SPRINT with two or more read counts are of high quality. In comparison, the A-to-G rate of the RESs called by filtering all SNVs is only 76.5%, suggesting that many one-read-count SNVs may be system errors.
SPRINT identifies RESs from SNVs without the need to filter out SNPs. To illustrate its working strategy, we identify the SNPs from the high-quality SNVs called by SPRINT in Alu regions from GM12878 (cytosolic) (those with two or more read counts), and then call the remaining SNVs as RESs. By defining a SNV duplet as two consecutive SNVs that have the same type of variation (e.g. both are A-to-G or both are A-to-C), we find that almost all RES-based SNV duplets (both SNVs are RESs) are within 400 nt, while almost no SNP-based SNV duplets (both SNVs are SNPs) are within 1600 nt (Fig. 1b). In fact, the fraction of RES-based SNV duplets is almost all above 95% when they are within 1600 nt (Fig. 1b). This thus inspires us to identify RESs by clustering SNV duplets (see Supplementary Fig. S1c for the clustering procedure). We first identify all SNV duplets by implementing a specific distance cutoff (e.g. with 200 nt). Note that a SNV can be included into two adjacent SNV duplets. Then, we scan the genome from the start position of each chromosome to merge SNV duplets that share a common SNV. The merging stops if the next SNV duplet does not share a common SNV with the previous one, and a cluster of SNV duplets is identified. A new merging process is initiated from the next SNV duplet. After the scanning and merging are completed, we obtain a large number of SNV duplet clusters. Finally, we select those SNV duplet clusters whose cluster size (defined as the number of SNVs within the SNV duplet cluster) is above a certain cutoff, and consider all SNVs within these clusters as RESs. When the cluster size and the distance cutoffs are set at 2 and 200 nt, respectively, the precision [1—percentage of the identified RESs that are SNPs (Zhang and Xiao, 2015)] approximating 95% while the recall (the percentage of true RESs that are identified) is reasonably high (80.3%) (Fig. 1c). The corresponding number of RESs is 116 488, and the A-to-G rate is 98.8%. It’s worth noting that the A-to-G rate is almost constantly above 98% at all tested cluster size and distance cutoffs (Fig. 1d). To investigate whether we may further increase the number of called RESs, we apply the clustering strategy to all SNVs. By choosing the cluster size and the distance cutoffs are set at 3 and 200 nt, respectively, we obtain 335 499 RESs. The A-to-G rate is 99% (Supplementary Fig. S2a), suggesting that the clustering strategy can effectively remove those one-read-count SNVs that are likely system errors. Combining the RESs called from high-quality SNVs and from all SNVs together, we obtain 359 725 RES (98.7% A-to-G) in Alu regions from the mapped reads of GM12878 (cytosolic).
Identifying RESs in non-Alu regions is a challenging task (Ramaswami et al., 2012; Zhang and Xiao, 2015). Here, we use a summed editing rate (A-to-G plus C-to-U rate) to indicate the quality of the called SNVs. By fixing the distance cutoff at 200 nt and setting the cluster size cutoff at 5 and 7 for repetitive and non-repetitive SNVs, respectively, we obtain 5469 and 2081 RESs with a summed editing rate of 99.3% (96% A-to-I; 3.3% C-to-U) and 95.6% (75.9% A-to-I; 19.7% C-to-U) in these two regions, respectively (Supplementary Fig. S2b and c). Combining them with the RESs called in Alu regions, we obtain 367 275 RESs (98.6% A-to-G) from the mapped reads in GM12878 (cytosolic).
The clustering strategy is also applied for identifying hyper-RESs from remapped reads. By setting the cluster size and the distance cutoffs at 5 and 200 nt, respectively (Supplementary Fig. S2d), we obtain 422 068 hyper-RESs (98.9% A-to-G) in GM12878 (cytosolic). Among them, only 59 566 are also identified from mapped reads, suggesting that most hyper-RESs are different from regular-RESs. Finally, we combine regular-RESs and hyper-RESs together, and obtain a total number of 729 777 RESs from GM12878 (cytosolic) without filtering out SNPs (Supplementary Table S1).
3.2 The validation of SPRINT’s effectiveness in identifying RESs
To examine SPRINT’s effectiveness in identifying A-to-I RESs, we apply SPRINT to two RNA-seq datasets: U87MG dataset that includes both wild-type and ADAR1 knockdown cell lines (Bahn et al., 2012), and a C.elegans dataset that includes both wild-type and adrs (adr-1 and adr-2, ADARs of C.elegans) knockdown samples (Zhao et al., 2015). In both datasets, we observe a significant reduction in the number of both regular and hyper A-to-I RESs detected by SPRINT when ADARs are knock down (Fig. 2a–d and Supplementary Table S1). For C-to-U RESs, we apply SPRINT to two datasets: the first consists of mouse liver and intestine RNA-seq data from both wild-type and Apobec-1 knockdown conditions (Blanc et al., 2014); the second is adenoviral delivery of Apobec1 (ad-Apobec-1) mouse liver RNA-seq data in which Apobec-1 was overexpressed (Blanc et al., 2014). In the first dataset, we also observe a significant reduction in the number of C-to-U RESs (wild type: 166; Apobec-1 knockdown: 6) called by SPRINT when Apobec-1 is knockdown (Fig. 2e and f and Supplementary Table S1), and the percentage of SNP among the C-to-U RESs called by SPRINT in mouse wild type liver and intestine was both lower than 2.5% (mouse dbSNP, version 128, downloaded from UCSC Table Browser). While in the second dataset, SPRINT identifies 62 788 C-to-U RESs when Apobec-1 is overexpressed (Supplementary Table S1), much greater than that in wild type mouse liver (166 C-to-U RESs in the first dataset), and the percentage of SNP among the C-to-U RESs called by SPRINT in Apobec-1 overexpressed liver was less than 1%. The significant reduction and increase of the number of RESs called in Apobec-1 knockdown and overexpression samples demonstrated SPRINT's effectiveness in calling C-to-U RESs, while the very low percentage of SNPs suggests that the C-to-U RESs called by SPRINT are likely of high quality. We further find that there are only very few C-to-U RESs in Alu regions in both wild type and ad-Apobec-1 samples (wild type intestine: 15, 0.8%; wild type liver: 8, 0.5%; ad-Apobec-1 liver: 992, 1.56%), while all C-to-U RESs found when Apobec-1 is knockdown are in Alu regions (Supplementary Table S1). This suggests that the C-to-U RESs found in Alu regions are likely false positives, and they are therefore removed from the report of C-to-U RESs from here on.
Fig. 2.
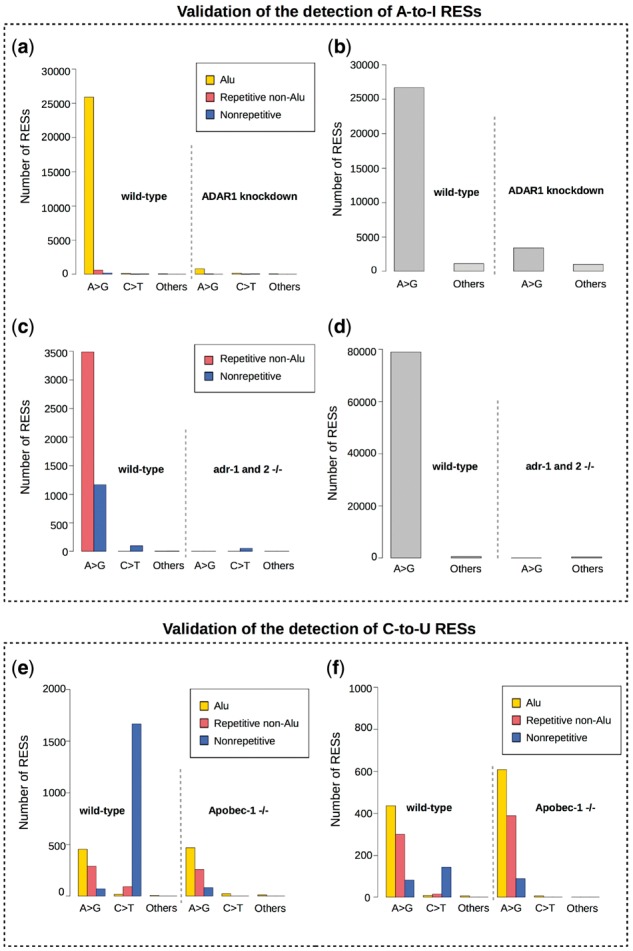
The validation of SPRINT’s effectiveness in detecting RESs. (a) The number of regular-RESs and (b) the number of hyper-RESs identified by SPRINT in wild-type and ADAR1 knockdown U87MG cell line (Bahn et al., 2012). (c) The number of regular-RESs and (d) the number of hyper-RESs identified by SPRINT in wild-type and ADARs (adr-1 and adr2) knockdown C.elegans embryos (strand-specific) (Zhao et al., 2015). (e) The number of C-to-U RESs identified by SPRINT in wild-type and Apobec-1 knockdown mouse intestine (Blanc et al., 2014). (f) Similar to (e) except that the samples are from mouse liver. In (a–f), ‘>’means ‘to’, A-to-I is detected as A-to-G, and C-to-U is detected as C-to-T. Others refer to all types of variations except A-to-I and C-to-U. Because U87MG and mouse (liver and intestine) RNA-seq datasets are not strand-specific (Bahn et al., 2012, 2014), A-to-G mismatches might be detected as T-to-C mismatches when reads are mapped to opposite strand, and C-to-T mismatches might be detected as G-to-A. Therefore, A-to-G and T-to-C editing sites are combined to represent A-to-I editing sites, while C-to-T and G-to-A RES are combined to represent C-to-U editing sites in those two datasets
By applying SPRINT to a randomly selected portion (e.g. 10%, 20%, etc.) of reads in GM12878 (cytosolic) that mimic different sequencing depths, we find that SPRINT is able to identify RESs with high quality regardless of sequencing depth, though higher sequencing depth is needed in order to achieve a more complete coverage of RESs (the number of called RESs is almost linearly correlated with sequencing depths) (Supplementary Fig. S3). The simulation results also suggest that the number of RESs cannot be directly compared across samples unless they are under similar sequencing depths. Note that in all datasets used to validate SPRINT, the RNA-seq data being compared are under similar sequencing depth. In addition, since most RESs have only one or two read counts (Supplementary Fig. S4), it implies that many RESs may fail to be identified under current sequencing depth.
3.3 The comparison of SPRINT with other methods
We first compare the RESs called by SPRINT with the previously reported RESs in the studies of Ramaswami et al. (2012), Zhang and Xiao (2015), Porath et al. (2014) (Table 1), Blanc et al. (2014) and Zhao et al. (2015). For regular-RESs, in GM12878 (cell), SPRINT identifies 355 730 RESs (96.6% A-to-G), much greater than that (150 865, 95.7% A-to-G) obtained by Ramaswami et al. In both of Alu and non-Alu regions, SPRINT identify 2.3 fold and 5.1 fold number of RESs comparing to the number of RESs called by Ramaswami et al. with consistently high precision [above 96.5%, defined as 1 − percentage of the identified RESs that are SNPs (Zhang and Xiao, 2015)]. Here, it can be noted that SPRINT has a lower A-to-G rate in non-repetitive RESs, which is highly likely to be caused by the sequencing artefacts (clustered G-to-T sites) in the RNA-seq data of ENCODE project. For all ENCODE's RNA-seq data analyzed in this study, after the removal of A-to-G and C-to-U sites from the RESs called by SPRINT there are a very high frequency of G-to-T sites (36–95%) in the remaining sites, which is in significant contrast to that of the RESs called from the RNA-seq data generated by the other studies (9–28% G-to-T sites after the removal of A-to-G and C-to-U sites). For the RESs called by Ramaswami et al., 47% of the remaining sites after the removal of A-to-G and C-to-T sites are also G-to-T sites. See Supplementary Figure S5 for details, and this kind of bias shows that users of SPRINT should check the rate of observed changes before any conclusion. If G-to-T sites are removed from the RESs called by SPRINT, then A-to-G rate in nonrepetitive regions is over 98.6%. We further compare the RESs called by Ramaswami et al. with those called by SPRINT, and find 78.9% of the RESs identified by Ramaswami et al. are also found by SPRINT (82.3% for A-to-I RESs), partly validating the quality of the RESs identified by SPRINT. In GM12878 (cytosolic), SPRINT identifies 367 275 RESs (98.6% A-to-G), much greater than that (genome-aware: 41 027, 99% A-to-G; 70% SNP known: 37 591, 98.4% A-to-G) reported by Zhang et al. (GIREMI). SPRINT also achieves higher precision in repetitive non-Alu and nonrepetitive regions (96.8% and 95.5%; 0% SNP known) than that reported by Zhang et al. (84.3% and 73.8%; 70% SNP known).
Table 1.
The comparison of SPRINT with other methods on identifying RES
| Alu sites |
Repetitive non-Alu sites |
Nonrepetitive sites |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell lines | Tools | Known SNP (%) | Total | A-to-G (%) | Precision (%) | FDR (%) | Total | A-to-G (%) | Precision (%) | FDR (%) | Total | A-to-G (%) | Precision (%) | FDR (%) | |||
| GM12878, cell | SPRINT | 0 | 336 304 | 97.7 | 96.5 | – | 14 019 | 87.8 | 97.2 | – | 5407 | 49.8a | 96.8 | – | |||
| Ramaswami et al.* | 100 | 147 029 | 95.8 | – | – | 2385 | 97.4 | – | – | 1451 | 86.6 | – | – | ||||
| Regular-RESs | GM12878, cytosolic | SPRINT | 0 | 359 725 | 98.9 | 96.9 | – | 5469 | 96 | 96.8 | – | 2081 | 75.9 | 95.5 | – | ||
| GIREMI* | 70 | 36 131 | 99 | 99.4 | – | 267 | 83.7 | 84.3 | – | 1193 | 82.8 | 73.8 | – | ||||
| GIREMI* | 100 | 39 757 | 99.7 | – | – | 260 | 88.6 | – | – | 1010 | 73.5 | – | – | ||||
| U87MG | SPRINT | 0 | 48 085 | 99.6 | 96.2 | 3.2 | 988 | 99.5 | 97.1 | 4.5 | 296 | 87.8 | 91.2 | 0 | |||
| GIREMI | 100 | 2152 | 99.8 | – | 0.7 | 114 | 96.5 | – | 9 | 509 | 88.6 | – | 53 | ||||
| RNAEditor | 100 | 62 979 | – | – | 8.2 | 6142 | – | – | 42.3 | 155 | – | – | 55.5 | ||||
| REDItools (de novo) | 100 | 628 | 96.5 | – | 3.6 | 238 | 46.2 | – | 80 | 14 949 | 39.7 | – | 100 | ||||
| JACUSA (RRD) | 100 | 2154 | 94.7 | – | – | 331 | 39 | – | – | 4527 | 20.8 | – | – | ||||
| Total | A-to-G (%) | ||||||||||||||||
| GM12878, cell | SPRINT | 328 762 | 97.9 | ||||||||||||||
| Hyper-RESs | Porath et al.* | 157 077 | 96 | ||||||||||||||
| U87MG | SPRINT | 57 913 | 96 | ||||||||||||||
| Porath et al.* | 27 124 | 94.6 | |||||||||||||||
Note: ‘*’ means the data are derived from the corresponding study. The details for running competing tools are described in Methods. ‘–’ means not assessed: For A-to-G rate, RNAEditor only outputs A-to-G changes, and therefore cannot be assessed; For precision, in U87MG dataset all methods except SPRINT call RESs by removing SNPs in dbSNP from the called SNVs, and therefore cannot be assessed; For FDR, only U87MG dataset is used for assessment, because it has ADAR knockdown sample. Since JACUSA (RRD) compares variants of RNA (CTRL) with that of RNA (KD), the FDR of JACUSA is unavailable.
The lower A-to-G rate is attributed to the presence of many clustered G-to-T changes which may be sequencing artefacts of the RNA-seq data in ENCODE project (see Supplementary Fig. S5 for details).
We next compare SPRINT with the method used by Blanc et al. (2014) on identifying C-to-U RESs. In wild-type mouse intestine and liver, SPRINT identifies 1757 and 158 C-to-U RESs, respectively, both of which are much greater than that (438 and 39 in intestine and liver, respectively) reported by Blanc et al. (2014). The C-to-U RESs found by SPRINT in mouse liver and intestine are mainly in 3’-UTR, which is consistent with what Blanc et al. reported (Blanc et al., 2014; Rosenberg et al., 2011) (Supplementary Fig. S6a).
For hyper-RESs, we compare SPRINT with the method developed by Porath et al. (2014). Porath et al. reported 27 124 hyper-RESs (94.6% A-to-G) in wild-type U87MG. In comparison, SPRINT identifies 57 913 hyper-RESs with higher A-to-G rate (Table 1). Porath et al. (2014) also analyzed GM12878 (cell), and identified 157 077 hyper-RESs (96% A-to-G). SPRINT identifies 328 762 hyper-RESs (97.9% A-to-G) in the same data (Table 1). Recently, Zhao et al. (2015) investigated both regular- and hyper-RESs in C.elegans, and reported a combined number of 50 740 A-to-I RESs. From the same data, SPRINT identifies a much higher number (232 133) of A-to-I RESs (regular: 11 958; hyper: 227 703) (Table 1). The variation pattern of the number of A-to-I RESs identified by SPRINT at different development stage of C.elegans is similar to that reported by Zhao et al. (2015) (Supplementary Fig. S6b).
Currently, there are a number of RES detection tools available for running locally, including GIREMI (Zhang and Xiao, 2015), RNAEditor (John et al., 2016), REDItools (Picardi and Pesole, 2013), RED (Sun et al., 2016), RES-Scanner (Wang et al., 2016) and JACUSA (Piechotta et al., 2017). Note that all these tools require the input of either complete or incomplete SNP annotations, and most of them are not fully automated, requiring users to conduct reads mapping and SNV calling; in addition, none of them can detect hyper-RESs (Supplementary Table S2). Here we use U87MG dataset to compare SPRINT with some of these tools for detecting regular-RESs. The U87MG dataset includes the RNA-seq data of U87MG ADAR knockdown sample (Bahn et al., 2012), which can be used as a negative control to assess the false discovery rate (FDR) of different methods. By assuming that all RNA editing sites (RESs) detected in U87MG ADAR knockdown sample are false positives, and assuming that a method detects comparable number of false positive RESs in U87MG ADAR sample (because they have similar sequencing depth), we can compute the FDR of a given method as the ratio of the number of A-to-I RESs detected from U87MG ADAR knockdown sample to the number of A-to-I RESs detected from U87MG ADAR control sample (no more than 100%). As RES-Scanner requires the use of paired DNA-seq data that are not available for U87MG in the study of Bahn et al. (2012) [though the DNA-seq data of U87 cell line has been released by a previous study (Clark et al., 2010), this data has been generated using a different sequencing platform and at low coverage], while we encounter difficulties to run RED locally, we compare SPRINT with GIREMI, RNAEditor, REDItools (de novo) and JACUSA (RRD) using U87MG as a benchmark dataset (see Section 2 for details on running these tools) (Table 1). Note that all these methods being compared with SPRINT use complete SNP annotations. Because JACUSA (RRD) detects RESs by detecting the differences between the SNV called in paired RNA-seq data, its FDR cannot be calculated. Among all methods reporting FDR, SPRINT achieves the lowest FDR in all three categories (Alu, repetitive non-Alu and non-repetitive regions: 3.2%, 4.5% and 0%, respectively) except for GIREMI in Alu regions (0.9%). In Alu regions, although GIREMI has a lower FDR than SPRINT does while REDItools (de novo)'s FDR (3.6%) is close to SPRINT's, the number of RESs detected by both GIREMI (2152) and REDItools (628) is significantly much lower than that by SPRINT (48 085). RNAEditor detects more number of RESs than SPRINT does in Alu, and repetitive non-Alu regions, yet it has higher FDR, especially in repetitive non-Alu regions. It's worth noting that in non-repetitive regions, all the methods being compared with SPRINT have much higher FDR (53–100% FDR for the other methods, compared to 0% FDR by SPRINT). Because the FDR of JACUSA is unavailable, we compare its A-to-G rate with SPRINT's. JACUSA detects less number of RESs, and has lower A-to-G rate than SPRINT does, especially in repetitive non-Alu and nonrepetitive regions [SPRINT: 99.6%, 99.5% and 87.8%; JACUSA (RRD): 94.7%, 39% and 20.8%]. We further compare the RESs called by different methods, and find that many RESs called by SPRINT are not found by the other methods (Supplementary Fig. S7). By using U87MG as a benchmark dataset, we conclude that SPRINT achieves lowest FDRs on detecting RESs among the four methods. We also evaluate the computational speed of different methods using U87MG. SPRINT spends much less time than RNAEditor, and is comparable to the other two methods in terms of computational time (Supplementary Table S3). Reads mapping is the most time-costing step in SPRINT. Once reads mapping is completed, less than half hour is needed for SNV calling and RES calling. A binary executable version and a python package of SPRINT along with install instructions are available for downloading at http://sprint.tianlab.cn/SPRINT/.
3.4 RNA editing across tissues and species
Here, we apply SPRINT to a recently published dataset (Ruiz-Orera et al., 2015) that includes deeply sequenced RNA-seq data of brain, liver, heart and testis from four species—human, chimpanzee, rhesus and mouse (Section 2 and Supplementary Table S1). The A-to-G rate of both regular and hyper RES is all above 95% in these samples (Supplementary Table S1), indicating that the RESs identified by SPRINT in these samples are of high quality. In all four tissues of the four species, regular A-to-I RESs occur most frequently in intronic and intergenic regions, infrequently in 5’ UTR, and seldom in CDS (Fig. 3a). The proportion of regular A-to-I RESs in 3’ UTR is significant (3–32%) in both human and mouse, and varies in between tissues and ages. Though it is smaller (1–5%) and less variable in chimpanzee and rhesus, this may be attributed to the incomplete transcript annotations in these two species. In addition, there is a significant enrichment of RESs in 3’ UTR for regular A-to-I RESs that are common to all tissues for all four species (Fig. 3a). A-to-I RESs is also highly enriched in 3’ UTR compared to randomly selected ‘A’ sites in Alu regions (Fig. 3a). These lines of evidence thus highlight the significance of A-to-I RESs in 3’ UTR. Hyper A-to-I RESs have similar distributions to that of regular A-to-I RESs. We then use Variant Effect Predictor (VEP, http://www.ensembl.org/info/docs/tools/vep/) to annotate the potential coding consequences of human CDS regular and hyper A-to-I RESs, and find the proportion of missense variants is around 60% in all four tissues with variations (ranging from 60–67%) (Fig. 3c), suggesting that A-to-I RESs might have great influence on protein translation, which have also been reported by a previous study (Picardi et al., 2015). C-to-U RESs show different distributions in between species, especially in between mouse and human. For example, a significant proportion of C-to-U RESs are in CDS in human; in contrast, they are mainly in 3’ UTR in mouse (Fig. 3a). Further inspection of the CDS C-to-U RESs detected by SPRINT in human and chimpanzee reveals that they mainly occur in mitochondria (Fig. 3b), which remains further validation and investigation.
Fig. 3.
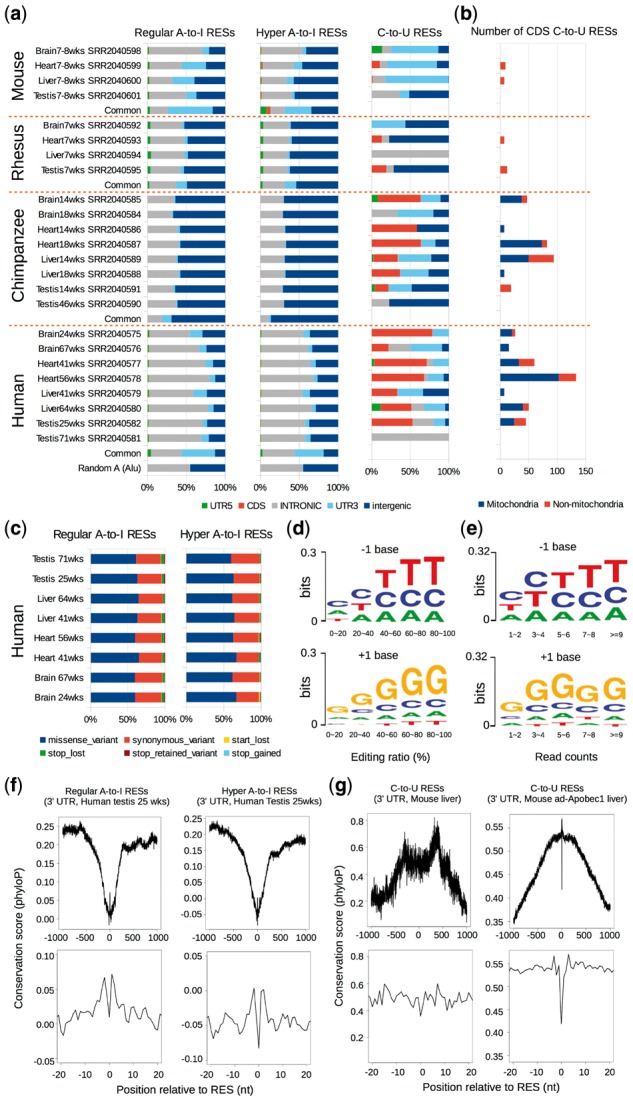
RNA editing in different tissues of human, chimpanzee, rhesus and mouse. (a) The proportions of different categories of regular A-to-I RESs (left), hyper A-to-I RESs (middle) and C-to-U RESs (right) called by SPRINT in different tissues of human, chimpanzee, rhesus and mouse. ‘wks’ refers to ‘weeks’. (b) The number of CDS C-to-U RESs detected in different tissues of the four species. (c) The fractions of potential coding consequences (e.g. missense, synonymous, etc.) of CDS A-to-I RESs called by SPRINT in human. We used Variant Effect Predictor (VEP, http://www.ensembl.org/info/docs/tools/vep/) to annotate the potential coding consequences of human CDS regular and hyper A-to-I RESs. (d) The preference for the nucleotide before (−1, upper) and after (+1, lower) a regular A-to-I RES for RESs with different editing ratios. Here, the read depth of a RES is required to be greater than or equal to five. The nucleotide preference is plotted using WebLogo(Crooks et al., 2004). (e) Similar to (d) except that RESs with different read counts are investigated. (f) The averaged phyloP conservation scores (Pollard et al., 2010) at different positions relative to a regular A-to-I RES (left) or a hyper A-to-I RES (right) in 3’ UTR, with the position ranging from −1000 to +1000 (upper) and from -20 to +20 (lower). (The sequence patterns of A-to-I RESs in other categories can be found in Supplementary Fig. S7). In (d–f), the A-to-I RESs used are from human testis (25 weeks), because human testis has the most number of detected A-to-I RESs among all tissues investigated in this study. (g) Similar to (f), except that C-to-U RESs are investigated. The left and right sub-figures are plotted using the C-to-U RESs identified from mouse liver (7–8 weeks) and mouse ad-Apobec1 liver, respectively
Previous studies (Bahn et al., 2012; Ramaswami et al., 2012) have reported that the nucleotide before and after an A-to-I RES prefers to be T and G, respectively. Here, we find such preferences become more significant when either the read counts or the editing ratio of A-to-I RESs increases (Fig. 3d, e). This tendency is observed in all samples in this dataset (Supplementary Fig. S8a and b), and by using the RESs identified by Ramaswami et al. (2012) (Supplementary Fig. S8c). It’s worth noting that previous studies typically used A-to-I RESs with higher read counts (e.g. 5) to derive the nucleotide preference (Bahn et al., 2012). Here, even though we do not find significant pattern of nucleotide preferences for RESs with low read counts, there is no evidence that they may be noise, given that these RESs also have high A-to-G rate (Supplementary Fig. S4c and d) and that we have demonstrated the effectiveness of SPRINT in detecting RESs by using ADARs knock out RNA-seq data. Blanc et al. (2014) found that both A and T are preferred for the nucleotide surrounding a C-to-U RES. Here, we find similar pattern in mouse liver (the other samples are not analyzed because of the lack of enough C-to-U RESs) and also in ad-Apobec-1 mouse liver (Supplementary Fig. S8e). It has been reported that A-to-I RESs are less conserved than their neighbor sites (Zhang and Xiao, 2015). Here, we find similar conservation patterns for regular A-to-I RESs in human and mouse (Fig. 3f and Supplementary Fig. S9). Chimpanzee and rhesus are not analyzed here because of unavailable phyloP scores (Pollard et al., 2010). For C-to-U RESs, we find in mouse liver that they are also less conserved than their neighbor sites, though the nucleotides that are proximate to C-to-U RESs are more conserved than those further away (Fig. 3g). Similar yet more significant conservation pattern is found for C-to-U RESs in ad-Apobec-1 mouse (Fig. 3g).
So far, we have analyzed regular and hyper A-to-I RESs separately. By normalizing the number of regular and hyper A-to-I RESs using the total reads in each sample, we find they are almost linearly positively correlated with each other (Fig. 4a). In addition, the number of regular and hyper A-to-I RESs are also linearly positively correlated at gene level (Fig. 4b and Supplementary Fig. S10). Therefore, we combine them together when comparing the number of A-to-I RESs across tissues and species. In general, primates have many more A-to-I RESs [817.4–6057.5 RESs per one million (1 M) reads] than mouse (45.9–471.5 per 1 M reads) has (Fig. 4c). Considering that the normalized number of A-to-I RESs varies during the development, as shown in the C.elegans data (Supplementary Fig. S7b), we select the samples in chimpanzee, rhesus and mouse that are at relatively similar age in order to conduct comparison in between tissues. In all these three species, brain has the highest normalized number of A-to-I RESs among the four tissues investigated (Fig. 4c), especially in mouse (Supplementary Table S1 and Supplementary Fig. S11). For example, the normalized number of A-to-I RESs in mouse brain is over 10 times to that in heart and 3.5 times to that in liver, in contrast to 5.1 and 1.7 in chimpanzee, and 2.4 and 2.0 in rhesus, respectively. The above lines of evidence highlight the importance of RNA editing in brain, especially for mouse.
Fig. 4.
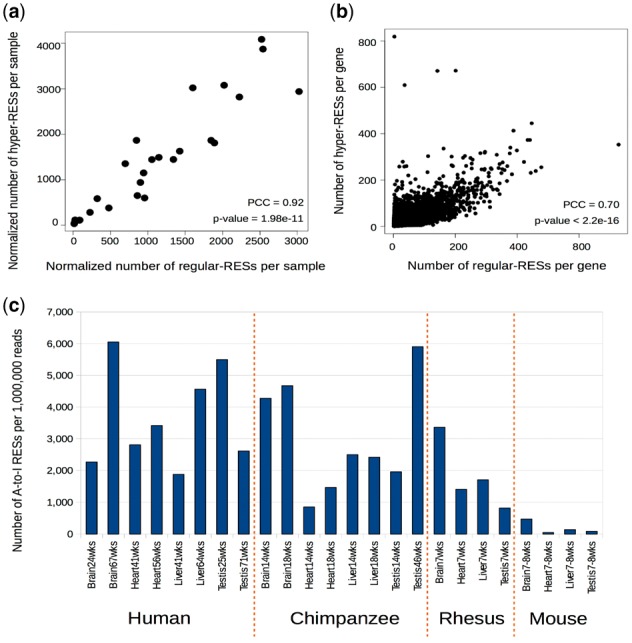
The normalized number of A-to-I RESs across different tissues in the four species. (a) The normalized number of regular A-to-I RESs versus the normalized number of hyper A-to-I RESs for all samples investigated in this study. The normalized number refers to the number of RESs per one million reads. (b) The number of regular A-to-I RESs versus the number of hyper A-to-I RESs for all genes in human testis (25 weeks). PCC refers to Pearson Correlation Coefficient. R (version 3.2.2) is used to calculate PCC and p-value with the command options of ‘cor.test (x, y, alternative=’greater’)’. (c) The normalized number of A-to-I RESs (the union of regular and hyper A-to-I RESs) in different tissues of the four species. ‘wks’ refers to weeks
3.5 RNA editing in embryonic mouse central nervous system
We further apply SPRINT to the embryonic and adult mouse dataset that includes the RNA-seq data from CnsE11half (11.5 days, Cns refers to central nervous system), CnsE14, CnsE18, CbellumAdult (adult cerebellum), FlobeAdult (adult frontal lobe), LiverE14, LiverE18 and LiverAdult (adult liver) from the Mouse ENCODE project [all strand-specific, poly (A)+] (Supplementary Table S1 and Section 2). The A-to-G rate of both regular and hyper RESs is 97% in adult cerebellum and frontal lobe, and is above 90% in adult liver. It is only above 80% in embryonic samples, which is because the number of RESs found in these samples is much smaller than that in adult cerebellum and frontal lobe (around 6–12 times smaller). When the number of A-to-I RESs increases during the development of embryonic CNS (Fig. 5a), the A-to-G rate also increases. Different from embryonic CNS, the normalized number of A-to-I RESs in embryonic liver even slightly decreases during the development (Fig. 5a), though adult liver still has about two times of A-to-I RESs to that in embryonic liver. For C-to-U RESs, the normalized number does not change much during the development of either embryonic CNS or liver, and is in similar range in between embryonic and adult tissues (Supplementary Table S4). Consistent with the increase of the number of A-to-I RESs, the expression level of three ADARs (Adar, Adarb1 and Adarb2) all significantly increase during the development of embryonic CNS (Supplementary Table S5). In comparison, Adarb2 is not expressed during the development of embryonic liver, and even though the expression level of both Adar and Adarb1 increases, it’s much lower in embryonic liver than in embryonic CNS.
Fig. 5.
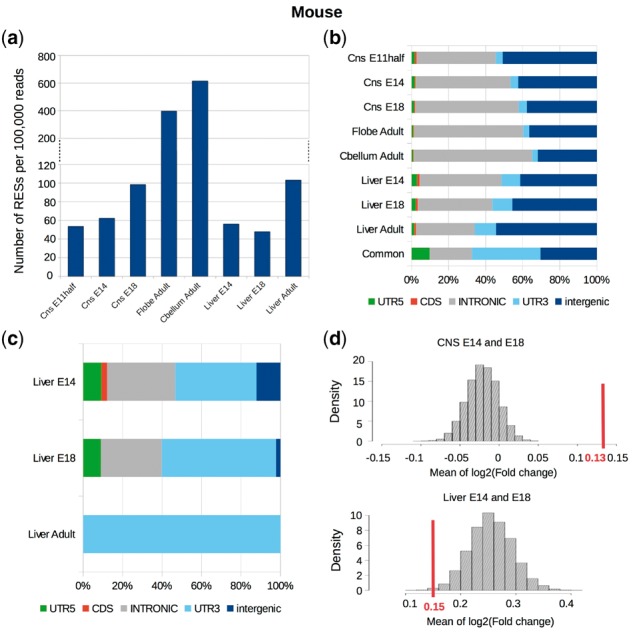
RNA editing in embryonic and adult mouse tissues. (a) The normalized number and (b) the proportions of different categories of A-to-I RESs, and (c) the proportions of different categories of C-to-U RESs in mouse embryonic and adult tissues. (d) The mean expression level change of newly edited genes during the development of embryonic CNS (upper) and liver (lower). The line segment represents the mean expression level changes [log2 (fold change)] of newly edited genes (1822 and 1139 genes in CNS and liver, respectively), while the null distributions are plotted by computing the mean expression level change of randomly selected genes (the same number as the newly edited genes) in CNS and liver (10 000 times of randomization)
Next, we investigate the distribution of RESs in the development of embryonic CNS and liver. We find the proportion of 3’-UTR A-to-I RESs is very different in between embryonic CNS and liver (3.8–4.4% for CNS, and 10.2–10.8% for liver), while it is similar in between similar tissues (Fig. 5b), suggesting it may be tissue-specific. In addition, A-to-I RESs that are common to all seven samples investigated here are significantly enriched in UTR, particularly in 3’ UTR (Fig. 5b). For C-to-U RESs, the proportion of 3’ UTR C-to-U RESs significantly increases during the development of embryonic liver, and almost all C-to-U RESs are in 3’ UTR in adult liver (Fig. 5c). The distributions of C-to-U RESs in embryonic CNS are not investigated because of very small number of C-to-U RES in these samples (Supplementary Table S4).
We identify genes that are under A-to-I editing only in a later development stage of embryonic CNS, and inspect their expression level change. We find that newly edited genes are often accompanied by the elevation of gene expression levels (p-value significant by permutation) (Fig. 5d). In contrast, this association is not observed in embryonic liver (Fig. 5d). We further identify those genes that are newly edited with at least 10 A-to-I RESs from CnsE11.5 to CnsE18, and find they are significantly enriched in functions of nervous system (e.g. GO: 0007268—synaptic transmission, GO: 0007611—learning and memory, etc.) (Supplementary Table S6), suggesting that RNA editing might play important roles in the development of CNS. For example, Grik2 whose expression level has a fold change of 5.5 has 51 new A-to-I RESs in CNSE18. It encodes a subunit of kainite glutamate receptors that are the predominant excitatory neurotransmitter receptors in mammalian brain, and play important roles in a variety of normal neurophysiologic processes (Lanore et al., 2012; Li et al., 2009). Another example is Grin2a that has 38 new A-to-I RESs and a fold-change of 4.7 in expression level in CNSE18. This gene encodes a subunit of N-methyl-D-aspartate (NMDA) receptors that are involved in long-term potentiation in synaptic transmission (Lal et al., 2015; Turner et al., 2015; Zhong et al., 2014). In comparison, those genes that are edited throughout the development of CNS are enriched with more general functions, such as GO: 0009057—macromolecule catabolic process, GO: 0007049—cell cycle, etc.) (Supplementary Table S7). These genes include Btrc that functioned in cell cycle checkpoints (Busino et al., 2003; Jin et al., 2003), and Mad2l2 that is required by pluripotent embryonic stem cells (Pirouz et al., 2015), etc. It is therefore likely that RNA editing may not only be of importance for CNS development, but also be involved in embryonic cell development.
4 Discussion
Current methods on detecting RESs typically require the use of SNP annotations to filter SNVs (Bahn et al., 2012; Ramaswami et al., 2012; Zhang and Xiao, 2015). For organisms that do not have SNP annotations available, matched DNA-seq data are currently needed for filtering out SNPs, which are costly and cannot be generalized. It is therefore highly desirable to have a method that detects RESs without the need to filter out SNPs. In this study, we have developed a novel method named SPRINT that identifies RESs by clustering SNV duplets, bypassing the need of SNP annotations. The clustering approach not only distinguishes RESs from SNPs, but also effectively removes the one-read-count SNVs that are likely system errors, allowing the utilization of all SNVs that significantly increases the number of called RESs. The use of clustering is nothing new. For example, RNAEditor implemented clustering only after SNPs have been filtered out and an initial set of RESs have been identified, making it still rely on the use of SNP annotations. However, the introduction of SNV duplets is novel, and is based on the fact that SNV duplets of SNPs and SNV duplets of RESs have very different distributions that was not discovered before. This is the foundation of SPRINT, and is the major reason why SPRINT is a novel method. This approach has also been applied for detecting hyper-RESs, making SPRINT a comprehensive tool for analyzing RNA editing. The quality of RESs called by SPRINT is well demonstrated by their high A-to-G rate in almost all samples analyzed in this study. In addition, SPRINT’s effectiveness in detecting A-to-I and C-to-U RESs has been validated by the significant reduction in the number of RESs called from samples where genes encoding the respective editing enzymes are knockdown. Besides the SNP-free advantage over existing methods, SPRINT is also able to identify significantly more number of RESs than existing methods do (Porath et al., 2014; Ramaswami et al., 2012; Zhang and Xiao, 2015; Zhao et al., 2015), and appears to have lower FDR than the other methods by using U87MG as the benchmark dataset. Finally, SPRINT has been fully automated to be applicable to any RNA-seq data that have reference genome sequences available. As such, SPRINT should be of great use for accelerating the study of RNA editing. A website of SPRINT (http://sprint.tianlab.cn/) has been constructed to store the RESs detected by SPRINT in this study.
We have applied SPRINT to investigate RNA editing in four tissues from human, chimp, rhesus and mouse, and also in mouse embryonic and adult tissues. On the one hand, these applications have demonstrated that SPRINT is applicable to any RNA-seq data without the need of SNP annotations. On the other hand, besides confirming previous reports on RESs’ distributions, sequence and conservation patterns in more conditions and more species (Bahn et al., 2012; Blanc et al., 2014; Porath et al., 2014; Ramaswami et al., 2012; Zhang and Xiao, 2015), we also obtain a number of novel findings about RNA editing. First of all, we provide more lines of evidence that 3’ UTR A-to-I RESs are likely of functional significance, particularly its significant enrichment among the common RESs in all four species. Secondly, we find the numbers of regular A-to-I RESs and hyper A-to-I RESs are almost linearly positively correlated with each other at both sample and gene level, suggesting that they may not have mechanistic difference. Thirdly, we find that in human a significant proportion of C-to-U RESs are in CDS, with many located in mitochondria. In addition, C-to-U RESs are located in more conserved regions than those nucleotides further way from the RESs, suggesting that C-to-U RESs may play significant functional roles. Fourthly, we find in all four species that brain is under more extensive RNA editing than the other three tissues are, especially for mouse. Finally, we find that the number of A-to-I RESs significantly increases during the development of mouse embryonic CNS, while the newly edited genes in a later development stage are not only coupled with the increase in expression level, but also are significantly enriched in functions involved in the development of CNS, suggesting that RNA editing plays an important role in the development of CNS.
In SPRINT, we use BWA (Li and Durbin, 2009a,b; Li and Homer, 2010) for reads mapping. It has been noted that different mapping strategy has an effect on RNA editing detection (Picardi and Pesole, 2013; Ramaswami et al., 2012). The use of splice-aware aligners such as Tophat2 (Kim et al., 2013), may further improve SPRINT's performance. However, this may require significant modifications on current workflow of SPRINT, as we will need to test the efficacy of different splice-aware aligners and explore how to merge (cluster) SNV duplets in a single transcript. Nevertheless, as SPRINT is SNP-free, fully automated, excellent in detecting RNA editing and applicable to a broad range of organisms, it is already of great use for assisting in in understanding the mechanisms and functional roles of RNA editing. The addition of splice-aware aligners in SPRINT and the extension to RNA-seq data without available reference genome sequence will be explored in the future development of SPRINT.
Funding
This work was supported by the National Natural Science Foundation of China [31671367, 31471245]; the Shanghai Municipal Commission of Science and Technology Program [14DJ1400100]; the National Key Research and Development Program of China [2016YFC1000505]; the Specialized Research Fund for the Doctoral Program of Higher Education of China [20120071110018]; the Innovation Program of Shanghai Municipal Education Commission [13ZZ006]; Shanghai Shuguang Program [13SG05].
Conflict of Interest: none declared.
Supplementary Material
References
- Bahn J.H. et al. (2012) Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res., 22, 142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benne R. (1996) RNA editing. The long and the short of it. Nature, 380, 391–392. [DOI] [PubMed] [Google Scholar]
- Blanc V., Davidson N.O. (2003) C-to-U RNA editing: mechanisms leading to genetic diversity. J. Biol. Chem., 278, 1395–1398. [DOI] [PubMed] [Google Scholar]
- Blanc V. et al. (2014) Genome-wide identification and functional analysis of Apobec-1-mediated C-to-U RNA editing in mouse small intestine and liver. Genome Biol., 15, R79.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchert G.M. et al. (2009) Adenosine deamination in human transcripts generates novel microRNA binding sites. Hum. Mol. Genet., 18, 4801–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busino L. et al. (2003) Degradation of Cdc25A by beta-TrCP during S phase and in response to DNA damage. Nature, 426, 87–91. [DOI] [PubMed] [Google Scholar]
- Clark M.J. et al. (2010) U87MG decoded: the genomic sequence of a cytogenetically aberrant human cancer cell line. PLoS Genet., 6, e1000832.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks G.E. et al. (2004) WebLogo: a sequence logo generator. Genome Res., 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S. et al. (2012) Landscape of transcription in human cells. Nature, 489, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farajollahi S., Maas S. (2010) Molecular diversity through RNA editing: a balancing act. Trends Genet., 26, 221–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli D. et al. (2015) Principles governing A-to-I RNA editing in the breast cancer transcriptome. Cell Rep., 13, 277–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaisler-Salomon I. et al. (2014) Hippocampus-specific deficiency in RNA editing of GluA2 in Alzheimer's disease. Neurobiol. Aging, 35, 1785–1791. [DOI] [PubMed] [Google Scholar]
- Grice L.F., Degnan B.M. (2015) The origin of the ADAR gene family and animal RNA editing. BMC Evol. Biol., 15, 4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L. et al. (2015) The genomic landscape and clinical relevance of A-to-I RNA editing in human cancers. Cancer Cell, 28, 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hideyama T. et al. (2012) Profound downregulation of the RNA editing enzyme ADAR2 in ALS spinal motor neurons. Neurobiol. Dis., 45, 1121–1128. [DOI] [PubMed] [Google Scholar]
- Huang da W. et al. (2009a) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res., 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W. et al. (2009b) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc., 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Jin J. et al. (2003) SCFbeta-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev., 17, 3062–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John D. et al. (2016) RNAEditor: easy detection of RNA editing events and the introduction of editing islands. Brief. Bioinf [Epub ahead of print]. doi: 10.1093/bib/bbw087. [DOI] [PubMed] [Google Scholar]
- Kim D. et al. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol., 14, R36.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal D. et al. (2015) Investigation of GRIN2A in common epilepsy phenotypes. Epilepsy Res., 115, 95–99. [DOI] [PubMed] [Google Scholar]
- Lanore F. et al. (2012) Deficits in morphofunctional maturation of hippocampal mossy fiber synapses in a mouse model of intellectual disability. J. Neurosci. Off. J. Soc. Neurosci., 32, 17882–17893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B. et al. (2009) Down-regulation of GluK2 kainate receptor expression by chronic treatment with mood-stabilizing anti-convulsants or lithium in cultured astrocytes and brain, but not in neurons. Neuropharmacology, 57, 375–385. [DOI] [PubMed] [Google Scholar]
- Li H., Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Homer N. (2010) A survey of sequence alignment algorithms for next-generation sequencing. Brief. Bioinf., 11, 473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. et al. (2011) Widespread RNA and DNA sequence differences in the human transcriptome. Science, 333, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maydanovych O., Beal P.A. (2006) Breaking the central dogma by RNA editing. Chem. Rev., 106, 3397–3411. [DOI] [PubMed] [Google Scholar]
- Nishikura K. (2010) Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem., 79, 321–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Yaacov N. et al. (2015) Elevated RNA editing activity is a major contributor to transcriptomic diversity in tumors. Cell Rep., 13, 267–276. [DOI] [PubMed] [Google Scholar]
- Peng Z. et al. (2012) Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat. Biotechnol., 30, 253–260. [DOI] [PubMed] [Google Scholar]
- Picardi E. et al. (2015) Profiling RNA editing in human tissues: towards the inosinome Atlas. Sci. Rep., 5, 14941.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picardi E., Pesole G. (2013) REDItools: high-throughput RNA editing detection made easy. Bioinformatics, 29, 1813–1814. [DOI] [PubMed] [Google Scholar]
- Piechotta M. et al. (2017) JACUSA: site-specific identification of RNA editing events from replicate sequencing data. BMC Bioinformatics, 18, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirouz M. et al. (2015) Destabilization of pluripotency in the absence of Mad2l2. Cell Cycle, 14, 1596–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard K.S. et al. (2010) Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res., 20, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porath H.T. et al. (2014) A genome-wide map of hyper-edited RNA reveals numerous new sites. Nat. Commun., 5, 4726.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G. et al. (2012) Accurate identification of human Alu and non-Alu RNA editing sites. Nat. Methods, 9, 579–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G. et al. (2013) Identifying RNA editing sites using RNA sequencing data alone. Nat. Methods, 10, 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg B.R. et al. (2011) Transcriptome-wide sequencing reveals numerous APOBEC1 mRNA-editing targets in transcript 3' UTRs. Nat. Struct. Mol. Biol., 18, 230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueter S.M. et al. (1999) Regulation of alternative splicing by RNA editing. Nature, 399, 75–80. [DOI] [PubMed] [Google Scholar]
- Ruiz-Orera J. et al. (2015) Origins of de novo genes in human and chimpanzee. PLoS Genet., 11, e1005721.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikanai T. (2015) RNA editing in plants: machinery and flexibility of site recognition. Biochim. Biophys. Acta, 1847, 779–785. [DOI] [PubMed] [Google Scholar]
- Slotkin W., Nishikura K. (2013) Adenosine-to-inosine RNA editing and human disease. Genome Med., 5, 105.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y. et al. (2016) RED: a Java-MySQL software for identifying and visualizing RNA editing sites using rule-based and statistical filters. PloS One, 11, e0150465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tempel S. (2012) Using and understanding RepeatMasker. Methods Mol. Biol., 859, 29–51. [DOI] [PubMed] [Google Scholar]
- Trapnell C. et al. (2012) Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc., 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner S.J. et al. (2015) GRIN2A: an aptly named gene for speech dysfunction. Neurology, 84, 586–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlstedt H. et al. (2009) Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res., 19, 978–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. et al. (2016) RES-Scanner: a software package for genome-wide identification of RNA-editing sites. GigaScience, 5, 37.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Xiao X. (2015) Genome sequence-independent identification of RNA editing sites. Nat. Methods, 12, 347–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H.Q. et al. (2015) Profiling the RNA editomes of wild-type C.elegans and ADAR mutants. Genome Res., 25, 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H.J. et al. (2014) Functional polymorphisms of the glutamate receptor N-methyl D-aspartate 2A gene are associated with heroin addiction. Genet. Mol. Res. GMR, 13, 8714–8721. [DOI] [PubMed] [Google Scholar]
- Zimin A.V. et al. (2014) A new rhesus macaque assembly and annotation for next-generation sequencing analyses. Biol. Direct, 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipeto M.A. et al. (2015) RNA rewriting, recoding, and rewiring in human disease. Trends Mol. Med., 21, 549–559. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.