

Abstract
Motivation
Moonlighting proteins (MPs) are an important class of proteins that perform more than one independent cellular function. MPs are gaining more attention in recent years as they are found to play important roles in various systems including disease developments. MPs also have a significant impact in computational function prediction and annotation in databases. Currently MPs are not labeled as such in biological databases even in cases where multiple distinct functions are known for the proteins. In this work, we propose a novel method named DextMP, which predicts whether a protein is a MP or not based on its textual features extracted from scientific literature and the UniProt database.
Results
DextMP extracts three categories of textual information for a protein: titles, abstracts from literature, and function description in UniProt. Three language models were applied and compared: a state-of-the-art deep unsupervised learning algorithm along with two other language models of different types, Term Frequency-Inverse Document Frequency in the bag-of-words and Latent Dirichlet Allocation in the topic modeling category. Cross-validation results on a dataset of known MPs and non-MPs showed that DextMP successfully predicted MPs with over 91% accuracy with significant improvement over existing MP prediction methods. Lastly, we ran DextMP with the best performing language models and text-based feature combinations on three genomes, human, yeast and Xenopus laevis, and found that about 2.5–35% of the proteomes are potential MPs.
Availability and Implementation
Code available at http://kiharalab.org/DextMP.
1 Introduction
Investigation of function of proteins is a central problem in bioinformatics as it is an essential step for unfolding obscurities of cellular processes. Although a majority of proteins are speculated to perform a single function, over the past decade a significant number of multifunctional, or more popularly called ‘moonlighting’ proteins are emerging into attention in the biology community (Campbell and Scanes, 1995; Jeffery, 1999; Weaver, 1998). Moonlighting proteins (MPs) are defined as proteins that perform multiple independent cellular functions within a single polypeptide chain. Functional diversity of these proteins are not due to gene fusions, multiple domains in the same protein chain, multiple RNA splice variants or proteolytic fragments, families of homologous proteins or pleotropic effects (Huberts and Vander Klei, 2010; Jeffery, 1999, 2004; Mani et al., 2014). Most prominent examples of MPs are enzymes (Jeffery, 1999, 2004). The first of such findings was in late 1980s, crystallins (Piatigorsky and Wistow, 1989; Wistow and Kim, 1991), which are structural eye lens proteins that also have enzyme function. Since then, MPs are continued to be found in a wide variety of genomes with diverse cellular functions and molecular mechanisms for switching functions.
In parallel to serendipitous findings of MPs through experiments, bioinformatics approaches have been applied to characterize MPs in recent years (Khan and Kihara, 2014). Existing studies investigated different aspects of MPs such as sequence similarity (Gomez et al., 2003; Khan et al., 2012), conserved motifs/domains, structural disorder (Hernández et al., 2011), and protein-protein interaction (PPI) patterns (Chapple et al., 2015; Gómez et al., 2011; Pritykin et al., 2015). We have recently developed a computational prediction method named MPFit, which predicts MPs and non-MPs using a diverse set of proteomics data (Khan and Kihara, 2016). Development of MPFit was based on our previous study where we presented a systematic characterizations of MPs in a computational framework (Khan et al., 2014). However, all these existing studies overlook a major resource of information of protein function, i.e. text-based information that underlies in scientific literature and textual description of protein annotation in databases such as UniProt (UniProt Consortium, 2014). In most cases MPs are not explicitly labelled in the database with ‘moonlighting’, ‘dual function’, ‘multitasking’, or other related words, even in cases where two distinct functions are known and clearly stated in its database entry. To accommodate the current limited knowledge of MPs, two online repositories of MPs (Hernández et al., 2014; Mani et al., 2014) were built on expert knowledge with manual curation from literature. This situation convinced us that application of text mining techniques on MP literature would provide a major boost towards automatic MP annotation. In this work we propose a first text mining-based approach for predicting MPs, named DextMP (Deep dive into tEXT for predicting Moonlighting Proteins).
For the last decade, text mining techniques has been extensively developed to unravel non-trivial knowledge from structured/unstructured text data (Manning et al., 2008). Most of the existing works are based on bag-of-words that leverages word-related statistics in the text (Joachims, 1998). The next generation of text-based feature learning models represent each text with a distribution of latent topics (Hoffman et al., 2010). In recent years, unsupervised deep learning-based feature construction has become popular in text mining (Mikolov et al., 2013). Such deep-learning-based methods map text into a condensed d-dimensional continuous vector space such that semantically similar texts are embedded nearby each other.
DextMP consists of four logical steps: first, it extracts textual information of proteins from literature (publication titles or abstracts) and functional description in UniProt. Next, it constructs a k-dimensional feature vector from each text. In this step, a state-of-the-art deep unsupervised learning algorithm is applied, which is called paragraph vector (Le and Mikolov, 2014), along with two other widely used language models, Term Frequency-Inverse Document Frequency (TFIDF) in the bag-of-words category (Manning et al., 2008) and Latent Dirichlet Allocation (LDA) in the topic modeling category (Hoffman et al., 2010). Third, using four machine learning classifiers, a text is classified to MP or to non-MP based on the text features. Finally, prediction made to each literature for a protein is summarized to make a prediction to the protein. Cross-validation results on the dataset of known MPs and non-MPs (control dataset) show that DextMP can successfully predict MPs with over 91% accuracy, with a significant improvement over existing MP prediction methods. Among the different forms of text information, abstracts taken from literature and function description in UniProt showed better performance than the title of literature. Lastly, we ran DextMP with the best performing language models and text-based feature combinations on three genomes, Saccharomyces cerevisiae (yeast), Homo sapiens (human) and Xenopus laevis (African clawed frog), and found that about 2.5–35% of the proteomes are potential MPs.
2 Materials and methods
We first explain text data and features used, then describe learning models of DextMP.
2.1 Dataset of MPs and non-MPs
The dataset of MPs and non-MPs (i.e. negative example of moonlighting proteins) were taken from our previous work (Khan and Kihara, 2016). The dataset contains 263 MPs selected from a manually curated MP database, MoonProt (Mani et al., 2014). Proteins that do not have a UniProt ID were discarded. In addition, five MPs were discarded because they have over 25% sequence identity to other proteins in the dataset. Non-MPs were selected using the following Gene Ontology (GO) function annotation-based criteria developed in our previous works (Khan et al., 2014; Khan and Kihara, 2016). From the four most dominant genomes in the MP dataset, namely, human (45 MP, 17.1%), E.coli (29 MPs, 11%), yeast (23 MPs, 8.7%) and mouse (11 MPs, 4.2%), a protein was selected as a non-MP if a) it has at least eight GO term annotations, b) when GO terms in the Biological Process (BP) category were clustered using the semantic similarity score (Schlicker et al., 2006) no more than one cluster was obtained at either the 0.1 threshold or the 0.5 threshold, and c) no more than one cluster of Molecular Function (MF) GO terms at semantic similarity scores of 0.1 and 0.5 were formed. In essence, a protein is considered as a non-MP if it has a sufficient number of GO annotations but they are not functionally diverse. We further ruled out non-MPs that had above 25% sequence identity with another non-MP sequences, and finally selected 162 non-MPs, among which 60 are from human (37.0%), 52 from mouse (32.1%), 34 from yeast (20.9%) and 16 from E.coli (9.88%). In summary, 263 MP and 162 non-MP were selected as the control dataset for the DextMP model.
2.2 Text extraction
For each of the proteins in the control dataset, we extracted three categories of text information from UniProt: a) the title of each reference paper of the protein entry; b) the abstract of each reference; and c) the summary description of the protein’s function in the FUNCTION field in UniProt. The text data for a) and c) were directly collected from the UniProt data dump (http://www.uniprot.org/downloads), and b) was collected by crawling web links in the PUBLICATION list of the entry. Table 1 shows the statistics of the data size. Note that while one protein can have multiple titles and abstracts associated with it, it only has one function description. For 49 MPs, no publication title was found, whereas 105 MPs did not have a hyperlink directed to a publication abstract (Table 1). Figure 1 shows the distribution of the number of abstracts per MP and non-MP in the dataset.
Table 1.
Data size for the MP/non-MP dataset
| #Proteins | #Titlesa | #Abstractsa | #Functions | |
|---|---|---|---|---|
| MP | 263 | 2496 (214) | 1450 (158) | 194 |
| non-MP | 162 | 1665 (162) | 1624 (162) | 162 |
In the parenthesis the number of proteins is shown for which the text data was found. For example, out of 263 MPs, at least one publication title was found for 214 MPs.
Fig. 1.

Distribution of the number of abstracts per protein. Black, MP; gray, non-MP in the control dataset. The first bar is for 1 and 2 abstracts, next bar is for 3 and 4 and so on
Obtained text data underwent three layers of data clean-up. First, redundant literature that appears both in MPs and non-MPs were discarded (this typically happens for papers that describe many proteins, e.g. genome annotation). Second, from each text data, all stop words, punctuations, and special symbols including Greek letters were removed. Finally, stemming and lemmatization were performed (Manning et al., 2008) using the nltk package (Bird, 2006).
As an example we briefly describe text data for a MP, phosphoglucose isomerase (PGI) in mouse (UniProt ID: P06745). It primarily acts as an enzyme in the second step of glycolysis. This protein moonlights by acting as a cytokine/growth factor, and causes pre-B cells to mature into antibody secreting cells, supports survival of embryonal neurons, and causes differentiation of some leukemia cell lines. Among 14 references for this protein, one is entitled ‘tumor cell autocrine motility factor is the neuroleukin/phosphohexose isomerase polypeptide’, which implies that this protein is an MP. It becomes apparent if one reads the abstract of this paper (http://www.uniprot.org/citations/8674049) or the function description of the entry, which says ‘besides its role as a glycolytic enzyme, mammalian PGI can function as a tumor-secreted cytokine and an angiogenic factor (AMF) that stimulates endothelial cell motility. PGI is also a neurotrophic factor (Neuroleukin) for spinal and sensory neurons’. Despite this clear knowledge of moonlightness of this protein, the UniProt entry does not use any exact keyword, e.g. moonlighting proteins, multifunction, etc., which clearly indicates that this is an MP.
2.3 Framework of DextMP
The overall framework of DextMP is shown in Figure 2. It is split in two parts, MP/non-MP prediction to a text (the text prediction model, the top panel in Fig. 2) and prediction made to a query protein by combining prediction made for each text of the protein (the bottom panel).
Fig. 2.
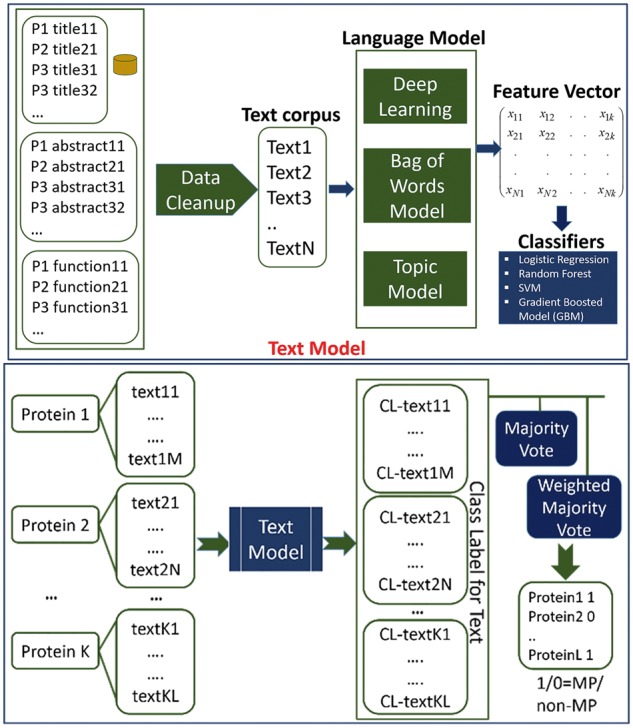
Schematic diagram of DextMP. The upper panel shows the text prediction process while the bottom panel is for the prediction model that uses predicted text labels to make the final MP/non-MP classification. P1, Protein 1, CL: Class Label
In the text prediction model, first, three different types of text information, titles, abstracts and function descriptions, are extracted. Then the data clean-up step is carried out. Next, the texts in the dataset are applied to a deep unsupervised feature construction technique (Le and Mikolov, 2014), a bag of words model (Manning et al., 2008), and topic modeling (Hoffman et al., 2010) to construct text features. Finally, machine learning classification algorithms, namely, logistic regression (LR), random forest (RF), Support Vector Machine (SVM) and gradient boosted machine (GBM) (Pedregosa et al., 2011) are applied to the learned features to provide a MP/non-MP prediction on each text data.
Once we have a MP/non-MP class prediction for each text, we use the model shown in bottom panel of Figure 2 to obtain a class prediction for proteins (protein-level prediction). Each protein is associated with a certain number of texts (titles/abstracts) that have predicted class labels. To make the final MP/non-MP classification, two heuristics were applied: (i) A majority vote, where we simply take the binary class label votes for the protein using different majority cutoffs, 50%, 70%, 80% and 90%. (ii) A weighted majority vote, where a weight for a text is from the class prediction probability from the text level prediction. The weighted majority vote was applied to three classifiers, LR, RF and GBM. This latter part of DextMP is not applied when function description of proteins was used, since there is only one description for a protein and voting is not needed.
2.4 Learning features from text
Here we explain the three language models used for feature construction from text (Fig. 2, top panel).
1. Bag-of-words with TFIDF: Given a text corpus (collection of sentences/texts), the bag-of-words model first computes the dictionary that contains all the words in the text corpus. Given a dictionary of size N, a text can be represented as an N-dimensional real-valued vector with TFIDF values for each word in the dictionary. For a word w, TFIDF is be computed as follows: TFIDF(w) = TF(w) * IDF(w), where Term Frequency, TF(w) = (number of times word w appears in a text)/(total number of words in the text); and Inverse Document Frequency, IDF(w) = loge(total number of texts in the corpus/number of texts with word w); Intuitively, TFIDF measures the importance of a keyword to a sentence with respect to its entire dictionary corpus.
2. Topic Modeling with LDA: In principle, the bag-of-words model has two critical limitations: for a large dictionary, the size of the feature vector for each text can be huge, which makes it computationally expensive, and it does not take consideration of the word ordering in a text. To alleviate above two challenges, in topic modeling a text is modeled as a distribution of words for latent topics, where the number of topics is a user-defined parameter. Latent Dirichlet Allocation (LDA) is one of the most popular topic modeling algorithms, which uses two Dirichlet-multinomial distributions to model the mappings between documents and topics, and topics and words. We used an open source Python implementation of LDA (Rurek and Sojka, 2010).
3. Unsupervised Deep Language Model, DEEP and PDEEP: As the third language model, we used a deep learning-based unsupervised feature construction algorithm (Le and Mikolov, 2014). This model maps texts into a continuous vector space of a dimension d, such that semantically similar texts appear close in the space. For a sequence of words W = (w0, w1, …, wn), where wi ∈ D (D is the dictionary), suppose wi is an input word and rest of the words in the dictionary form the context wc = (w0, w1, . .wi-1, wi+1, . wn) . A neural network with a single hidden layer is trained so that for a vector of D dimension with 1 for wi and 0 otherwise, the network outputs the conditional probability that the other words co-appear in the neighborhood of wi in a text. Using the conditional probabilities that each word co-appears with wi in the training dataset of texts as known outputs, weights of hidden layers are trained with back-propagation, and the weights of hidden layers are considered as a feature vector of wi.
Mikolov et al. extended the above model by modifying the probability expression to Pr[W|T]. Here, T is the text containing the sequence of words, and can be thought as another word. Similar to the above model along with the task of maximizing the conditional probability, it outputs d-dimensional feature vector representation of each text, T. We used an open source Python implementation of the ‘paragraph vector’ deep learning model (Rurek and Sojka, 2010).
Using the deep learning method, we computed two models, DEEP and PDEEP (Pre-trained deep learning model). For DEEP, features were constructed on texts from the control dataset of MP and non-MP only. For PDEEP, we used the entire text data from UniProt. Concretely, we extracted a total of 1 060 520 titles and 551 056 function descriptions from the UniProt data dump. Since publication abstracts are not available in the data dump, we omitted PDEEP training for abstracts.
2.5 Parameter tuning of DextMP
We used a grid search to determine hyper-parameters for LDA and DEEP. In LDA, the ‘number of topics’ parameter was tuned by a grid search performed between 10 and 100 with a step size of 10 for each text type. In DEEP, we tuned three hyper-parameters: the ‘minimum count’ parameter was tuned within a range of 1–5, ‘window size’ was tuned within 2–8, both with a step size of 1, and ‘dimension size’ was tuned in a range from 20 to 200 with an increasing step size of 20. The parameter ‘minimum count’ indicates the minimum number of texts that the word must appear in, ‘window size’ is the size of the convolution context, and ‘dimension size’ indicates the length of the feature vector representation. For PDEEP, we used the same parameters as DEEP.
The hyper-parameters associated with the four classifiers of DextMP were also determined using a grid search. For LR and SVM we tuned the regularization, a cost parameter and a kernel function (linear or radial basis function), and used the default values for the other parameters in the models in the sklearn package (Pedregosa et al., 2011). For RF and GBM, we tuned the ‘number of trees’ parameter, the ‘learning rate’ parameter for GBM, and used the default for the others.
We performed a five-fold cross-validation. The dataset was split into five sub-groups, among which three sub-groups were used for training, another sub-group was used for validation, and the last sub-group was used for testing. Given a vector of hyper-parameters for a combination of a model and a classifier, where a model is either LDA/DEEP and a classifier be LR/RF/SVM/GBM, we performed a grid search for the optimal hyper-parameters over the training set, used the validation set to find the best hyper-parameter vector, and ran the optimized model with the hyper-parameter values for the test set to report results. For example, when LDA and LR combination was to be trained, for each of all the combinations of the ‘number of topics’ parameter for LDA and the regularization parameter for LR, model parameters were optimized on a training set that consists of three sub-groups. Once the model was optimized for each of the all hyper-parameter combinations on the training set, the optimized models were tested on the validation set to determine the best hyper-parameter combination and the model optimized under the hyper-parameters. Then, the selected model was tested on the testing set to report the F-score for that parameter setting. Each sub-group was used once for testing. We performed the above procedure for five test sub-groups independently, and finally reported the average F-score computed for the 5 test sub-groups.
3 Results
3.1 Text features of MPs
To begin with, we browsed abundant words in texts of MPs in the control dataset. In Figure 3, word clouds of the three categories of texts, publication titles, function descriptions and abstracts, are shown.
Fig. 3.

Word clouds of text information of moonlighting protein dataset. The size of a word in the visualization is proportional to the number of times the word appears in the input text. (A–C): titles, function descriptions and abstracts, respectively. The images were generated at http://www.wordle.net/
From the word clouds, a few points came to light: the words ‘enzyme’, ‘kinase’ and ‘transcription’ appear in all three text types in Figure 3 (red circles). Word counts of ‘enzyme’ in titles, abstracts, UniProt function descriptions are 108/34, 562/107, 89/26, respectively for MP/non-MP. Counts for ‘kinase’ and ‘transcription’ were (102/16, 210/105, 44/9) and (87/59, 431/331, 75/34), respectively, for (titles, abstracts, UniProt function descriptions) of MP/non-MP. This is consistent with previous reports that many MPs were known primarily as enzymes when their secondary function, such as transcription factor, was discovered (Hernández et al., 2014; Jeffery, 2003; Khan and Kihara, 2016; Mani et al., 2014). The word ‘ribosome’, which appeared as the top word in Figure 3A (green circles), also agrees with our previous finding (Khan and Kihara, 2016) that predicted MPs were enriched in ribosomal pathways in the KEGG database (Kanehisa and Goto, 2000), and found in literature (Wool, 1996). Additionally, words that are clear indicators of MPs also appeared, such as ‘bifunctional’ (counts were 21/0, 29/5, 6/0 for MP/non-MP in titles, abstracts, function descriptions, respectively; blue circle in Fig. 3A) and ‘multifunctional’ (12/0, 19/4, 4/0 for MP/non-MP in titles, abstracts, function descriptions, respectively).
3.2 DextMP performance on text level prediction
We now show prediction results of the text-level MP prediction by DextMP on the control dataset (Table 2). A schematic diagram of this part of the DextMP model is described in the top panel of Figure 2 and explained in Section 2.2. Along with the two different deep learning based models (DEEP and PDEEP), we used two other methods in popular language model categories, TFIDF in the ‘bag-of-words’ category and LDA in the ‘topic modelling’ category. For each language model, three forms of text information, titles, abstracts and UniProt function descriptions, were used and compared. Note that the abstracts-PDEEP combination was omitted, as it requires all publication abstracts for the entire protein corpus in UniProt for model training. Since UniProt does not maintain any file dump for publication abstracts, it requires running a web-crawler and downloading abstract texts for all proteins in UniProt, which became computationally very expensive. For learned features by each language model, we further used four classifiers, LR, RF, SVM and GBM to make MP/non-MP classification (shown in right columns of Table 2).
Table 2.
Summary of the text-level prediction with different combinations of text types, language models and classifiers
| Text Type | Language Model | Classifiers |
|||
|---|---|---|---|---|---|
| LR | RF | SVM | GBM | ||
| Titles | TFIDF | 0.7774 | 0.7942 | 0.8751 | 0.7218 |
| LDA | 0.6128 | 0.6829 | 0.6584 | 0.7065 | |
| DEEP | 0.7696 | 0.7402 | 0.8429 | 0.8029 | |
| PDEEP | 0.6262 | 0.5482 | 0.4836 | 0.6445 | |
| Abstracts | TFIDF | 0.9220 | 0.8682 | 0.9371 | 0.8396 |
| LDA | 0.6419 | 0.6936 | 0.6512 | 0.7349 | |
| DEEP | 0.7775 | 0.8119 | 0.8480 | 0.7987 | |
| PDEEP | – | – | – | – | |
| Function Descriptions | TFIDF | 0.7412 | 0.7439 | 0.7715 | 0.6947 |
| LDA | 0.6128 | 0.6829 | 0.6582 | 0.7065 | |
| DEEP | 0.8929 | 0.8962 | 0.9184 | 0.8788 | |
| PDEEP | 0.7017 | 0.7211 | 0.3474 | 0.6917 | |
Two-class weighted F-score was reported, where F-score of MP and non-MP was calculated and weighted average of them was taken, where the weights are the number of data points of each class. The values shown are the average of the test sets in the Five-fold cross-validation. LR, Logistic Regression; RF, Random Forest; SVM, Support Vector Machine; GBM, Gradient Boosted Machine.
Among all the text-language_model-classifier combinations tested in Table 2, the highest F-score, 0.9371, was recorded by the combination of TFIDF and SVM when it was applied to literature abstracts (abstracts-TFIDF-SVM). The precision was 0.8920 and the recall was 0.8640. Besides this best combination, seven more combinations showed an F-score over 0.850. Comparing the three text types, abstracts had the highest F-score (0.9371), and UniProt function descriptions was second highest (0.9184), and using titles had the lowest (0.8751).This order was consistent when the average F-score across different model-classifier combinations for each text type was considered: the abstracts again showed the highest value of 0.8053 in comparison to the function descriptions (0.7138) and the titles (0.7141). We further counted which text type showed the highest F-score for combinations of language models (PDEEP was excluded) and classifiers, e.g. TFIDF-LR. Six combinations showed the highest F-score when applied to abstracts, four combinations with function descriptions, and two were best with literature titles. These analyses show that MP detection can be done better by using abstracts or UniProt function descriptions than simply using literature titles.
Next we compare four language models. In Table 2, for each text type, the best performing language model under four classifiers is highlighted in bold. Surprisingly, that the simple TFIDF worked well with titles and abstracts. This is likely because titles are simpler so that TFIDF can easily capture MP-specific words to make correct predictions. DEEP showed superior performance in the function description category, which implies that complex semantic inter-word relations in the function descriptions require a complex model to correctly identify characteristics of MPs and non-MPs. DEEP clearly outperforms LDA in all three text information categories and all four classifiers, while PDEEP had 3 (out of 8) wins over LDA. PDEEP was built as an extension from DEEP by enlarging its training set to the whole corpus in UniProt. This model showed a lower F-score consistently for both the title and the function description categories. The reason for the lower accuracy of PDEEP is maybe because the training data used for PDEEP is too large and somewhat generalized textual features unique for MPs.
Comparing the four classifiers, SVM showed the best result in six cases among the eleven different settings (rows in Table 2), GBM won for four cases and RF for one case. LR did not win a single setting.
We have also tested DextMP’s prediction accuracy when text summaries were used as input. A summary of each abstract and each UniProt function description was computed using a well-known algorithm, TextRank (Rada and Tarau, 2004) implemented in a general- purpose python text summarization package, sumy (https://pypi.python.org/pypi/sumy). On average a summary reduced word counts of an abstract from 178.3 to 152.7 and from 172.2 to 145.4 for a function description. Using computed summaries of abstracts, F-score for the TFIDF model with the LR, SVM, GBM and RF classifiers reduced to 0.7937, 0.8043, 0.7259 and 0.7692, respectively. We used the TFIDF model for this comparison because it performed best among the language models used on the original abstract texts (Table 2). For functional descriptions, using summaries also reduced F-score of the DEEP model with the four classifies to 0.8525, 0.8395, 0.7494 and 0.8210, respectively. DEEP was used here since it performed best for function descriptions. The reduction of the F-score was about 5 to 20% relative to when the original texts were used.
In terms of computational time, DEEP takes substantially more time in training relative to TFIDF and LDA, because the neural network needs to be trained (Table 3). However, since training can be pre-computed using a training dataset and reused later, in practice DEEP does not take much time when applied to predictions of new data relative to the time needed for training. In contrast, although TFIDF takes a short time for training, it takes a substantially longer time in prediction because the size of a feature vector becomes large as more unique words appear, which exceeded 16 000.
Table 3.
Computational time (seconds)
| Phase | Text Type | Language model |
||
|---|---|---|---|---|
| TFIDF | LDA | DEEP | ||
| Training | Titles | 5.8*10−5 | 1.0*10−3 | 4.4*10−1 |
| Abstracts | 3.3*10−4 | 2.9*10−3 | 9.0*10−1 | |
| Function Dsc. | 6.3*10−4 | 1.5*10−2 | 1.2 | |
| Feature generation | Titles | 7.8*10−4 | 3.3*10−4 | 1.8*10−4 |
| Abstracts | 3.2*10−3 | 6.6*10−4 | 2.4*10−4 | |
| Function Dsc. | 2.2*10−3 | 1.0*10−3 | 2.0*10−4 | |
| Classification | Titles | 5.1*10−2 | 3.8*10−3 | 9.2*10−3 |
| Abstracts | 1.2*10−1 | 4.0*10−3 | 1.3*10−2 | |
| Function Dsc. | 7.0*10−2 | 5.3*10−3 | 6.1*10−3 | |
Computational time is classified into three steps of the DextMP algorithm, training, feature generation and classification. Training is the time needed on average for processing a text to compute parameters of a language model. Feature generation is the time needed to compute a feature vector of a text to be classified. Classification is the average time that each classifier took to make a classification to a text.
3.3 DextMP performance on protein level prediction
Next, we discuss the performance of DextMP on the final protein-level MP/non-MP classification using predictions made to each text that belongs to proteins. This process is represented in the bottom panel in Figure 2. When UniProt function descriptions are used, a protein-level prediction is identical to the text-level prediction, because a query protein has only one UniProt description. When titles or abstracts of literature were used as text information, classification labels assigned to texts of a query protein were summarized using a simple majority vote or a weighted majority vote. As mentioned in Section 2.3, for a simple majority vote, four majority cutoffs, 50%, 70%, 80%, 90%, were tested in cross-validation for each combination of (text type)-(language model)-(classifier), and the cutoff that gave the largest F-score in the validation set was chosen and applied to the testing set. In Figure 4 we compared F-scores of protein level classification of the 21 (text type)-(language model)-(classifier) combinations using the simple majority votes and the weighted majority votes. Out of all 44 combinations in Table 2, the 21 combinations used in Figure 4 were LR, RF, and GBM classifiers applied to the title and abstract categories. The function descriptions category was excluded as a text type because it does not need voting. Among the 21 combinations, the simple majority votes showed a larger F-score for 15 cases than the counterpart, although margins are not very large. Therefore, we only show the results with the simple voting for the rest of this work.
Fig. 4.

Protein-level cross-validation F-scores for weighted and non-weighted majority votes. Results for 21 (text type)-(language model)-(classifier) combinations are compared
Table 4 summarizes F-scores of the protein-level MP prediction. The highest F-score was achieved when function descriptions were used by a combination of DEEP-SVM (0.9184). Note that values for function descriptions are identical to the text-level accuracy (Table 2) because one protein has only one description in UniProt. Comparing with the text-level prediction results in Table 2, a similar order of performance by different setting combinations was observed. However, a difference is that in almost all the cases the text-level accuracy was higher than the protein-level, which indicates the voting step in the protein-level prediction decreased the accuracy. Following the highest F-score combination of function descriptions-DEEP-SVM, the next three top combinations were all with function descriptions, which kept the same values as the text-level prediction, using the DEEP language model. Similar to what was observed in Table 2, TFIDF showed the best results with all the classifiers in the titles category, and also the best with three classifiers in the abstracts category, as highlighted in bold. Precision and recall values were well balanced for the F-score results in Table 4. For example, for the titles-TFIDF-SVM combination, which showed an F-score of 0.8330, precision and recall were 0.8316 and 0.8479, respectively.
Table 4.
Summary of the protein-level prediction
| Text Type | Language Model | Classifiers |
|||
|---|---|---|---|---|---|
| LR | RF | SVM | GBM | ||
| Titles | TFIDF | 0.7703 | 0.7474 | 0.8330 | 0.6901 |
| LDA | 0.5654 | 0.5723 | 0.5836 | 0.6227 | |
| DEEP | 0.6651 | 0.6698 | 0.7557 | 0.6826 | |
| PDEEP | 0.6611 | 0.5278 | 0.4314 | 0.6021 | |
| Abstracts | TFIDF | 0.8132 | 0.8225 | 0.8208 | 0.7833 |
| LDA | 0.5459 | 0.5739 | 0.5342 | 0.5713 | |
| DEEP | 0.7650 | 0.8105 | 0.7747 | 0.7909 | |
| PDEEP | – | – | – | – | |
| Function Descriptions | TFIDF | 0.7412 | 0.7439 | 0.7715 | 0.6947 |
| LDA | 0.6128 | 0.6829 | 0.6582 | 0.7065 | |
| DEEP | 0.8929 | 0.8962 | 0.9184 | 0.8788 | |
| PDEEP | 0.7017 | 0.7211 | 0.3474 | 0.6917 | |
F-score was reported. The values shown are the average of the test sets in the five-fold cross validation. LR, Logistic Regression; RF, Random Forest; SVM, Support Vector Machine; GBM, Gradient Boosted Machine. For each text type, titles, abstracts and function descriptions, the best performing language model under four classifiers is highlighted in bold.
For comparison, we ran a sequence-based function prediction method, PFP (Hawkins et al., 2006, 2009) on the control dataset and classified the proteins to MP/non-MP based on predicted GO terms. Following the GO term-based MP/non-MP classification performed in our previous study (Khan et al., 2014), a protein was classified into MP if more than one GO term in the BP category was predicted with moderate or higher confidence scores (a PFP raw score > 500), and if the GO terms were classified into more than two clusters using the relevance semantic similarity (SS_Rel) score (Schlicker et al., 2006) of 0.1 and more than four clusters at SS_Rel of 0.5. This protocol predicted 127/52 MPs/non-MPs correctly out of 263/162 MPs/non-MPs, resulting in an F-score of 0.4472. Thus, DextMP showed a higher accuracy than the function prediction-based results.
We think the prediction accuracy shown in Table 4 is sufficiently high for practical use, particularly for a large scale screening considering that an alternative for finding MPs from text is for someone to read texts one by one.
3.4 Genome-scale MP prediction using DextMP
Finally, we applied DextMP for predicting MPs in three genomes. Two genomes, S. cerevisiae (yeast) and H. sapiens (human), were chosen because MP prediction by MPFit (Khan and Kihara, 2016) were previously tested on them, so that we can compare DextMP with MPFit. One more genome, X.laevis, was chosen because omics-data, such as gene expression and protein-protein interaction data, are not available for this organism, and thus existing MP prediction methods (Chapple et al., 2015; Gómez et al., 2011), which rely on omics-data, cannot be used. Therefore DextMP can make unique contributions. Among the eleven settings we tested in Table 3, we used the top two models in the titles category and the top two in the function descriptions category from Table 4, i.e. titles-TFIDF-SVM, titles-TFIDF-LR, function_descriptions-DEEP-RF and function_descriptions-DEEP-SVM, and took the consensus of the four predictions. We did not use abstracts-based methods because abstracts were not directly available at UniProt and were not convenient for a large-scale prediction.
In our previous work, we developed an omics-data-based MP prediction method, MPFit (Khan and Kihara, 2016) and demonstrated that it outperformed two existing methods, one of which uses a target organism’s PPI network (Chapple et al., 2015) and another method that is based on GO term annotation of proteins (Pritykin et al., 2015). Therefore, in this section, we compare DextMP mainly with MPFit on the yeast and human genomes for which MPFit was applied.
Table 5 summarizes predictions to the three genomes. For the yeast genome, out of 6721 proteins, 6500 had both title and function description in UniProt so that DextMP can run on them (coverage 96.73%). Among these proteins, 2316 (34.46% of the entire proteins in the genome) were predicted as MP by DextMP when a consensus of three settings are considered, and 896 (13.33%) if a consensus of the all four settings was considered. In our previous work, MPFit predicted 10.97% of the yeast proteins are MPs, which is similar to the current prediction with the full (four) consensus. Since yeast has 27 known MPs in the MoonProt database (Mani et al., 2014), we computed recall based on them. Out of 27 known MPs, 24 and 20 are detected as MPs when a consensus of ≥3 and 4 settings are considered, which give recall value of 0.889 and 0.741, respectively. MPFit recorded a recall of 0.8146 (22 out of 27) in the previous work, which is between the two values in the current work. These two recall values are significantly higher than the GO term-based prediction by Pritykin et al. (2015), which was 0.4815. Besides the high recall value, DextMP also has a strong advantage of having a higher coverage than both MPFit and the method by Pritykin et al., because text information is in general more available than omics-data or GO annotations, which the two methods use as input. The coverage for DextMP was 96.73%, while MPFit and Pritykin et al. had a coverage of 69.56% and 68.69%, respectively.
Table 5.
Genome-scale prediction by DextMP
| Yeast | Human | X.laevis | |
|---|---|---|---|
| # Proteins | 6721 | 20 104 | 11 078 |
| Coverage | 96.73% | 98.06% | 30.54% |
| # MPs (%) (vote ≥ 3) | 2316 | 4781 | 600 |
| (34.46%) | (23.78%) | (5.42%) | |
| # MPs (%) (vote = 4) | 896 | 1682 | 279 |
| (13.33%) | (8.37%) | (2.51%) | |
| # known MPs | 23 | 45 | – |
| recall (vote ≥ 3) | 0.889 | 0.933 | – |
| recall (vote = 4) | 0.741 | 0.689 | – |
Coverage, the percentage of proteins in a genome that have both literature title and function descriptions, so that DextMP can run on them. Two prediction results are shown: the number of predicted MP proteins which are detected by three or more settings (vote ≥ 3) and the number of MPs detected by the all four settings unanimously (vote = 4). X.laevis does not have known MPs. The fraction in parentheses was computed for predicted MPs among all the proteins in the genome.
The human genome has a very high coverage of 98.06% (19 713 proteins out of 20, 104 proteins), which have text information and were subject to DextMP’s prediction. This is much higher than the coverage for both MPFit (67.91%), the GO-based method by Pritykin et al. (48.08%) and the PPI-based method (Chapple et al., 2015) (64.01%). Out of 45 known MPs in human, 42 were predicted correctly by DextMP (recall: 0.9333) when a consensus with three or more settings was considered. With the full consensus of the four settings, 31 MPs were correctly detected (recall: 0.689). These two recall values are higher than the two existing methods, the GO-based method (recall: 0.4889) and the PPI-based method (recall: 0.0667). Our previous method, MPFit, had a recall of 0.7333, which is between the two recall values recorded in the current work. DextMP predicted that 23.78% to 8.37% of human proteins are MPs with the two cutoffs of consensus voting. The lower value, 8.37%, is close to the MPFit’s prediction of 7.82% (Khan and Kihara, 2016).
As discussed above, a major advantage of DextMP is that it solely relies on text information of proteins, unlike the other methods that cannot be applied for proteins that lack experimental studies (e.g. PPI) or well-curated GO term annotations. Capitalizing on this aspect, we ran another genome, X.laevis with DextMP as it is not applicable for MPFit or the two other existing prediction methods because the genome lacks experimental studies. For X.laevis, out of 11 078 proteins 30.5% have literature information in UniProt. Due to the smaller number of proteins with literature information as compared with human and yeast, the fraction of predicted MPs in X.laevis, 2.51-5.42%, seems small, but the fraction against the 11 078 proteins with literature is in accordance with the results for yeast and human.
We now discuss three case studies where DextMP made correct prediction to known MPs while our previous method, MPFit, failed. The first example is a band 3 anion transport protein in human (UniProt ID: P02730). The primary function of this protein is transportation of inorganic anions across the plasma membrane while the moonlighting function is a scaffold providing binding sites for glycolytic enzymes (Low et al., 1993). MPFit failed to predict this protein as an MP because this protein lacks four out of six omics-data features (i.e. PPI, phylogenetic profile, genetic interaction), which MPFit imputes to complete input feature values but apparently it did not work. In contrast, this protein has functional description in UniProt, which clearly depicts its two functions as follows: functions both as a transporter that mediates electroneutral anion exchange across the cell membrane and as a structural protein, and interactions of its cytoplasmic domain with cytoskeletal proteins, glycolytic enzymes, and hemoglobin. Based on this text, it was easy for DextMP to make a correct MP prediction.
The second example is protein PHGPx (UniProt ID: P36969) in human. The primary function of this MP is cell protection against membrane lipid peroxidation and cell death while the moonlighting function is the protein’s structural role in mature spermatozoa (Scheerer et al., 2007). MPFit could not see characteristics of MPs in this protein’s omics data, because some input features were not available and moreover, an important feature, functional divergence of interacting proteins in its PPI network, was not observed. However, the protein’s functional description in UniProt indicates two functions, protects cells against membrane lipid peroxidation and required for normal sperm development and male fertility, which resulted in a correct MP prediction by DextMP.
The last example is gephyrin (UniProt ID: Q9NQX3) in human. This protein anchors transmembrane receptors by connecting membrane proteins to cytoskeleton microtubule binding proteins. Its moonlighting function is biosynthesis of the molybdenum cofactor (Stallmeyer et al., 1999). Similar to the previous two examples, this protein lacks several omics-data that are used as features in MPFit. Gene expression data showed that this protein has a similar expression pattern with genes with different functional classes, which is an indicator of an MP, but this information was diluted in combination with other omics-data. On the other hand, it is clear from its functional description that it has two functions: It says microtubule-associated protein involved in membrane protein-cytoskeleton interactions related to its first function, and catalyzes two steps in the biosynthesis of the molybdenum cofactor, which is related to the second function.
Lastly, we provide two examples where DextMP made novel MP prediction (i.e. proteins which are not recorded as known MPs in the MoonProt database). The first example is aminoacyl tRNA synthase complex-interacting protein 1 (UniProt ID: Q12904) in human. This protein is a non-catalytic component of the multi-synthase complex, and among other multi-functionalities, it moonlights by binding to tRNA, possesses inflammatory cytokine activity, and is involved in glucose homeostasis, angiogenesis and wound repair (Han et al., 2006). The second example is exoribonuclease in yeast (UniProt ID: P22147). According to its function description in UniProt, this is also a multi-functional protein that exhibits several independent functions at different levels of the cellular processes. It is a 5′–3′ exonuclease component of the nonsense-mediated mRNA decay (NMD), and has a role in multiple processes including DNA strand exchange and exonuclease activities, preventing accumulation of potentially harmful truncated proteins, regulating the decay of wild-type mRNAs, degradation of mature tRNA, and defense against viruses, among other functions (Johnson and Kolodner, 1999; Käslin and Heyer, 1994).
4 Discussion
We developed DextMP that predicts MPs from text information, which is the first work of this kind. DextMP complements our earlier work, MPFit, which predicts MPs from their omics data-based features. DextMP showed significant improvement of predictions over existing methods. Moreover, it is widely applicable because it only needs the text information of target proteins. Since the study of MPs is still in its early stage, even in cases that proteins are known to have multiple distinct functions, they are not explicitly labeled as MPs in databases. DextMP will be a very useful tool for detecting potential MPs from a vast amount of UniProt entries.
It would be appropriate to discuss implication and technical nature of the provided genome-scale MP prediction. Since DextMP uses literature information of genes, the quantity and the quality of available literature directly affects to prediction results. A smaller number of MPs were detected in X. laevis than yeast and human apparently because only 30.54% of genes in the genome have literature information. It is also noticed that the predicted fraction of MPs in yeast is larger than human, but this result is also at least partly reflecting the fact that yeast has one of the most well studied and annotated genomes as it is a model organism for systems biology. Another technical point to note is that the accuracy of DexMP was confirmed on the control set, where the numbers of positive and negative data are balanced. Since this MP/non-MP distribution is different in genomes from the control set, the accuracy of the genome-scale prediction may be affected by that. Also the negative data used in the control set have unavoidable uncertainty, because non-MPs in the dataset may be found as MPs in the future.
In Table 5, we provided two MP estimations by using cutoffs of three or four votes. Using the three-vote criterion showed a better recall against known MPs by design, however, it is difficult to determine which estimations should be more trusted since the number of known MPs are currently very limited. Thus these two criteria should be considered as confidence levels of a prediction. The current prediction provides a rough estimates of MPs in genomes, which itself would be informative and useful to gain a large perspective of MPs. Ultimately, literature of all genes in genomes needs to be manually checked to obtain the precise number of MPs, where DextMP’s prediction can help in prioritizing genes to examine.
The results of the genome-scale prediction nevertheless suggest that MPs are not mere exceptions but common in organisms. This observation triggers various interesting biological questions, for example, how proteins gain moonlighting functions during evolution and biophysical mechanisms that enable a protein to have multiple functions. Correct annotation to proteins with dual functions also affects to functional enrichment analysis (Wei et al., 2017), which is commonly used in systems and network biology (Dotan-Cohen et al., 2009; Hawkins et al., 2010; Rachlin et al., 2006). This work is also relevant to computational biologists, particularly those who are working on developing function prediction methods (Hawkins and Kihara, 2007), genome annotation, function analysis on networks and curation of functional annotation in databases.
Overall this work will help our understanding of the multi-functional nature of proteins at the systems level, and will aid in exploring the complex functional interplay of proteins in a cellular process.
Acknowledgements
The authors thank Sarah Rodenbeck for proofreading the manuscript.
Funding
This work was partly supported by the National Science Foundation (DBI1262189). DK also acknowledged supports from the National Institutes of Health (R01GM097528) and the National Science Foundation (IIS1319551, IOS1127027, DMS1614777).
Conflict of Interest: none declared.
References
- Bird S. (2006) NLTK: the natural language toolkit COLING/ACL Interact. Present. Sessions, 69–72. [Google Scholar]
- Campbell R.M., Scanes C.G. (1995) Endocrine peptides ′moonlighting′ as immune modulators: roles for somatostatin and GH-releasing factor. J. Endocrinol., 147, 383–396. [DOI] [PubMed] [Google Scholar]
- Chapple C.E. et al. (2015) Extreme multifunctional proteins identified from a human protein interaction network. Nat. Commnun., 6, 7412.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotan-Cohen D. et al. (2009) Biological process linkage networks. PLoS ONE, 4, e5313.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez A. et al. (2011) Do protein-protein interaction databases identify moonlighting proteins? Mol. BioSyst., 7, 2379–2382. [DOI] [PubMed] [Google Scholar]
- Gomez A. et al. (2003) Do current sequence analysis algorithms disclose multifunctional (moonlighting) proteins? Bioinformatics, 19, 895–896. [DOI] [PubMed] [Google Scholar]
- Han J.M. et al. (2006) Structural separation of different extracellular activities in aminoacyl-tRNA synthetase-interacting multi-functional protein, p43/AIMP1. Biochem. Biophys. Res. Commun., 342, 113–118. [DOI] [PubMed] [Google Scholar]
- Hawkins T. et al. (2006) Enhanced automated function prediction using distantly related sequences and contextual association by PFP. Protein Sci., 15, 1550–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins T. et al. (2010) Functional enrichment analyses and construction of functional similarity networks with high confidence function prediction by PFP. BMC Bioinformatics, 11, 265–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins T. et al. (2009) PFP: Automated prediction of gene ontology functional annotations with confidence scores using protein sequence data. Proteins Struct. Funct. Bioinf., 74, 566–582. [DOI] [PubMed] [Google Scholar]
- Hawkins T., Kihara D. (2007) Function prediction of uncharacterized proteins. J. Bioinf. Comput. Biol., 5, 1–30. [DOI] [PubMed] [Google Scholar]
- Hernández S. et al. (2011) Do moonlighting proteins belong to the intrinsically disordered protein class? J. Proteomics Bioinf., 5, 262–264. [Google Scholar]
- Hernández S. et al. (2014) MultitaskProtDB: a database of multitasking proteins. Nucleic Acids Res., 42, D517–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman M. et al. (2010) Online learning for latent dirichlet allocation. Adv. Neural Inf. Process. Syst., 23, 856–864. [Google Scholar]
- Huberts D.H., Vander Klei I.J. (2010) Moonlighting proteins: an intriguing mode of multitasking. Biochim. Biophys. Acta, 1803, 520–525. [DOI] [PubMed] [Google Scholar]
- Jeffery C.J. (2003) Moonlighting proteins: old proteins learning new tricks. Trends Genet., 19, 415–417. [DOI] [PubMed] [Google Scholar]
- Jeffery C. (1999) Moonlighting proteins. Trends Biochem. Sci., 24, 8–11. [DOI] [PubMed] [Google Scholar]
- Jeffery C. (2004) Moonlighting proteins: complications and implications for proteomics research. Drug Discov. Today TARGETS, 3, 71–78. [Google Scholar]
- Joachims T. (1998) Text categorization with support vector machines: Learning with many relevant features. Eur. Conf. Mach. Learn., 10, 137–142. [Google Scholar]
- Johnson A.W., Kolodner R.D. (1999) Strand exchange protein 1 from Saccharomyces cerevisiae. A novel multifunctional protein that contains DNA strand exchange and exonuclease activities. J. Biol. Chem., 266, 14046–14054. [PubMed] [Google Scholar]
- Kanehisa M., Goto S. (2000) KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res., 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käslin E., Heyer W.D. (1994) A multifunctional exonuclease from vegetative Schizosaccharomyces pombe cells exhibiting in vitro strand exchange activity. J. Biol. Chem., 269, 14094–14102. [PubMed] [Google Scholar]
- Khan I. et al. (2014) Genome-scale identification and characterization of moonlighting proteins. Biol. Direct., 9, 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan I., Kihara D. (2014) Computational characterization of moonlighting proteins. Biochem. Soc. Trans., 42, 1780–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan I., Kihara D. (2016) Genome-scale prediction of moonlighting proteins using diverse protein association information. Bioinformatics, 32, 2281–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan I. et al. (2012) Evaluation of function predictions by PFP, ESG, and PSI-BLAST for moonlighting proteins. BMC Proceedings, 6, S5.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Q.V., Mikolov T. (2014) Distributed representations of sentences and documents. arXiv Preprint, 1405.4053. [Google Scholar]
- Low P.S. et al. (1993) Regulation of glycolysis via reversible enzyme binding to the membrane protein, band 3. J. Biol. Chem., 268, 14627–14631. [PubMed] [Google Scholar]
- Mani M. et al. (2014) MoonProt: a database for proteins that are known to moonlight. Nucleic Acids Res., 43, D277–D282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning C.D. et al. (2008) Introduction to Information Retrieval. Cambridge University Press, Cambridge. [Google Scholar]
- Mikolov T. et al. (2013) Distributed representations of words and phrases and their compositionality. Adv. Neural Inf. Process. Syst., 26, 3111–3119. [Google Scholar]
- Pedregosa F. et al. (2011) Scikit-learn: machine learning in Python. J. Mach. Learn. Res., 12, 2825–2830. [Google Scholar]
- Piatigorsky J., WistowG J. (1989) Enzyme/crystallins: gene sharing as an evolutionary strategy. Cell, 57, 197–199. [DOI] [PubMed] [Google Scholar]
- Pritykin Y. et al. (2015) Genome-wide detection and analysis of multifunctional genes. PLoS Comput. Biol., 11, e1004467.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachlin J. et al. (2006) Biological context networks: a mosaic view of the interactome. Mol. Syst. Biol., 2, 66.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada M., Tarau P. (2004) TextRank: Bringing order into texts. In: Proceedings of EMNLP, Association for Computational Linguistics, Barcelona, Spain, pp. 404–411.
- Rurek R., Sojka P. (2010) Software Framework for Topic Modelling with Large Corpora. In: Proceedings of LREC 2010 workshop New Challenges for NLP Frameworks, University of Malta, Valletta, Malta, pp. 45–50.
- Scheerer P. et al. (2007) Structural basis for catalytic activity and enzyme polymerization of phospholipid hydroperoxide glutathione peroxidase-4 (GPx4). Biochemistry, 46, 9041–9049. [DOI] [PubMed] [Google Scholar]
- Schlicker A. et al. (2006) A new measure for functional similarity of gene products based on Gene Ontology. BMC Bioinformatics, 7, 322.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallmeyer B. et al. (1999) The neurotransmitter receptor-anchoring protein gephyrin reconstitutes molybdenum cofactor biosynthesis in bacteria, plants, and mammalian cells. Proc. Natl. Acad. Sci. U. S. A., 96, 1333–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium. (2014) Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res., 42, D191–D198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver D.T. (1998) Telomeres: moonlighting by DNA repair proteins. Curr. Biol., 8, R492–R494. [DOI] [PubMed] [Google Scholar]
- Wei Q. et al. (2017) NaviGO: Interactive tool for visualization and functional similarity and coherence analysis with gene ontology. BMC Bioinformatics, 18, 177.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wistow G.J., Kim H. (1991) Lens protein expression in mammals:taxon-specificity and the recruitment of crystallins. J. Mol. Evol., 32, 262–269. [DOI] [PubMed] [Google Scholar]
- Wool I.G. (1996) Extraribosomal functions of ribosomal proteins. Trends Biochem. Sci., 21, 164–165. [PubMed] [Google Scholar]