

Abstract
Summary
Here we present open-source software for the analysis of high-dimensional cytometry data using state of the art algorithms. Importantly, use of the software requires no programming ability, and output files can either be interrogated directly in CymeR or they can be used downstream with any other cytometric data analysis platform. Also, because we use Docker to integrate the multitude of components that form the basis of CymeR, we have additionally developed a proof-of-concept of how future open-source bioinformatic programs with graphical user interfaces could be developed.
Availability and Implementation
CymeR is open-source software that ties several components into a single program that is perhaps best thought of as a self-contained data analysis operating system. Please see https://github.com/bmuchmore/CymeR/wiki for detailed installation instructions.
1 Introduction
In recent years, the complexity of cytometry data has quickly grown due to the advent of new technologies such as mass cytometry and the undertaking of large multi-center collaborations like the PRECISESADS project that gathers cytometry data from thousands of patients. Although a number of new algorithms have been proposed to help analyze either these high-dimensional datasets or datasets with large sample sizes, access to these tools generally require either programming abilities or paid-subscriptions to proprietary software as a recent review of computational cytometry has pointed out (Saeys et al., 2016). We have therefore developed open-source software for the analysis of cytometry data using state of the art algorithms. Importantly, CymeR provides the user a simple graphical user interface (GUI), however, the code underlying the functions is easily accessible, and an interface to RStudio and Jupyter Notebook pre-installed with many R and Python packages is also made available. In addition, because we use the KNIME data analysis framework (Berthold et al., 2008) as the graphical front-end to CymeR, the user can apply a number of transformations and data mining techniques on their data outside of the functions we have written. KNIME also provides seamless integration with the Business Intelligence and Reporting Tool (BIRT) for the creation of sophisticated reports.
2 Available functionality
2.1 Pre-analysis of cytometry files
CymeR expects files to be in the FCS format, however, if they are in the LMD format there is a function provided to convert from LMD files to FCS files. In addition, many of the same functions that can be performed in proprietary software such as FlowJo can also be performed in CymeR such as compensation, transformations, quality assessment and others. While we provide a large number of pre-analysis functions that other software lack, however, such as row down-sampling using CUR matrix decomposition (Mahoney et al., 2009) and detection of anomalous events using flowAI (Monaco et al., 2016), we realize that traditional cytometric analysis software is highly optimized for operations such as manual gating, which we do not try to supplant. Thus, we provide functions to import and export data to and from FlowJo and Cytobank (Chen et al., 2014) through a graphical front-end to flowWorkspace (Finak et al., 2012) and CytoML, respectively.
2.2 State-of-the-art analysis of cytometry files
CymeR is mainly designed for the analysis of high-dimensional data. This high-dimensionality can either be dozens of antibodies as is the case with CyTOF data or thousands of files as is the case with large multi-center flow cytometry experiments. In the first case, we currently provide access to Cytofkit (Becher et al., 2014), Destiny (Angerer et al., 2015), FlowSOM (Van Gassen et al., 2015), PhenoGraph (Levine et al., 2015), SCAFoLLD (Spitzer et al., 2015), SPADE (Qiu et al., 2011), t-SNE (van der Maaten, 2008), VorteX (Samusik et al., 2016) and Wishbone (Setty et al., 2016), and in the latter case we have provided a simple GUI for OpenCyto (Finak et al., 2014). For almost all CymeR functions, the main output is a new FCS file with new columns corresponding to new dimensions, cluster membership or whatever else is applicable. Thus, one could, for example, run t-SNE and then Destiny and the final file would contain eight new columns: Both t-SNE dimensions and the first five diffusion components found by Destiny, which could be subsequently visualized in a CymeR 2D/3D scatterplot (Fig. 1) or another program that accepts FCS files as input. In addition, the resulting FCS expression matrix can easily be written out as a CSV or XLS file for further analysis.
Fig. 1.
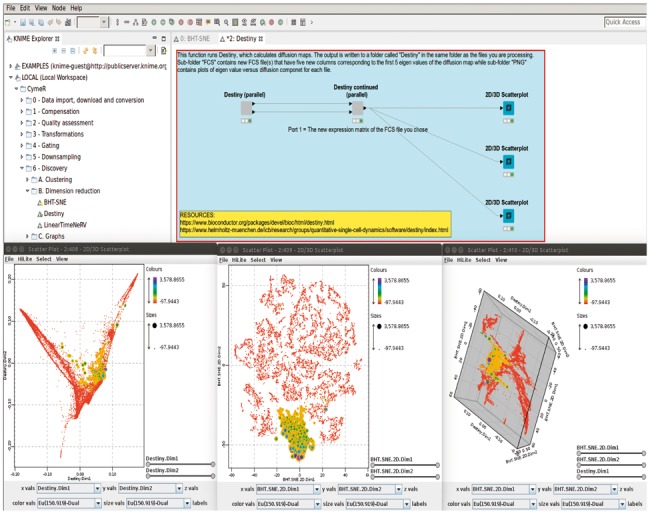
A screen shot showing the CymeR interface for Destiny on the top and three CymeR scatterplots of the same mass cytometry data colored and sized by a single attribute on the bottom. The bottom-left scatterplot shows the data represented by the first two diffusion components found by the Destiny algorithm while the bottom-center scatterplot shows the data represented by two t-SNE dimensions. The bottom-right scatterplot shows the data represented by the two t-SNE dimensions in the x and y dimensions and by the first diffusion component in the z dimension. The bottom-center scatterplot was also highlighted for cells positive for the chosen attribute, which immediately propagated to all other views of the same data (Color version of this figure is available at Bioinformatics online.)
2.3 New functionality and parallel implementation
We have included Neighbor Retrieval Visualizer (NeRV) (Venna et al., 2010), which is conceptually related to t-SNE, but which has the potential to give an alternate view of the analyzed data and therefore new insight. Also, while a few programs such as OpenCyto and SPADE allow the use of multiple cores during analysis, most programs remain confined to the use of a single core. Thus, we have coded most of the functions to utilize the parallelization offered by R's foreach package. Therefore, for many of our functions as many files can be processed at a time as the computer has cores, which greatly facilitates analysis of large multi-file experiments.
2.4 Data mining, visualization and report generation using KNIME
The use of KNIME as CymeR’s graphical front-end has the added benefit that once the FCS data is loaded into CymeR any of KNIME's 1000+ functions can be easily applied. In addition, KNIME provides a host of refined visualization tools such as a conditional box plot, a parallel coordinates viewer, a radar plot viewer and many more. Furthermore, KNIME is tightly integrated with BIRT, which allows easy report generation of results from within CymeR.
3 Discussion and conclusion
We have attempted to further the democratization of cytometric analysis by providing a simple, intuitive and powerful open-source GUI. Thus, our target audiences are the biologists with limited resources or programming ability who wants to apply new and innovative algorithms on their own data. We hope though, that our program can still be of great use to the immunobioinformatician by providing transparent access to the underlying code and easy access to the underlying packages through RStudio and Jupyter Notebook. We also provide a compelling proof-of-concept of how a graphical, multi-faceted program can be built by leveraging rich bioinformatic ecosystems like R/Bioconductor (Gentleman et al., 2004) with custom code and then subsequently distributed using the power of software compartmentalization provided by Docker containers.
Acknowledgements
We would like to thank Concepción Marañón and Olivia Santiago for feedback.
Funding
Funding has been provided by the Innovative Medicines Initiative and the pharmaceutical EFPIA members UCB, Bayer, Eli Lilly, IRIS Services and Genzyme Sanofi for the project PRECISESADS 6A #115565.
Conflict of Interest: none declared.
References
- Angerer P. et al. (2015) Destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics, 25, 2078–2079. [DOI] [PubMed] [Google Scholar]
- Becher B. et al. (2014) High-dimensional analysis of the murine myeloid cell system. Nat. Immunol., 12, 1181–1189. [DOI] [PubMed] [Google Scholar]
- Berthold M.R. et al. (2008) KNIME: the Konstanz information miner In: Preisach C., et al (eds.) Data Analysis, Machine Learning and Applications: Studies in Classification, Data Analysis, and Knowledge Organization, vol. 1 Springer, Berlin, Heidelberg, pp. 319–326. [Google Scholar]
- Chen T.J. et al. (2014) Cytobank: providing an analytics platform for community cytometry data analysis and collaboration. Curr. Top. Microbiol. Immunol., 377, 127–157. [DOI] [PubMed] [Google Scholar]
- Finak G. et al. (2012) QUAliFiER: an automated pipeline for quality assessment of gated flow cytometry data. BMC Bioinformatics, 13, 252.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G. et al. (2014) OpenCyto: an open source infrastructure for scalable, robust, reproducible, and automated, end-to-end flow cytometry data analysis. PLoS Comput. Biol., 10, 8.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman R.C. et al. (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol., 5, 10.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine J.H. et al. (2015) Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell, 1, 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney M.W. et al. (2009) CUR matrix decompositions for improved data analysis. Proc. Natl. Acad. Sci. U. S. A., 3, 697–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco G. et al. (2016) flowAI: automatic and interactive anomaly discerning tools for flow cytometry data. Bioinformatics, 32, 2473–2480. [DOI] [PubMed] [Google Scholar]
- Qiu P. et al. (2011) Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat. Biotechnol., 29, 886–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeys Y. et al. (2016) Computational flow cytometry: helping to make sense of high-dimensional immunology data. Nat. Rev. Immunol., 16, 449–462. [DOI] [PubMed] [Google Scholar]
- Samusik N. et al. (2016) Automated mapping of phenotype space with single-cell data. Nat. Methods, 6, 493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setty M. et al. (2016) Wishbone identifies bifurcating developmental trajectories from single-cell data. Nat. Biotechnol., 34, 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer M.H. et al. (2015) An interactive reference framework for modeling a dynamic immune system. Science, 349, 6244.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L. (2008) Accelerating t-SNE using Tree-Based Algorithms. J. Mach. Learn. Res., 15, 3221–3245. [Google Scholar]
- Van Gassen S. et al. (2015) FlowSOM: using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A, 7, 636–645. [DOI] [PubMed] [Google Scholar]
- Venna J. et al. (2010) Information retrieval perspective to nonlinear dimensionality reduction for data visualization. J. Mach. Learn. Res., 11, 451–490. [Google Scholar]