

Abstract
Motivation
Large-scale molecular profiling data have offered extraordinary opportunities to improve survival prediction of cancers and other diseases and to detect disease associated genes. However, there are considerable challenges in analyzing large-scale molecular data.
Results
We propose new Bayesian hierarchical Cox proportional hazards models, called the spike-and-slab lasso Cox, for predicting survival outcomes and detecting associated genes. We also develop an efficient algorithm to fit the proposed models by incorporating Expectation-Maximization steps into the extremely fast cyclic coordinate descent algorithm. The performance of the proposed method is assessed via extensive simulations and compared with the lasso Cox regression. We demonstrate the proposed procedure on two cancer datasets with censored survival outcomes and thousands of molecular features. Our analyses suggest that the proposed procedure can generate powerful prognostic models for predicting cancer survival and can detect associated genes.
Availability and implementation
The methods have been implemented in a freely available R package BhGLM (http://www.ssg.uab.edu/bhglm/).
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Precision medicine needs accurate prognostic prediction (predicting the risk of future relapse after an initial treatment or responsiveness to different treatments) (Barillot et al. 2012; Chin et al. 2011). Many traditional clinical prognostic and predictive factors have been known for diseases (e.g. cancers) for years, however, usually provide poor prognosis and prediction (Barillot et al. 2012). There is therefore a need for new prognostic and predictive factors with better reproducibility and better discriminatory power between different prognosis and drug responsiveness groups. Precision medicine is often based on the genetic background of the patients (Barillot et al. 2012; Collins and Varmus 2015). Recent high-throughput technologies can easily and robustly generate large-scale molecular profiling data, offering extraordinary opportunities to search for new biomarkers and to build accurate prognostic and predictive models (Barillot et al. 2012; Collins and Varmus 2015). However, significant challenges exist, including: (i) how to select predictive molecular factors among numerous candidates, (ii) how to precisely estimate the effects of predictors and (iii) how to combine many predictive factors into accurate predictive models.
Several methods have been applied to address the challenges, mainly including traditional variable selection, principal components method and penalized regressions. Extensive studies have demonstrated that penalized regressions usually have good predictive performance and can result in powerful and interpretable predictive models (Bovelstad et al. 2007, 2009). Various penalized survival models have recently been proposed to analyze large-scale data, including lasso, ridge or elastic-net Cox proportional hazards models (Hastie et al. 2015; Simon et al. 2011; Tibshirani 1997; van Houwelingen et al. 2006) and parametric models (Li et al. 2016; Mittal et al. 2013). The penalized survival models can be fit by fast algorithms, and can achieve both predicting survival and identifying important predictors (Hastie et al. 2015; Simon et al. 2011; Tibshirani 1997). Thus these penalized methods have widely been used for analyzing high-dimensional molecular data (Barillot et al. 2012; Gerstung et al. 2015; Sohn and Sung 2013; Yuan et al. 2014; Zhang et al. 2013; Zhao et al. 2015). Recently, Bayesian survival models and spike-and-slab variable selection methods have been applied to molecular predictive modeling (Bonato et al. 2011; Lee et al. 2011, 2015; Li and Zhang 2010; Monni and Li 2010; Peng et al. 2013; Stingo et al. 2010; Tai et al. 2009). These methods use Markov Chain Monte Carlo (MCMC) algorithms to fully explore the joint posterior distribution. Although statistically sophisticated, these MCMC-based methods are computationally intensive for analyzing large-scale molecular data.
In this article, we propose new hierarchical Cox survival models with spike-and-slab double-exponential priors for jointly analyzing large-scale molecular data for building powerful predictive models and identifying important predictors. The spike-and-slab prior is the fundamental basis for most Bayesian variable selection approaches, and has proved remarkably successful (Chipman 1996; Chipman et al. 2001; George and McCulloch 1993, 1997; Ročková and George, 2014; Ročková and George, 2016, unpublished manuscript). Most previous spike-and-slab variable selection approaches use the spike-and-slab normal priors on coefficients and employ computationally intensive MCMC algorithms to search for high posterior models. Recently, Ročková and George (2016, unpublished manuscript) proposed a new framework, called the spike-and-slab lasso, for high-dimensional normal linear models, and showed that it has remarkable properties(Ročková and George, 2016, unpublished manuscript).
We extend the spike-and-slab lasso framework to Cox survival models. The proposed spike-and-slab lasso Cox models can adaptively shrink coefficients and thus can result in accurate estimates and predictions. We propose an efficient algorithm to fit the spike-and-slab lasso Cox models by incorporating EM steps (Expectation-Maximization) into the extremely fast cyclic coordinate descent algorithm. The proposed approach integrates two popular methods, i.e. lasso and Bayesian spike-and-slab hierarchical modeling, into one unifying framework, and thus combines the remarkable features of the two popular methods while diminishing their shortcomings. The performance of the proposed method is assessed via extensive simulations and compared with the commonly used lasso Cox regression. We apply the proposed procedure to two cancer datasets with censored survival outcomes and thousands of molecular features: breast cancer and myelodysplastic syndromes (MDS). Our results show that the proposed method can generate powerful prognostic models for predicting cancer survival and can detect survival associated genes.
2 Materials and methods
2.1 Cox proportional hazards models
For censored survival outcomes, we observe a pair of response variables yi = (ti, di) for each individual, where the censoring indicator di takes 1 if the observed survival time tifor individual i is uncensored and 0 if it is censored. Denote the true survival time by Ti for individual i. Thus, when di = 1, Ti = ti, whereas when di = 0, Ti > ti. Cox proportional hazards model is the most widely used method for studying the relationship between the censored survival response and some explanatory variables X, and assumes that the hazard function of survival time T takes the form (Ibrahim et al. 2001; Klein and Moeschberger 2003):
| (1) |
where the baseline hazard function is unspecified, X and are the vectors of explanatory variables and coefficients, respectively, and is the linear predictor or called the prognostic index.
Fitting classical Cox models is to estimate by maximizing the partial log-likelihood (Cox 1972):
| (2) |
where is the risk set at time . In the presence of ties, the partial log-likelihood can be approximated by the Breslow or the Efron methods (Breslow 1972; Efron 1977). The standard algorithm for maximizing the partial log-likelihood is the Newton–Raphson algorithm (Klein and Moeschberger 2003; van Houwelinggen and Putter 2012).
The lasso uses the L1 penalty to the partial log-likelihood function and estimates the parameters by maximizing the penalized log-likelihood (Friedman et al. 2010; Hastie et al. 2015; Simon et al. 2011; Tibshirani 1997; Zou and Hastie 2005):
| (3) |
The overall penalty parameter controls the overall strength of penalty and the size of the all coefficients. The lasso Cox model can be fit by the extremely fast cyclic coordinate descent algorithm(Simon, et al., 2011).
Ideally, one should use small penalty values for important predictors and large penalties for irrelevant predictors. We here propose a new approach, i.e., the spike-and-slab lasso, which can induce different shrinkage scales for different coefficients and allows us to estimate the shrinkage scales from the data.
2.2 Spike-and-slab lasso Cox models
The spike-and-slab lasso Cox models are more easily interpreted and handled from Bayesian hierarchical modeling framework (Ročková and George, unpublished manuscript; Ročková and George, 2016). It is well known that the lasso can be expressed as a hierarchical model with double-exponential prior on coefficients (Kyung, et al., 2010; Park and Casella, 2008; Tibshirani, 1996; Yi and Xu, 2008):
| (4) |
where the scale parameter s equals . The scale s controls the amount of shrinkage; smaller scale induces stronger shrinkage and forces the estimates of towards 0.
We develop the spike-and-slab lasso Cox models by extending the double-exponential prior to the following spike-and-slab mixture double-exponential prior:
or equivalently
| (5) |
where is the indicator variable, = 1 or 0, and the scale equals one of two preset positive values s0 and s1 (s1 > s0 > 0), i.e. . The scale value s0 is chosen to be small and serves a “spike scale” for modeling irrelevant (0) coefficients and inducing strong shrinkage on estimation, and s1 is set to be relatively large and thus serves as a “slab scale” for modeling large coefficients and inducing no or weak shrinkage on estimation. If we set s0 = s1, the spike-and-slab double-exponential prior becomes the double-exponential prior. Therefore, the spike-and-slab lasso includes the lasso as a special case.
The indicator variables play an essential role on linking the scale parameters with the coefficients. For our development, we confine attention to exchangeable priors on the indicator variables, that is, the indicator variables are assumed to follow the independent binomial distribution:
| (6) |
where θ is the probability parameter. However, the scope of the spike-and-slab mixture priors is greatly enhanced by the flexibility of the model space prior for the indicator variables , which can be used to concentrate on preferred and meaningful models.
For the probability parameter θ, we assume the uniform prior: θ ∼U(0, 1). The probability parameter θ can be viewed as the overall shrinkage parameter that equals the prior probability . The prior expectation of the scale equals , which lies in the range [s0, s1]. As will be seen, the scale for each coefficient can be estimated, leading to different shrinkage for different predictors.
2.3 Algorithm for fitting the spike-and-slab lasso Cox models
We here develop a fast algorithm to fit the spike-and-slab lasso Cox models by finding the posterior modes of the parameters, i.e. estimating the parameters by maximizing the posterior density. Our algorithm, called the EM coordinate descent algorithm, incorporates EM steps into the cyclic coordinate descent procedure for fitting the penalized lasso Cox regression. We derive the EM coordinate descent algorithm based on the log joint posterior density of the parameters ():
The log-likelihood function, , is proportional to the partial log-likelihood defined in Equation (1) or the Breslow or the Efron approximation in the presence of ties (Breslow 1972; Efron 1977), if the baseline hazard function h0 is replaced by the Breslow estimator (Breslow 1974; van Houwelinggen and Putter 2012). Therefore, the log joint posterior density can be expressed as
| (7) |
where is the partial likelihood described in (2), and .
The EM coordinate decent algorithm treats the indicator variables as ‘missing values’ and estimates the parameters () by averaging the missing values over their posterior distributions. For the E-step, we calculate the expectation of the log joint posterior density with respect to the conditional posterior distributions of the missing data . The conditional posterior expectation of the indicator variable can be derived as
| (8) |
where , and . Therefore, the conditional posterior expectation of can be obtained by
| (9) |
It can be seen that the estimates of pj and Sj are larger for larger coefficients , leading to different shrinkage for different coefficients.
For the M-step, we update () by maximizing the posterior expectation of the log joint posterior density with and replaced by their conditional posterior expectations. From the log joint posterior density, we can see that and can be updated separately, because the coefficients are only involved in and the probability parameter is only in . Therefore, the coefficients are updated by maximizing the expression:
| (10) |
where is replaced by its conditional posterior expectation derived above. Given the scale parameters Sj, the term serves as the L1 lasso penalty with as the penalty factors, and thus the coefficients can be updated by maximizing using the cyclic coordinate decent algorithm. Thus, the coefficients can be estimated to be 0. The probability parameter is updated by maximizing the expression:
| (11) |
We can easily obtain: .
In summary, the EM coordinate decent algorithm for fitting the spike-and-slab lasso Cox models proceeds as follows:
Choose a starting value, and . For example, we can initialize = 0 and = 0.5.
For t = 1, 2, 3, …,
E-step: Update and by their conditional posterior expectations.
M-step:
Update using the cyclic coordinate decent algorithm;
Update .
We assess convergence by the criterion: , where = is the estimate of deviance at the tth iteration, and is a small value (say 10−5).
2.4 Selecting optimal scale values
The performance of the spike-and-slab lasso Cox model approach can depend on the scale parameters (s0, s1). We can usually analyze data with reasonably preset scale values, for example, setting the slab scale s1 to a value (say s1 = 0.5) that induces weak shrinkage and the spike scale s0 to a small value (say s0 = 0.04) that induces strong shrinkage. Rather than restricting attention to a single model, however, our fast algorithm allows us to quickly fit a sequence of models, from which we can choose an optimal one based on some criteria. Our strategy is to fix the slab scale s1 = 0.5, and consider a sequence of L decreasing values {}: , for the spike scale s0. We then fit L models with scales {}. Increasing the spike scale s0 tends to include more non-zero coefficients in the model. This procedure is similar to the lasso implemented in the widely-used R package glmnet, which quickly fits the lasso Cox models over a grid of values of covering its entire range, giving a sequence of models for users to choose from (Hastie et al. 2015; Simon et al. 2011).
There are several ways to measure the performance of a fitted Cox model, including the partial log-likelihood (PL), the concordance index (C-index), the survival curves and the survival prediction error (van Houwelinggen and Putter 2012). The partial log-likelihood function measures the overall quality of a fitted Cox model, and thus is usually used to choose an optimal model (Simon et al. 2011; van Houwelinggen and Putter 2012; van Houwelingen et al. 2006). To evaluate the predictive performance of a Cox model, a general way is to fit the model using a dataset and then calculate the above measures with independent data. We use the pre-validation method, a variant of cross-validation (Hastie et al. 2015; Tibshirani and Efron 2002). We randomly split the data to K subsets of roughly the same size, and use (K−1) subsets to fit a model. Denote the estimate of coefficients from the data excluding the kth subset by . We calculate the prognostic indices for all individuals in the kth subset of the data, called the cross-validated or pre-validated prognostic index. Cycling through K parts, we obtain the cross-validated prognostic indices for all individuals. We then use () to compute the measures described above. The cross-validated prognostic score for each patient is derived independently of the observed response of the patient, and hence the ‘pre-validated’ dataset {} can essentially be treated as a ‘new dataset’. Therefore, this procedure provides valid assessment of the predictive performance of the model (Hastie et al. 2015; Tibshirani and Efron 2002).
We also use an alternative way to evaluate the partial log-likelihood, i.e., the so-called cross-validated partial likelihood (CVPL), defined as (Simon et al. 2011; van Houwelinggen and Putter 2012; van Houwelingen et al. 2006)
| (12) |
where is the estimate of from all the data except the kth part, is the partial likelihood of all the data points and is the partial likelihood excluding part k of the data. By subtracting the log-partial likelihood evaluated on the non-left out data from that evaluated on the full data, we can make efficient use of the death times of the left out data in relation to the death times of all the data.
2.5 Implementation
We have created an R function bmlasso() for setting up and fitting the spike-and-slab lasso Cox, and several other R functions (e.g. summary.bh, plot.bh, predict.bh, cv.bh) for summarizing the fitted models and for evaluating the predictive performance. We have incorporated these functions into the freely available R package BhGLM (http://www.ssg.uab.edu/bhglm/).
3 Simulation study
3.1 Simulation design
We used simulations to validate the proposed spike-and-slab lasso approach, and to compare with the lasso in the R package glmnet. In each situation, we simulated two datasets, and used the first one as the training data to fit the models and the second one as the test data to evaluate the predictive values. For each simulation setting, we replicated the simulation 50 times and summarized the results over these replicates. We reported the results on the predictive values including partial log-likelihood and C-index in the test data, the accuracy of parameter estimates and the proportions of coefficients included in the model.
For each dataset, we generated n (=500) observations, each with a survival response, consisting of an observed censored survival time ti and a censoring indicator di, and a vector of m (=200, 1000) continuous predictors. The vector Xi, was generated with 50 elements at a time, i.e. the sub-vector , , was randomly sampled from multivariate normal distribution , where with . Thus, the predictors within a group were correlated and between groups were independent. We generated “true” survival time Ti for each individual from the exponential distribution: , where the coefficients were preset as described below. We then generated censoring time Ci for each individual from the exponential distribution: , where ri were randomly sampled from a standard normal distribution. The observed censored survival time ti was set to be the minimum of the “true” survival and censoring times, , and the censoring indicator diwas set to be 1 if Ci > Ti and 0 otherwise.
We set five coefficients β5, β20, β40, βm–50 and βm–5 to be non-zero, two of which were negative, and all others to be 0. Table 1 shows the preset non-zero coefficient values for six simulation scenarios.
Table 1.
The simulated effect sizes of five coefficients under different scenarios (n = 500)
| β5 | β20 | β40 | βm-50 | βm-5 | ||
|---|---|---|---|---|---|---|
| Scenario 1 | m = 200 | 0.214 | 0.261 | 0.262 | −0.296 | −0.422 |
| Scenario 2 | m = 200 | 0.359 | 0.451 | −0.445 | −0.518 | 0.725 |
| Scenario 3 | m = 200 | −0.522 | −0.635 | 0.630 | 0.744 | 1.017 |
| Scenario 4 | m = 1000 | −0.209 | −0.257 | 0.258 | 0.298 | 0.422 |
| Scenario 5 | m = 1000 | 0.366 | 0.450 | 0.446 | −0.518 | −0.725 |
| Scenario 6 | m = 1000 | 0.514 | −0.625 | 0.631 | 0.722 | −1.037 |
We analyzed each simulated dataset using the lasso Cox model implemented in the R package glmnet and the proposed spike-and-slab lasso Cox model. For the lasso Cox approach, we used 10-fold cross-validation to select an optimal value of λ, which determines an optimal lasso Cox model, and reported the results based on the optimal lasso Cox model. For the spike-and-slab lasso Cox approach, we mainly considered a slab scale s1 = 0.5, and six spike scales: s0 = sλ + 0.02, sλ + 0.01, sλ, sλ − 0.01, sλ − 0.02, sλ − 0.03, where sλ is the scale of the double-exponential prior corresponding to the optimal lasso Cox model, i.e. sλ = 1/λ as described in Equation (4). To investigate the impact of the scales (s0, s1) on the results, we fit the simulated datasets under scenarios 3 and 6 with 81 combinations when s0 changed from 0.01 to 0.09 and s1 from 0.1 to 0.9.
3.2 Simulation results
3.2.1. Predictive performance
Supplementary Table S1 shows the partial log-likelihood and C-index in the test data from the lasso and the spike-and-slab lasso under different s0 values. Table 2 shows the selected optimal model under different simulated scenarios. From these results, we can see that the spike-and-slab lasso Cox with an appropriate value of s0 performed better than the lasso. Table 2 also shows that the spike-and-slab lasso had slightly higher C-index value than the lasso. Thus, the spike-and-slab lasso approach can generate better discrimination.
Table 2.
The partial log-likelihood and C-index values over 50 simulated replicates under different simulated scenarios
| Simulated scenarios | Methods | Partial log-likelihood | C-index |
|---|---|---|---|
| Scenario 1 | lasso: sλ = 0.045a | −1200.49(62.769) | 0.680(0.022) |
| sslasso: s0 = sλ | −1201.039(63.520) | 0.679(0.022) | |
| Scenario 2 | lasso: sλ = 0.052a | −1204.975(49.157) | 0.735(0.019) |
| sslasso: s0 = sλ − 0.02 | −1198.005(49.900) | 0.740(0.020) | |
| Scenario 3 | lasso: sλ = 0.057a | −1150.189(60.427) | 0.795(0.016) |
| sslasso: s0 = sλ − 0.03 | −1141.872(61.211) | 0.800(0.016) | |
| Scenario 4 | lasso: sλ = 0.029a | −1195.046(52.336) | 0.624(0.025) |
| sslasso: s0 = sλ+0.01 | −1192.936(51.836) | 0.625(0.028) | |
| Scenario 5 | lasso: sλ = 0.034a | −1153.503(53.632) | 0.785(0.015) |
| sslasso: s0 = sλ − 0.02 | −1140.097(54.464) | 0.790(0.015) | |
| Scenario 6 | lasso: sλ = 0.042a | −1159.421(55.534) | 0.792(0.018) |
| sslasso: s0 = sλ − 0.02 | −1143.262(57.398) | 0.801(0.018) |
Note: Scenario 1, 2 and 3: n = 500, m = 200; Scenario 4, 5 and 6: n = 500, m = 1000. The slab scales, s1, are 0.5 in all scenarios. The optimal models for different scenarios are summarized here. More results can be found in Supplementary Table S1
sλ denotes the average value of penalty parameters sλ (sλ = 1/λ) of 50 repeated samples, where λ is the optimal value of the penalty parameter for the lasso. ‘sslasso’ represents the spike-and-slab lasso Cox model
From Table 2 and Supplementary Table S1, we found that the best models depended on the spike scale s0. Supplementary Figure S1 shows the profiles of the partial log-likelihood function on a grid of values of (s0, s1) for scenarios 3 and 6. It can be seen that the slab scale s1 within the range [0.1, 0.9] had little influence on the partial log-likelihood function, while the spike scale s0 strongly affected the model performance. These results show that our approach with a fixed slab scale s1 is reasonable.
3.2.2. Solution path and adaptive property for the spike-and-slab lasso
The performance of the proposed method strongly depended on the spike scale s0. To fully investigate the impact of the spike scale s0 on the results, Figure 1A and B presents the solution paths under scenario 3 by the proposed model with s1 = 0.5 and the lasso model, respectively. Figure 1C and D shows the profiles of CVPL by 10-fold cross-validation for the proposed model and the lasso model, respectively. Similar to the lasso, the spike-and-slab lasso Cox is a path-following strategy for fast dynamic posterior exploration. However, the solution path is essentially different from that of the lasso model. For the lasso solution as shown in Figure 1B, the number of non-zero coefficient could be a few, even zero if a strong penalty is adopted. However, in the spike-and-slab lasso, larger coefficients will be always included in the model with none or weak shrinkage, while irrelevant coefficients are removed. Supplementary Figure S2 shows the solution paths of the proposed model with s1 = 0.5 and the lasso model under scenario 6.
Fig. 1.
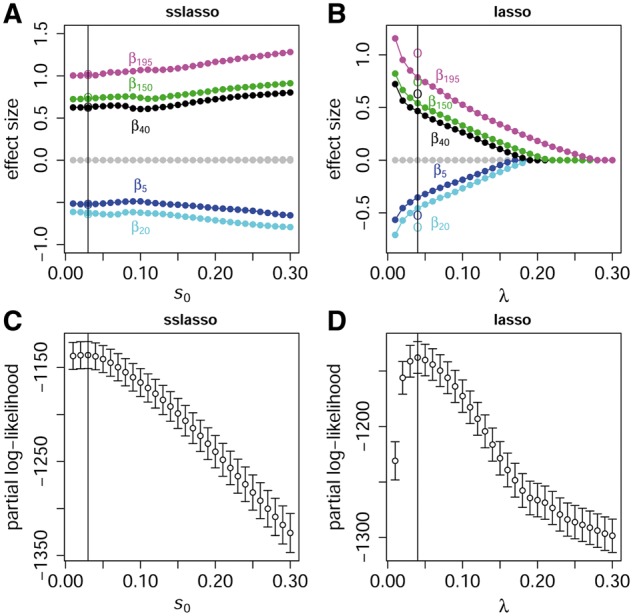
The solution paths and partial log-likelihood profiles of the proposed spike-and-slab lasso Cox model and the lasso Cox model under scenario 3 with n = 500 and m = 200. (A, C) Present the solution path and the CVPL profile for proposed spike-and-slab lasso Cox model, respectively. (B, D) Present the solution path and the CVPL profile for the lasso model, respectively. The points and circles on the solution paths represent the estimated values of five simulated non-zero coefficients over 50 replicates and the true non-zero coefficients. The vertical lines correspond to the optimal models with the largest CVPL
To show the adaptive property of the proposed method, we performed additional simulation study with (n, m) = (500, 1000) and (s0, s1) = (0.05, 0.5). Supplementary Figure S3 shows the adaptive shrinkage amount, along with the different effect size. It clearly shows that the proposed spike-and-slab lasso Cox model approach has self-adaptive and flexible characteristics.
3.2.3. Accuracy of parameter estimates
Figure 2 and Supplementary Figures S4 and S5 show the estimates of coefficients from the spike-and-slab lasso Cox model and the lasso Cox model over 50 replicates of training data. It can be seen that the spike-and-slab lasso provided more accurate estimation in most situations, especially for larger coefficients. In contrast, the lasso can over-shrink large coefficients, due to employing a single penalty on all the coefficients.
Fig. 2.
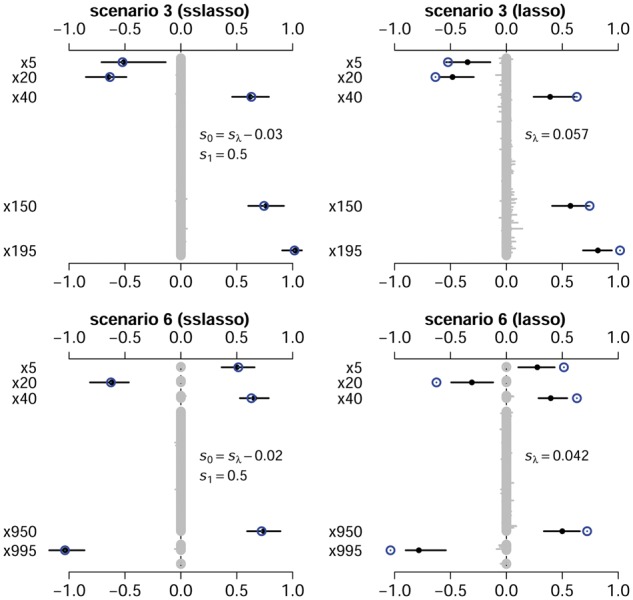
The parameter estimation averaged over 50 replicates for the spike-and-slab lasso Cox model and the lasso Cox model. The numbers of simulated predictors are m = 200 for scenario 3 and m = 1000 for scenario 6. The cycles are the assumed true values. The black points and lines represent the estimated values and the interval estimates of coefficients over 50 replicates
We also summarized the mean absolute errors (MAE) of coefficient estimates, defined as , in Table 3. A smaller MAE suggests more accurate parameter estimation. It can be seen that the proposed models gave smaller MAE in all scenarios.
Table 3.
Average number of non-zero coefficients and mean absolute error (MAE) of coefficient estimates over 50 simulations for the best spike-and-slab lasso Cox model and the best lasso model
| sslasso (s1 = 0.5) |
lasso |
|||
|---|---|---|---|---|
| Average number | MAE | Average number | MAE | |
| Scenario 1, s0 = sλ | 18.68 | 1.285(0.402) | 20.10 | 1.348(0.378) |
| Scenario 2, s0 = sλ − 0.02 | 7.82 | 0.605(0.319) | 20.52 | 1.531(0.308) |
| Scenario 3, s0 = sλ − 0.03 | 6.92 | 0.458(0.253) | 23.58 | 1.618(0.340) |
| Scenario 4, s0 = sλ + 0.01 | 35.08 | 1.708(0.432) | 16.34 | 1.374(0.257) |
| Scenario 5, s0 = sλ − 0.02 | 5.42 | 0.422(0.252) | 24.68 | 1.480(0.284) |
| Scenario 6, s0 = sλ − 0.02 | 8.22 | 0.457(0.316) | 37.36 | 2.183(0.404) |
3.2.4. Proportions of coefficients included in the model
We calculated the proportions for the simulated non-zero coefficients included in the model over the simulation replicates. Like the lasso approach, the proposed spike-and-slab lasso can estimate coefficients to be 0, and thus can easily return these proportions. Figure 3 and Supplementary Figure S6 show the inclusion proportions of the non-zero coefficients and the zero coefficients for the spike-and-slab lasso and the lasso Cox models. It can be seen that in most situations the inclusion proportions of the non-zero coefficients were similar for the two approaches. However, the lasso included zero coefficients in the model more frequently than the spike-and-slab lasso. This indicates that the spike-and-slab lasso approach can reduce noisy signals.
Fig. 3.
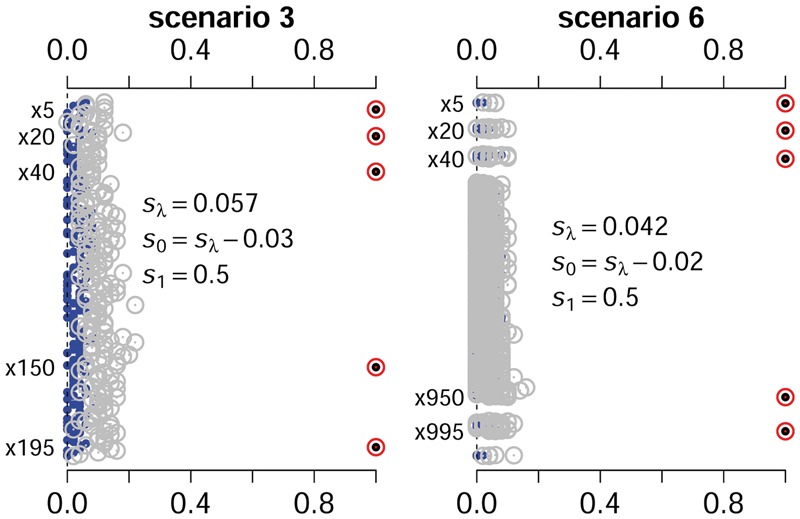
The inclusion proportions of the non-zero and zero coefficients in the model over the simulation replicates under scenarios 3 and 6. The points and cycles nearby the right part of the figure represent the proportions of non-zero coefficients for the spike-and-slab lasso Cox model and the lasso Cox model, respectively. The points and grey cycles nearby left axis represent the proportions of zero coefficients for the spike-and-slab lasso Cox model and the lasso Cox model, respectively
We summarized the average numbers of non-zero coefficients in Table 3. In most simulated scenarios, except for scenario 4, the average numbers of non-zero coefficients in the spike-and-slab lasso Cox models were much lower than those in the lasso Cox models. We also found that the average numbers of non-zero coefficients detected by the proposed models were close to the number of the simulated non-zero coefficients in most scenarios. However, the lasso always included many zero coefficients in the model.
4 Application to real data
4.1. Dutch breast cancer data
We applied our spike-and-slab lasso Cox model to analyze the well-known Dutch breast cancer data set. This dataset contains the microarray mRNA expression measurements of 4919 genes and the time of metastasis after adjuvant systemic therapy (a censored survival outcome) from 295 women with breast cancer (van de Vijver, et al. 2002; van't Veer et al. 2002). The 4919 genes were selected from 24,885 genes, for which reliable expression is available (van’t Veer et al. 2002). Among 295 tumors, 88 had distant metastases. Our analysis was to build a survival model for predicting the metastasis time using the 4919 gene-expression predictors. Prior to fitting the models, we standardized all the predictors.
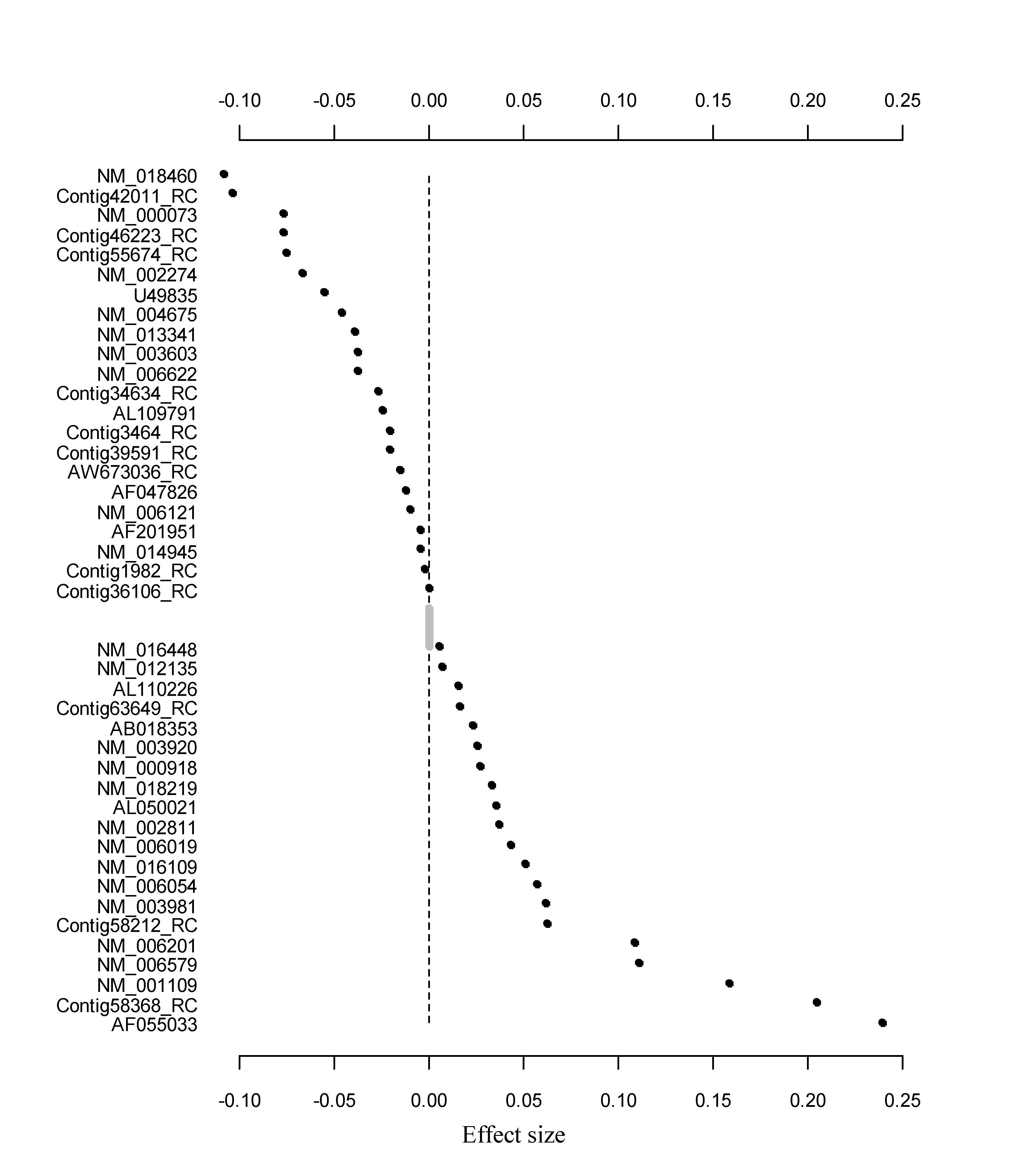
We fixed the slab scale s1 to 0.5, and varied the spike scale s0 over the grid of values: 0.005 + k × 0.005; k = 0, 1,…, 17, leading to 18 models. For each model, we performed 10-fold cross-validation with 10 replicates to select an optimal model based on the cross-validated partial log-likelihood (CVPL). Supplementary Figure S7 shows the profiles of CVPL. The largest value of CVPL appeared to be −541.237, when the spike scale s0 was 0.05. Therefore, the spike-and-slab lasso Cox model with the scale (0.05, 0.5) was chosen for the prediction. This proposed model detected 42 genes, and the effect sizes for most genes were small (Supplementary Fig. S8).
We performed 10-fold cross-validation with 10 replicates to evaluate the predictive values of the chosen model. The cross-validated C-index was estimated to be 0.670, which was significantly larger than 0.5, showing the discriminative ability of the final prognostic model. We estimated the cross-validated prognostic index, , for each patient, and then grouped the patients on the basis of the prognostic index into two groups of equal size by 50th percentile. The Kaplan–Meier survival curve in each subgroup was calculated to assess the predictive usefulness of the proposed model. Figure 4 shows the Kaplan–Meier survival curves for the chosen model fitted using the entire data (left panel) and the cross-validation (right panel). The Log-rank tests were performed to compare the survival estimates between two groups. As expected, the separation of the curves from cross-validation was not as strong as that of the model fitted using the entire data. However, it was still significant, indicating that the proposed model was very informative on prediction.
Fig. 4.
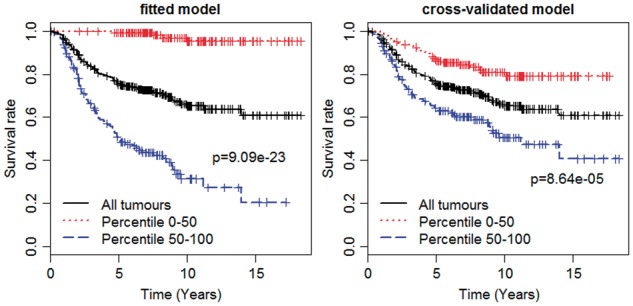
The Kaplan–Meier survival curves of the fitted spike-and-slab lasso Cox model (left panel) and the cross-validated model (right panel) for Dutch breast cancer data set. The Log-rank tests are performed to compare the two curves and obtain P values
Measuring survival prediction error is an alternative important way to evaluate the predictive performance of a survival model. Supplementary Figure S9 presents the prediction error curves for the fitted model, the cross-validated model and the null model (i.e. with no predictors). As expected, the proposed model had much lower prediction error than the cross-validated model and the null model. It also can be seen that the cross-validated model had lower prediction error than the null model. Thus, the detected genes provided valuable predictive information.
4.2. MDS data
The second dataset that we analyzed was a recently published data on MDS (Gerstung et al. 2015). Clinical data are available for 142 MDS patients, where 24 of them had 0 survival time and were excluded from the analysis. The outcome of interest is acute myeloid leukaemia (AML)-free survival. Among the 118 samples, the number of dead patients was 40. For gene expression (from CD34+ bone marrow cells), 124 samples have 21,762 features profiled. Even though we can analyze all the 21,762 features, considering that the number of genes related to AML free survival is not expected to be too large, we filtered the expression data using variance with cutoff 0.9 and selected 2177 gene expression for predictive modeling.
Gerstung et al. (2015) built a prognostic model for predicting AML-free survival using the first 20 principle components of all 21,762 gene expressions, and obtained an impressive prediction with C-index = 0.76. However, their model failed to identify important genes. Our analysis had advantages of not only providing prediction but also detecting associated genes.
Similar to the above analysis of the breast cancer dataset, we fixed the slab scale s1 to 0.5 and considered the spike scale s0 over the grid of values: 0.005 + k × 0.005; k = 0, 1,…, 17, and performed 10-fold cross-validation with 10 replicates to select an optimal model based on the CVPL. From Supplementary Figure S10, we can see that s0 = 0.075 resulted in the largest CVPL. Therefore, the scale (0.075, 0.5) was chosen for our spike-and-slab lasso Cox model. This model detected 19 genes, and the effect sizes for most genes were small (Supplementary Fig. S11).
The cross-validated C-index was estimated to be 0.695, which is close that of Gerstung et al. (2015). However, our prognostic model only included 19 genes, and thus was much more easily interpreted and clinically useful. We further estimated the Kaplan–Meier survival curves for two groups of equal size by 50th percentile of the cross-validated prognostic index values. Figure 5 shows that the Kaplan–Meier survival curves for the two groups were significantly different. We also estimated the prediction error curves for the fitted model, the cross-validated model and the null model. Supplementary Figure S12 shows that the fitted model and the cross-validated model had lower prediction error than the null model. Thus, the detected genes provided valuable predictive information.
Fig. 5.
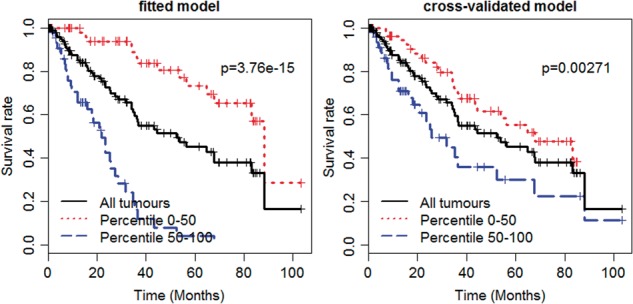
The Kaplan–Meier survival curves of the fitted spike-and-slab lasso Cox model (left panel) and the cross-validated models (right panel) for MDS dataset. The Log-rank tests are performed to compare the two survival curves and obtain P values
We also used the lasso Cox model to analyze these two datasets and compared the results of the lasso Cox model with the above results. The measures of predictive performance by the two methods were close, which may result from the small effect sizes of the detected genes. However, the lasso models included 46 and 31 genes for Dutch breast cancer and MDS datasets, respectively, while the proposed models included 42 and 19 genes for the two data, respectively, to achieve similar prediction accuracy. Most of the detected genes by the two approaches were overlapped. Although having not greatly improved the prediction, the proposed method generated simpler and more clinically useful prognostic models.
5 Discussion
We have developed a new hierarchical model approach, i.e. the spike-and-slab lasso Cox models, for detecting important variables and predicting survival outcomes (e.g. the time until an event such as tumor recurrence or death). Although focusing on molecular profiling data, the proposed approach also can be used for analyzing general large-scale survival data.
The key to our spike-and-slab lasso Cox models is proposing the new prior distribution, i.e. the spike-and-slab double-exponential prior, on the coefficients. The spike-and-slab double-exponential priors can induce different amounts of shrinkage for different predictors depending on their effect sizes, thus reducing the noises of irrelevant predictors and improving the accuracy of coefficient estimates and prognostic predictions. The spike-and-slab lasso Cox models can be effectively fitted by the proposed EM coordinate descent algorithm, which incorporates EM steps into the cyclic coordinate descent algorithm. The E-steps involve calculating the posterior expectations of the indicator variable γj and the scale Sj for each coefficient, and the M-steps employ the existing fast algorithm, i.e. the cyclic coordinate descent algorithm (Friedman et al. 2010; Hastie et al. 2015; Simon et al. 2011), to update the coefficients. As observed in our extensive data analyses, the EM coordinate descent algorithm converges rapidly, and is capable of identifying important predictors and building promising predictive models from numerous candidates. In our real data analysis, it took 0.014 and 0.004 min for Dutch breast cancer dataset and MDS dataset, respectively. The 10-fold cross-validation procedure took a little longer, but still less than one minute for both the two datasets.
The spike-and-slab lasso bridges two popular methods for high-dimensional data analysis, i.e. Bayesian variable selection (Chipman 1996; Chipman et al. 2001; George and McCulloch 1993, 1997; Ročková and George 2014) and the penalized lasso (Hastie et al. 2015; Tibshirani 1996, 1997), into one unifying framework, and thus remains the nice features of these two methods while diminishing their shortcomings (Ročková and George, 2016 unpublished manuscript). Similar to the penalized lasso, the spike-and-slab lasso can shrink irrelevant coefficients exactly to 0, thus automatically achieving variable selection and yielding easily interpretable results. More importantly, due to using the spike-and-slab mixture prior, the shrinkage scale for each predictor can be estimated from the data, yielding weak shrinkage on important predictors but strong shrinkage on irrelevant predictors and thus diminishing the well-known estimation bias of the lasso.
The spike-and-slab lasso Cox models include two scale values (s0, s1) which should be preset. A comprehensive approach is to perform a two-dimensional search on all plausible combinations of (s0, s1), and then to select an optimal model based on cross-validation. However, this approach can be time-consuming and inconvenient to use. Usually, the slab scale s1 has little influence on the fitted model, while the spike scale s0 can strongly affect the model performance. Similar to the approach of Ročková and George (2014, 2016), therefore, our strategy is to fix the slab scale s1 to be relatively large, and select an optimal spike scale s0 from a reasonable range, e.g. (0, 0.1), in real data analysis. One also can use the lasso model to guide the selection of s0. We found that the optimal value of s0 is usually close to sλ (=1/λ) obtained from the lasso. These findings can largely simplify our selection of the optimal spike scale.
The proposed spike-and-slab lasso approach may be easily extended to frameworks beyond normal linear regression and Cox survival models. We have already extended the spike-and-slab lasso to generalized linear models (Tang et al. 2017). With the spike-and-slab double-exponential prior, the conditional posterior expectations of missing values (i.e. the indicator variable γj and the scale Sj) depend only on the coefficients β, and thus the E-step developed can be applied to other models. Another important avenue for future research will be incorporating external information about the importance of predictors and the relationship between predictors into the proposed spike-and-slab lasso framework. The spike-and-slab mixture priors provide flexible and easy ways to incorporate structural information about the predictors into predictive modeling. These extensions would be essential for effectively integrating important biological information, and multi-level molecular profiling data.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank the three reviewers and the associate editor for their constructive suggestions and comments that have improved the manuscript.
Funding
This work was supported by the National Institutes of Health [NIH 2 R01GM069430, R03-DE024198, R03-DE025646], and was also supported by grants from China Scholarship Council, the National Natural Science Foundation of China [81573253], and project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions at Soochow University.
Conflict of Interest: none declared.
References
- Barillot E. et al. (2012) Computational Systems Biology of Cancer. Chapman & Hall/CRC Mathematical & Computational Biology. [Google Scholar]
- Bonato V. et al. (2011) Bayesian ensemble methods for survival prediction in gene expression data. Bioinformatics, 27, 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bovelstad H.M. et al. (2007) Predicting survival from microarray data–a comparative study. Bioinformatics, 23, 2080–2087. [DOI] [PubMed] [Google Scholar]
- Bovelstad H.M. et al. (2009) Survival prediction from clinico-genomic models–a comparative study. BMC Bioinform., 10, 413.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow N. (1974) Covariance analysis of censored survival data. Biometrics, 30, 89–99. [PubMed] [Google Scholar]
- Breslow N.E. (1972) Contribution to the discussion of the paper by D.R. Cox. J. R. Stat. Soc. B, 34, 216–217. [Google Scholar]
- Chin L. et al. (2011) Cancer genomics: from discovery science to personalized medicine. Nat. Med., 17, 297–303. [DOI] [PubMed] [Google Scholar]
- Chipman H. (1996) Bayesian variable selection with related predictions. Can. J. Stat, 24, 17–36. [Google Scholar]
- Chipman H. et al. (2001) The practical implementation of Bayesian model selection In: Lahiri P. (ed), Model Selection. Institute of Mathematical Statistics, Beachwood, Ohio. [Google Scholar]
- Collins F.S., Varmus H. (2015) A new initiative on precision medicine. N. Engl. J. Med., 372, 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox D.R. (1972) Regression models and life tables. J. R. Stat. Soc., 34, 187–220. [Google Scholar]
- Efron B. (1977) The efficiency of Cox's likelihood function for censored data. J. Am. Stat. Assoc., 72, 557–565. [Google Scholar]
- Friedman J. et al. (2010) Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw., 33, 1–22. [PMC free article] [PubMed] [Google Scholar]
- George E.I., McCulloch R.E. (1993) Variable selection via Gibbs sampling. J. Am. Stat. Assoc., 88, 881–889. [Google Scholar]
- George E.I., McCulloch R.E. (1997) Approaches for Bayesian variable selection. Statistica Sinica, 7, 339–373. [Google Scholar]
- Gerstung M. et al. (2015) Combining gene mutation with gene expression data improves outcome prediction in myelodysplastic syndromes. Nat. Commun., 6, 5901.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie T. et al. (2015) Statistical Learning with Sparsity—the Lasso and Generalization. CRC Press, New York. [Google Scholar]
- Ibrahim J. et al. (2001) Bayesian Survival Analysis. Springer, New York. [Google Scholar]
- Klein J., Moeschberger M. (2003) Survival Analysis. Springer, New York. [Google Scholar]
- Kyung M. et al. (2010) Penalized regression, standard errors, and Bayesian lassos. Bayesian Anal., 5, 369–412. [Google Scholar]
- Lee K.H. et al. (2011) Bayesian variable selection in semiparametric proportional hazards model for high dimensional survival data. Int. J. Biostat., 7, 21. [Google Scholar]
- Lee K.H. et al. (2015) Survival prediction and variable selection with simultaneous shrinkage and grouping priors. Stat. Anal. Data Min., 8, 114–127. [Google Scholar]
- Li F., Zhang N.R. (2010) Bayesian variable selection in structured high-dimensional covariate spaces with applications in genomics. J. Am. Stat. Assoc., 105, 1202–1214. [Google Scholar]
- Li Y. et al. (2016) Regularized Parametric Regression for High-dimensional Survival Analysis. Siam International Conference on Data Mining pp. 765–773.
- Mittal S. et al. (2013) Large-scale parametric survival analysis. Stat. Med., 32, 3955–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monni S., Li H. (2010) Bayesian methods for network-structured genomics data. In: Chen,M-H. et al. (eds) Frontiers of Statistical Decision Making and Bayesian Analysis In Honor of James O Berger. Springer, New York.
- Park T., Casella G. (2008) The Bayesian Lasso. J. Am. Stat. Assoc., 103, 681–686. [Google Scholar]
- Peng B. et al. (2013) An integrative framework for Bayesian variable selection with informative priors for identifying genes and pathways. PLoS One, 8, e67672.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ročková V., George E.I. (2014) EMVS: the EM approach to Bayesian variable selection. J. Am. Stat. Assoc., 109, 828–846. [Google Scholar]
- Ročková V. et al. (2016) Bayesian penalty mixing: the case of a non-separable penalty In: Frigessi A.et al. (eds.), Statistical Analysis for High-Dimensional Data: The Abel Symposium 2014. Cham, Springer International Publishing, pp. 233–254. [Google Scholar]
- Simon N. et al. (2011) Regularization paths for Cox's proportional hazards model via coordinate descent. J. Stat. Softw., 39, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn I., Sung C.O. (2013) Predictive modeling using a somatic mutational profile in ovarian high grade serous carcinoma. PLoS One, 8, e54089.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingo F. et al. (2010) A Bayesian graphical modeling approach to microRNA regulatory network inference. Annal. Appl. Stat., 4, 2024–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai F. et al. (2009) Bayesian variable selection in regression with networked predictors. University of Minnesota Biostatistics Technical Report. http://www.sph.umn.edu/faculty1/wpcontent/uploads/2012/11/rr2009-008.pdf.
- Tang Z. et al. (2017) The spike-and-slab lasso generalized linear models for prediction and associated genes detection. Genetics, 205, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R. (1996) Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B, 58, 267–288. [Google Scholar]
- Tibshirani R. (1997) The lasso method for variable selection in the Cox model. Stat. Med., 16, 385–395. [DOI] [PubMed] [Google Scholar]
- Tibshirani R.J., Efron B. (2002) Pre-validation and inference in microarrays. Stat. Appl. Genet. Mol. Biol., 1, 1–18. [DOI] [PubMed] [Google Scholar]
- van de Vijver M.J., et al. (2002) A gene-expression signatureas a predictor of survival in breast cancer. N. Engl. J. Med., 347, 1999– 2009. [DOI] [PubMed] [Google Scholar]
- van Houwelingen H.C. et al. (2006) Cross-validated Cox regression on microarray gene expression data. Stat. Med., 25, 3201–3216. [DOI] [PubMed] [Google Scholar]
- van Houwelinggen H.G., Putter H. (2012) Dynamic Prediction in Clinical Survival Analysis. CRC Press, Boca Raton. [Google Scholar]
- Van’T Veer L.J. et al. (2002) Gene expression profiling predicts clinical outcome of breast cancer. Nature, 415, 530–536. [DOI] [PubMed] [Google Scholar]
- Yi N., Xu S. (2008) Bayesian LASSO for quantitative trait loci mapping. Genetics, 179, 1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y. et al. (2014) Assessing the clinical utility of cancer genomic and proteomic data across tumor types. Nat. Biotechnol., 32, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W. et al. (2013) Network-based survival analysis reveals subnetwork signatures for predicting outcomes of ovarian cancer treatment. PLoS Comput. Biol., 9, e1002975.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q. et al. (2015) Combining multidimensional genomic measurements for predicting cancer prognosis: observations from TCGA. Brief. Bioinform., 16, 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H., Hastie T. (2005) Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B, 67, 301–320. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.