

Abstract
Antigen-specific treatments are highly desirable for autoimmune diseases in contrast to treatments which induce systemic immunosuppression. A novel antigen-specific therapy has been developed which, when administered semi-therapeutically, is highly efficacious in the treatment of the mouse model for multiple sclerosis, namely experimental autoimmune encephalomyelitis (EAE). The treatment uses dual-sized, polymeric microparticles (dMPs) loaded with specific antigen and tolerizing factors for intra- and extra-cellular delivery, designed to recruit and modulate dendritic cells toward a tolerogenic phenotype without systemic release. This approach demonstrated robust efficacy and provided complete protection against disease. Therapeutic efficacy required encapsulation of the factors in controlled-release microparticles and was antigen-specific. Disease blocking was associated with a reduction of infiltrating CD4+ T cells, inflammatory cytokine-producing pathogenic CD4+ T cells, and activated macrophages and microglia in the central nervous system. Furthermore, CD4+ T cells isolated from dMP-treated mice were anergic in response to disease-specific, antigen-loaded splenocytes. Additionally, the frequency of CD86hiMHCIIhi dendritic cells in draining lymph nodes of EAE mice treated with Ag-specific dMPs was reduced. Our findings highlight the efficacy of microparticle-based drug delivery to mediate antigen-specific tolerance, and suggest that such a multi-factor combinatorial approach can act to block autoimmunity.
Keywords: PLGA-Microparticles, drug delivery, Ag-specific, immunotherapy, EAE, multiple sclerosis
1. Introduction
Multiple sclerosis (MS) is an immune-mediated neurological disease that typically affects young adults with higher prevalence in females [1-3]. MS is a complex inflammatory disease of the central nervous system (CNS) where immune cells target and destroy oligodendrocytes and myelin sheath on nerve cells causing autoimmune demyelination [4, 5]. The precise instigating factor(s) that initiates MS remains unknown [4], but it is well established that proinflammatory CD4+ T cells are important in mediating MS pathogenesis, as well as that of experimental autoimmune encephalomyelitis (EAE), an animal model of MS [6]. Blood circulating CD4+ T cells from MS patients have been shown to recognize myelin oligodendrocyte glycoprotein (MOG) and myelin basic protein (MBP), two myelin-associated proteins shown to play a role in MS pathogenesis and used as basis for EAE induction [7-9]. Several subsets of proinflammatory CD4+ T cells have been implicated as crucial drivers of EAE, namely Th17 and Th1 cells. Th17 cells are CD4+ T cells that express the lineage transcription factor Rorγt and produce the proinflammatory cytokines IL-17A, IL-17F, and, in the setting of EAE, GM-CSF [10-14], while Th1 cells express the lineage transcription factor T-bet and produce the proinflammatory cytokine IFNγ, and were also demonstrated to be important in EAE disease pathogenesis [15]. Defects in Th17 and Th1 cells or GM-CSF production prevented disease in EAE, thus solidifying the central role of proinflammatory CD4+ T cells and the corresponding cytokines IL-17A, IFNγ, and GM-CSF in EAE [13, 15].
MS does not have a cure and current therapeutic options are limited. In the acute setting of MS exacerbation/relapse, methylprednisolone or other corticosteroids are used to provide immunosuppression [16]. Long-term management of MS involves disease-modifying therapies that may be poorly tolerated, inadequate in controlling disease, or incur life-threatening side effects and opportunistic infections [17]. Type 1 interferon monotherapy or in combination with glatiramer acetate is generally the safest and most tolerated therapy but often with inadequate response rate [18]. Natalizumab, a monoclonal antibody targeting the α4 integrin, thereby blocking leukocyte trafficking into the CNS, is highly effective in relapsing-remitting MS treatment, but it may cause progressive multifocal leukoencephalopathy (PML), a life-threatening opportunistic viral infection of the CNS [19]. Fingolimod, a sphingosine-1-phosphate receptor modulator, has been approved for oral treatment of MS, but it has multiple severe side effects, among which are PML and other infections [20]. Immunosuppressive regimens have also been used in MS treatment, including the purine analogue Cladribine, the immune modulator Laquinimod, anti-CD52 monoclonal antibody Alemtuzumab, anti-CD25 monoclonal antibody Daclizumab, and anti-CD20 monoclonal antibody Rituximab, but all have side effects related to immune suppression [16, 21, 22].
The goal in MS treatment is to develop therapies that induce tolerization of antigen-specific CD4+ T cells, without generalized non-specific immune suppression. Such a goal can be achieved by generation of tolerogenic dendritic cells (DCs), known to exhibit ‘semi-mature’ profiles and induce antigen-specific T cell anergy or generation of regulatory T (Treg) cells [23]. Exogenous generation of autologous DCs has been a major focus of the field, yet has numerous limitations including high cost, poor yield, and inefficient DC homing to regional lymph nodes upon re-administration [24-27]. As an alternative, polymeric particle vaccines are being pursued for the targeted delivery of conditioning factors to DCs in vivo [28-30]. Desirable key features of particle vaccines for immunotherapy include: control over phagocytosability, delivery of antigen to DCs, and local release of desired agents. Numerous materials have been investigated in particle-based biodegradable drug delivery, with a number of groups investigating vaccines consisting of antigen-loaded particles to target DCs [31-36]. Microparticles (MPs) fabricated from poly(lactic-co-glycolic acid) (PLGA) have been the most investigated vehicles for delivering immunotherapeutics. PLGA is approved by the U.S. Food and Drug Administration for use in numerous applications such as biodegradable surgical sutures and drug delivery products, which makes it extremely attractive for development of products quickly translatable to clinical use. PLGA is degraded in the body via bulk erosion and hydrolysis. PLGA MPs of ∼1 μm are phagocytosed efficiently by antigen presenting cells (APCs), providing directed delivery of antigens for immune recognition [37]. Following phagocytosis by APCs, sufficient levels of endosomal release of encapsulated antigens from PLGA microparticles have been demonstrated to generate both MHC II-directed, as well as MHC I-directed immune response through cross-presentation [38-40]. Given this capability along with a long-standing safety history, PLGA particles are therefore an excellent candidate as a carrier vehicle for vaccines utilizing encapsulated antigenic proteins or peptides along with immunomodulatory agents with intracellular targets.
We have developed a novel combinatorial dual-sized MP system (dMP), encapsulating, in addition to specific antigens, agents selected for their capacity to modulate DC function: both through intracellular delivery of agents encapsulated in small (∼1 μm) MPs, and subcutaneous local deposition of agents for controlled release in MPs too large to phagocytose (∼50 μm) [41, 42]. Our dMP system combines the attractive notion of antigen-specificity and combination therapy with our dual-sized controlled release scheme to provide immune modulation without systemic delivery. Specifically two sizes of MPs are used in the dual MP (dMP) system (Fig. 1): (1) phagocytosable ∼1 μm MPs for delivery of antigen (Ag) and drugs to intracellular targets within phagocytes, and (2) non-phagocytosable ∼50 μm MPs for controlled release of factors targeted to cell surface receptors in a localized microenvironment. Two phagocytosable MPs are used: (a) MPs encapsulating MS-specific antigens, myelin oligodendrocyte glycoprotein peptide (MOG35-55), or as a control, the irrelevant OVA323-339 peptide, and (b) MPs loaded with vitamin D3 (VD3). Two non-phagocytosable MPs are used: (c) MPs encapsulating TGF-β1, and (d) MPs encapsulating GM-CSF. These four MPs are mixed in equal mass and administered subcutaneously.
Figure 1. Dual microparticle (dMP) fabrication and characterization.
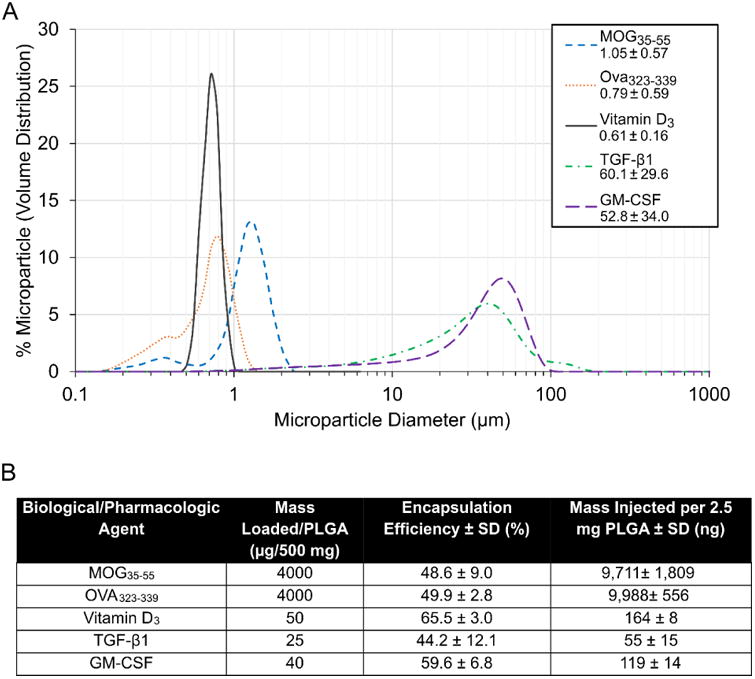
(A) Microparticle sizes are quantified by dynamic light scattering analyses with average ∼0.8 μm for vitamin D3, MOG35-55, and OVA323-339 MPs, and average ∼55 μm for TGF-β1 and GM-CSF MPs. (B) Loading amount, encapsulation efficiency, and dose per injection for MPs containing each biological/pharmacological agent.
The active metabolite of vitamin D3 (VD3) 1α,25(OH)2D3 has been shown to induce tolerogenic DCs through oxidative and glycolytic metabolic pathways, where glucose, glycolysis, and PI3K/Akt/mTOR are essential, and is intracellularly delivered to its nuclear receptor [43, 44]. Transforming growth factor-beta 1 (TGF-β1) is loaded into the 50 μm MPs to provide extracellular release to target its receptor on the DC surface. TGF-β1 treated-DCs show reduced expression of MHC II, costimulatory molecules and inflammatory cytokines, as well as increased production of IDO [45]. Exposure of T cells to TGF-β1 treated-DCs results in the induction of antigen-specific Treg cells, as well as deletion of antigen-reactive effector T cells [46]. Granulocyte-macrophage colony-stimulating factor (GM-CSF) is also separately loaded into a 50 μm MP for extracellular release, to locally attract and sustain DCs, as has been reported in a cancer vaccine approach [47]. Although it is the case that in EAE, GM-CSF is produced by pathogenic CD4+ T cells localized in the CNS [13, 14], and can potentiate activation of innate leukocytes [48], in low doses and provided locally, GM-CSF s a critical mediator in the differentiation and development of tolerogenic DCs with low expression of MHC-II/CD80/CD86, as well as low inflammatory cytokines, and increased PD-L1 expression [48-50]. In sum, the four factors are loaded into separate MPs, with those targeting intracellular pathways in phagocytosable MPs and those targeting surface receptors in non-phagocytosable MPs, hence, dual-sized. We have recently demonstrated the utility of a similar formulation of this multi-factor combinatorial dMP system in the early prevention of autoimmune type 1 diabetes in NOD mice, where a diabetes disease-relevant antigen insulin was incorporated [41].
Myelin antigen-coupled PLG sub-micron particles have previously been used in preventive and therapeutic regimens for relapsing/remitting EAE by intravenous administration, but was inefficient when administered subcutaneously [51]. Alternatively, intraperitoneally co-administered nanoparticles formulated with myelin antigen and aryl hydrocarbon receptor ligands promoted tolerogenic DCs and mitigated EAE, with expansion of Treg cells [52]. In testing treatment strategies for EAE, preventative/prophylactic treatment has been used when the treatment agents are administered to animals before EAE disease induction, therapeutic treatment is applied when agents are given to animals after appearance of clinical EAE disease signs, and semi-therapeutic regimen is used when treatment agents are administered to animals after EAE disease induction but before clinical disease signs [51, 53]. The combinatorial multi-factor dMP treatment investigated here offers the advantage of a subcutaneous localized administration, as opposed to systemic administration, with low-dose, localized, controlled release of specific factors designed to be retained at the injection site. We show that this dMP approach does not result in an increase of the tolerogenic factors systemically, efficiently treats EAE in a semi-therapeutic regimen, and is antigen-specific.
2. Materials and methods
2.1 Microparticle fabrication
Microparticles (MPs) were fabricated by standard oil-in-water single emulsion or water-in-oil-in-water double emulsion methods, as described previously [41]. All drugs were encapsulated in distinct MPs, as no two factors were loaded simultaneously. A 50:50 copolymer of poly(lactic-co-glycolic acid) (PLGA; MW ∼44,000 g/mol; Corbion Purac, Gorinchem, Netherlands) in methylene chloride (Themo Fisher Scientific, NJ, USA) was used to generate MPs. Ultrapure water (Barnstead GenPure, Thermo Fisher Scientific) was used as the aqueous phase, with dissolved surfactant, poly-vinyl alcohol (PVA; MW ∼15,000 g/mol; Themo Fisher Scientific), to stabilize the emulsions.
Phagocytosable MPs were fabricated by dissolving 500 mg of PLGA in methylene chloride at a 5% w/v ratio. 50 μg of vitamin D3 (Cayman Chemical) in 1 mL of methanol (Thermo Fisher Scientific) was loaded directly into the methylene chloride/PLGA solution and set to shake at 150 rpm for 10 mins. This solution was then added to 50 mL of 5% w/v PVA and homogenized at 35,000 rpm for 180 s using a tissue-miser homogenizer (Thermo Fisher Scientific) to form an oil-in-water emulsion. The microparticle solution was subsequently added to a beaker of 100 mL stirring 1% PVA and set to stir for 4-6 h for solvent evaporation and microparticle hardening to occur. For water-soluble MOG35-55- (Mimotopes, Victoria, Australia) and OVA323-339-encapsulated (Mimotopes) MPs, 4 mg of peptide in 200 μL PBS was added to the 5% methylene chloride/PLGA solution and homogenized at 35,000 rpm for 120 s to form a primary emulsion. This emulsion was added to 50 mL of 5% PVA and homogenized again at 35,000 rpm for 180 s to form the secondary emulsion, and added to the 100 mL of stirring 1% PVA.
Non-phagocytosable MPs encapsulating TGF-β1 and GM-CSF were fabricated by first dissolving 500 mg of PLGA in methylene chloride at a 20% w/v ratio. Human TGF-β1 (Peprotech) was reconstituted in 10 mM hydrochloric acid and 2 mg/ml bovine serum albumin in 250 μL PBS and recombinant mouse GM-CSF (Biolegend) was reconstituted in 400 μL PBS. Protein solutions were added to the methylene chloride/PLGA solution and vortexed (Thermo Fisher Scientific) at the highest setting (∼3,200 rpm) for 30 s to generate the primary emulsion. This emulsion was added to 5 mL of 2.5% PVA and vortexed again at 3,200 rpm for 60 s to form the secondary emulsion, and added to the 100 mL of stirring 1% PVA. Either methanol or PBS was used to generate unloaded MPs, depending on the control group being fabricated.
After 4-6 h, solutions were centrifuged at 10,000 ×g for 10 min to collect MPs and washed three times with ultrapure water. The resultant MPs were then flash-frozen in liquid nitrogen and lyophilized for 24 h. The MPs were stored at − 20 °C until their use.
2.2 Microparticle characterization
The size distributions of MPs were measured by the Beckman Coulter LS13320 (Beckman Coulter Inc., Brea, CA) and the Microtrac Nanotrac Dynamic Light Scattering Particle Analyzer (Microtrac, Montgomery, PA). The MP diameter is reported as mean ± standard deviation and displayed as a volume percentage.
Encapsulation efficiencies of proteins/peptides was assessed by μBCA (Thermo Fisher Scientific). Briefly, a known mass of MPs, as determined by the working range of the μBCA assay, was dissolved in a 0.2 M NaOH/5% sodium dodecyl sulfate (SDS) solution. An analogous process with unloaded MPs and soluble drug was performed. Solutions were neutralized with a small volume of HCl and protein/peptide concentration measured by μBCA. Serial dilutions of the unloaded MP/soluble drug solution determined the encapsulation efficiency. Vitamin D3 MPs were measured by dissolving 100 mg of MPs into 2 ml MC and re-precipitating the PLGA with a known volume of methanol. The suspension was centrifuged and the supernatant removed to a new tube. Following evaporation, residue remaining in the tube was concentrated in a known, small quantity of DMSO and the solution concentration measured by spectrophotometer.
2.3 C57BL/6 mice
C57BL/6 mice (B6NTac) were purchased from Taconic Biosciences. All animals were housed in specific pathogen free conditions in the Animal Care Service facilities of the University of Florida College of Medicine (UFCOM). All experiments were conducted on 8-20-week old male or female mice according to protocols approved by Institutional Animal Care and Use Committee at the University of Florida.
2.4 Site of injection analysis and microparticle trafficking
Characterization of nodules at the site of injection was carried out via flow cytometry and H&E staining. Initial studies characterizing DC recruitment and phenotype used a mixed cohort of 8-20-week-old male and female C57BL/6 mice. Animals were injected subcutaneously in the abdominal region using 20 G needles (BD Biosciences). MP injections consisted of 10 mg of MPs total (1:1:1:1 MP mass ratio) in 0.2 mL PBS. Nodules were excised 8 day after injection, enzymatically digested with 2 mg/mL collagenase type XI (Sigma-Aldrich, St. Louis, MO, USA) at 37 °C for 30 minutes, filtered through a 30 μm filter to remove large particulates, and cells stained for flow cytometry. For immunohistochemistry, nodules were fixed in 10% formalin overnight at 4 °C, processed and embedded in paraffin blocks and stained by the Molecular Pathology Core at the University of Florida.
Microparticle uptake in the nodule and trafficking to secondary lymphoid organs was assessed by loading the phagocytosable MPs concomitantly with Vybrant DiO (Invitrogen) fluorescent labelling dye and vitamin D3 or an irrelevant protein (denatured insulin). Non-drug loaded (unloaded) phagocytostable fluorescent MPs were also fabricated. Large, non-phagocytosable MPs were fabricated in the standard fashion, without the addition of fluorescent dye. At various timepoints (24 h, 48 h, and 8 d), after subcutaneous injection in the abdominal region, mice were euthanized and cells were isolated from various secondary lymphoid organs. Cells were stained with primary conjugated antibodies and analyzed via flow cytometry.
2.5 Evaluation of tolerogenic factors in the blood
A mixed cohort of 10 week old male and female C57BL/6 mice were injected subcutaneously in the abdominal region with dual-sized microparticle (dMP) formulation using 20 G needles (BD Biosciences). Blood was collected from submandibular vein of animals on days 2, 4, and 7 after subcutaneous dMP injection, processed for serum, and GM-CSF and TGF-β1 serum concentrations were measured using enzyme linked immunosorbent assay (ELISA) following manufacturer's protocol (BD Biosciences, cat# 555167, 559119). Negative control group was C57BL/6 mice with no treatment, and positive control group was C57BL/6 mice injected intravenously with 1/10 loading dose of GM-CSF and TGF-β1 immediately prior to blood collection. Absorbance was read at 450 nm using a SpectraMax M3 microplate reader (Molecular Devices) and serum concentrations of GM-CSF and TGF-β1 was calculated using a standard curve performed following manufacturer's protocol (BD Biosciences).
2.6 EAE induction
EAE was induced in 10-11-week old female C57BL/6 mice from Taconic Biosciences with Hooke Kit™ (Hooke Laboratories Inc., Cat# EK-2110). Briefly, 100 μL of MOG35-55/CFA emulsion was injected subcutaneously in each anterior back and posterior back for a total of 200 μL emulsion per mouse, according to manufacturer's protocol. Pertussis toxin (100 μL of 4 μg/mL) was injected intraperitoneally 2 hours and 24 hours following MOG35-55/CFA emulsion injection, according to manufacturer's protocol. Clinical scoring was established as follows: Score 1: flaccid tail, Score 2: weak hind limbs, Score 3: hind limb paralysis, Score 4: quadriplegia, Score 5: moribund, euthanasia.
2.7 dMP treatment in EAE mice
2.5 mg of each of the four MPs described in 2.1 were mixed for a total of 10 mg of dMP formulation per EAE mouse. dMPs were resuspended in 200 μL PBS per 10 mg of dMP. EAE mice were injected subcutaneously in the back between the two MOG35-55/CFA emulsion on the indicated days following EAE induction.
2.8 Isolation of mononuclear cells from CNS
Mononuclear cells were isolated from CNS as described previously [54, 55]. Briefly, cold PBS was used to perfuse CNS. Brain and spinal cord were homogenized with gentleMACS dissociator (Miltenyi Biotec), pressed through a 70 μm mesh, then suspended in final 30% isotonic Percoll (GE Healthcare) as described previously [54, 55]. This 30% isotonic Percoll solution containing homogenized CNS was layered on top of 70% isotonic Percoll and centrifuged for 30 min. at 500 ×g G as described previously [54, 55]. The 70%-30% interphase containing CNS mononuclear cells was collected then washed with 1× HBSS as described previously [54, 55]. Isolated mononuclear cells from CNS were subjected to fluorophore-conjugated antibody staining and flow cytometry analysis.
2.9 In vitro antigen re-stimulation assay
Total cells were isolated from draining lymph nodes of EAE mice treated with dMP MOG35-55 or dMP Ova323-339 then loaded with CFSE as described previously [56]. CFSE-loaded cells were then co-cultured with MOG35-55-loaded splenocytes isolated from congenic Rag1-/- mice for 72 hours. After 72 hours, cells were washed and surface stained with CD4, CD8, CD3, and analyzed by flow cytometry.
2.10 Antibodies
Cells were stained with the following antibodies: CD11b (APC, APC-eFluor 780, AF647, clone: M1/70), CD11c (Brilliant violet 650, PE-cyanine7, clone: N418, HL3), PD-L1 (CD274, B7-H1, Brilliant Violet 711, clone: 10F.9G2), B7-H2 (ICOS-L, CD275, eFluor 660, clone: HK5.3), CD272 (BTLA, PE, clone: 6A6), Fixable viability dye (eFluor 520, eFluor 780, FITC), IL-10 (PerCP-Cyanine5.5, clone: JES5-16E3), IL-27 p28 (PE-cyanine7, clone: MM27-7B1), Galectin-9 (Brilliant violet 421, clone: RG9-35), CD4 (Brilliant violet 711, clone: GK1.5), PD-1 (CD279, Brilliant violet 605, clone: 29F.1A12), Rorγt (APC, clone: AFKJS-9), T-bet (PE-cyanine7, clone: 4B10), IL-17a (eFluor 450, clone: eBio17B7), GM-CSF (PE, clone: MP1-22E9), IFNγ (FITC, clone: XMG1.2), Ly6C (eFluor 450, clone: HK1.4), Ly6G (GR-1, FITC, AF700, clone: 1A8-Ly6g), F4/80 (PE, clone: BM8), CD80 (Brilliant violet 605, clone: 16-10A1), CD86 (PE-cyanine7, Brilliant violet 605, clone: GL-1), MHC class II (I-A/I-E, PerCP-eFluor 710, clone: M5/114.15.2), CD25 (Brilliant violet 605, clone: PC61), HVEM (CD270, PE, clone: LH1), CD39 (PE-cyanine7, clone: 24DMS1), CD73 (eFluor 450, clone: TY/11.8), Foxp3 (eFluor 450, FITC, clone: FJK-16s), CTLA4 (CD152, APC, clone: UC10-4B9), GITR (CD357, PE-cyanine7, clone: DTA-1), CD103 (Integrin alpha E, APC, clone: 2E7), Lag-3 (CD223, PE, clone: eBioC9B7W), Granzyme B (FITC, clone: GB11), CD8a (PE, clone: 53-6.7), TCRβ (APC, clone: H57-597), CD3e (APC-cy7, clone: 145-2C11), CD45 (PE, APC-cyanine7, clone: 30-F11), and CD16/32 (FCγ III/II receptor, clone 2.4G2).
2.11 Intracellular/intranuclear staining
PMA/Ionomycin stimulation with Brefeldin A and intracellular/intranuclear staining were performed as described previously [55, 57]. Briefly, for detection of cytokines and transcription factors by intracellular/intranuclear flow cytometry, cells were cultured at 37°C and 5% CO2 for 4 hours in IMDM media (Gibco, Life Technologies) containing PMA (20 ng/ml) (Sigma) and Ionomycin (1 μg/ml) (Sigma). Brefeldin A (10 μg/mL) was added 1 hour following PMA/Ionomycin addition. Cells were washed and stained with Fixable Viability Dye (Affymetrix, Life Technologies) and surface markers following stimulation. Surface marker stained cells were fixed and permeabilized with Foxp3 Fix/Perm Kit (Affymetrix, Life Technologies) followed by cytokine and transcription factor staining as described previously [55, 57].
2.12 Flow cytometry and analysis
Flow cytometry was performed on a BD LSR II with BD FACS DIVA software for data acquisition (BD Biosciences). All flow cytometry data were analyzed with FlowJo software (Tree Star).
2.13. Statistical analysis
GraphPad Prism software version 5 was used for statistical analysis. Statistical significance was assessed by two-tailed unpaired Student's t tests for all analyses except figures 2H, 2I, 2K, 3B, and 8B where a one-way analysis of variance (ANOVA) with Tukey's post-hoc analysis was used. Statistical significance was considered when p < 0.05.
Figure 2. Subcutaneous DC recruitment and tolerization, and microparticle-associated cell trafficking to local lymph nodes in vivo.
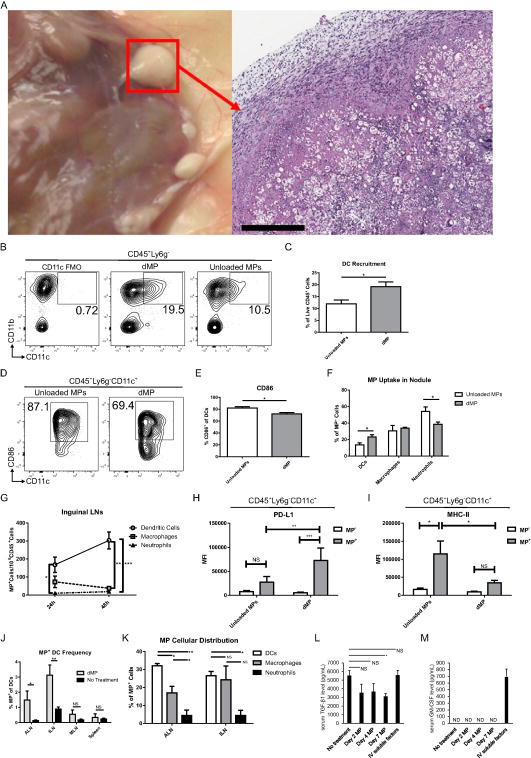
(A) Subcutaneous dMP injection sites (nodules) in B6 mice were excised 8 days after administration and characterized for cellular infiltration by hematoxylin and eosin staining; white bar represents 400 μm (B) Representative flow cytometry analysis of DC recruitment in dMP or unloaded MP nodules. A fluorescence minus one (FMO) control, containing all antibodies except CD11c, was assessed to determine gating of DCs. (C) Total frequency of DCs in respective nodules as a percent of live CD45+ cells. (D) Representative flow cytometry analysis for DC surface expression of co-stimulatory molecule CD86. (E) Total frequency of CD86+ expression on DCs isolated from respective nodules. (F) DC (Lyg6-CD11c+), macrophage (Lyg6-CD11c- CD11b+), and neutrophil (Lyg6+) MP uptake in dMP or unloaded MP nodules is characterized as a percentage of total phagcytosed MPs (CD45+DiO+). (G) Draining lymph nodes (inguinal) were excised at 24 and 48 h after dMP injection and analyzed for dMP+ phagocyte populations. (H) PD-L1 expression of DCs isolated from ILNs 24 h after MP injection and analyzed according to dMP or unloaded MP phagocytosis. (I) MHC-II expression of DCs isolated from ILNs 48 h after MP injection and analyzed according to dMP or unloaded MP phagocytosis. (J) Proximal draining lymph nodes (axillary [ALN] and inguinal [ILN]) and distal lymphoid organs (mesenteric lymph nodes [MLN] and spleen) were excised eight days after dMP injection and analyzed via flow cytometry for MP trafficking. Frequency of dMP+ DCs as a percent of total DCs is characterized in mice that received either no MPs (No Treatment) or the dMP. (K) dMP distribution across phagocyte populations at the eight day time point was characterized in proximal draining lymph nodes as a percent of total dMP + cells. (L) Serum TGF-β1 level (pg/mL) (M) Serum GM-CSF level (pg/mL) measured by ELISA from mice without treatment, mice days 2, 4, and 7 after subcutaneous dMP treatment, and mice with intravenous injection of TGF-β1 and GM-CSF immediately prior to blood collection. n=3-5 per group. p values were obtained from Student's t tests (C, E, F, G, and J) or one-way ANOVA with Tukey's post-hoc analysis (H, I, and K), * = p <0.05, ** = p < 0.01, *** = p < 0.001, n.s. = p >0.05 (not significant), ND (not detectable).
Figure 3. dMP-MOG35-55 formulation blocks experimental autoimmune encephalomyelitis in a semi-therapeutic treatment setting.
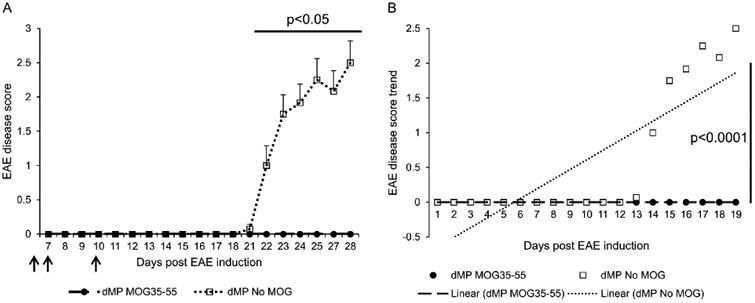
(A) EAE disease score (mean ± SEM) of B6 mice treated with either dMP MOG35-55 (filled circle) or dMP formulation without MOG35-55 (unloaded 0.8 μm MPs were substituted) (open square) on days 4, 7, and 10 (filled arrows) following EAE induction; n=10 per group. p value was obtained from Student's t test. (B) EAE disease score trend of B6 mice treated with either dMP MOG35-55 formulation (filled circle) or dMP formulation without MOG35-55 (open square) on days 4, 7, and 10 following EAE induction; n=10 per group. p value was obtained from ANOVA.
Figure 8. Efficacy of dMP treatment is dependent on antigen specificity.
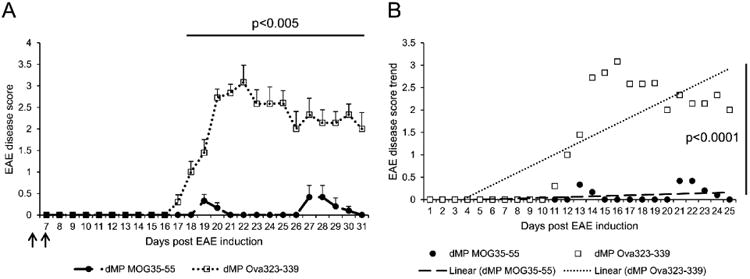
(A) EAE disease score (mean ± SEM) of B6 mice treated with either dMP MOG35-55 (filled circle) or dMP Ova323-339 (open square) on days 4 and 7 (filled arrows) following EAE induction; n=10 per group. p value was obtained from Student's t test. (B) EAE disease score trend of B6 mice treated with either dMP MOG35-55 (filled circle) or dMP Ova323-339 (open square) on days 4 and 7 following EAE induction; n=10 per group. p value was obtained from ANOVA.
3. Results
3.1 dMP fabrication and characterization
We have previously demonstrated key features and capabilities of the dMP system as a means of locally delivering loaded factors through large, non-phagocytosable MPs, and intracellularly-targeted factors to phagocytes through ∼1 μm diameter small MPs [42]. A similar formulation of the dMP system, encapsulating vitamin D3, TGF-β1, and GM-CSF was shown to prevent type 1 diabetes development in NOD mouse when co-delivered with MPs loaded with insulin peptide as antigen in small MPs [41]. This formulation has since been modified by increasing the loading of immunomodulatory factors. PLGA dMPs were fabricated using standard solvent evaporation methods to encapsulate factors. Following dMP fabrication, size distribution, loading amount, encapsulation efficiencies, and release profiles were quantified (Fig. 1). Consistent sizing of phagocytosable MPs, ∼0.8 μm-diameter, and non-phagocytosable MPs, ∼55 μm-diameter, irrespective of the drug loaded was demonstrated, highlighting the dual-sized nature of the dMP system (Fig. 1A). The encapsulation efficiencies for the small phagocytosable MPs were 48.6 ± 9.0%, 65.5 ± 3.0%, and 49.9 ± 2.8% for MOG35-55, vitamin D3, and Ova323-339 MPs, respectively (Fig. 1B) Comparable encapsulation efficiencies for the large non-phagocytosable MPs were observed, with 44.2 ± 12.1% and 58.3 ± 9.4% for TGF-β1 and GM-CSF MPs, respectively (Fig. 1B).
3.2. Subcutaneous dMP administration cause DC recruitment and tolerization, and microparticle-associated cell trafficking to local lymph nodes without systemic release
We investigated the capacity of the dMP formulation to recruit and tolerize DCs at the local injection site, and the ability of the cells that phagocytosed MPs to subsequently traffic to draining lymph nodes. Mice that received the dMP developed palpable nodules at the subcutaneous injection site a day after a single dMP injection. Surgical and histopathological analysis of the dMP nodules eight days after administration demonstrated high levels of proteinaceous deposition with significant nucleated cell infiltration surrounding the readily visible non-phagocytosable MPs (large white spheres) (Fig. 2A). Importantly, these nodules were resorbed within a month of injection as determined by palpation and surgical examination, approximately by the time the administered PLGA bolus completely degraded (data not shown). We assessed the composition of nodule-recruited cells by digestion of the nodule and flow cytometry analysis, and found that DC recruitment to the local subcutaneous MP nodule was improved when MPs were loaded with bioactive factors compared to unloaded MPs, with the total frequency of infiltrating DCs rising from 11.9% to 19.2% of total CD45+ cells (Fig. 2B-C). Furthermore, recruited DCs demonstrate characteristics of a non-activated phenotype, with decreased frequency of CD86+ DCs in the case of loaded MPs versus unloaded (Fig. 2D-E). Using fluorescently-loaded phagocytosable MPs in the dMP formulation, MP uptake in the nodule was assessed in phagocyte populations. Higher uptake of the phagocytosable dMP particles compared to unloaded MPs by DCs was evident, while the uptake of dMPs versus unloaded MPs was equivalent in macrophages and lower in neutrophils (Fig. 2F). Trafficking of phagocytes associated with microparticles (MP+ cells) was assessed in various peripheral lymphoid organs at multiple time points (Fig. 2G-K). At 24 and 48 h post-dMP injection, MP+ DCs were shown to drain to inguinal lymph nodes (ILNs) in the highest number compared to neutrophils (24 h) and both macrophages and neutrophils (48 h) (Fig. 2G). Notably, MP+ DCs isolated from ILNs 24 h after MP injection demonstrated upregulation of programmed death-ligand 1 (PD-L1) expression compared to dMP DCs or unloaded MP+ DCs, while PD-L1 expression between unloaded MP+ and unloaded MP- DCs remained unchanged (Fig. 2H). Similarly, dMP+ DCs isolated from ILNs 48 h after MP injection maintained immature phenotypes, whereas unloaded MP+ DCs significantly upregulated MHC-II compared to unloaded MP- DCs (Fig. 2I). At a later time point, eight days after dMP administration, MP+ DCs were present in proximal draining lymph nodes (axillary [ALNs] and ILNs), however not in distal lymphoid organs (mesenteric lymph nodes and spleen) (Fig. 2J), thus minimizing the potential for systemic immunosuppression. Upon further examination, MP+ DCs had the highest frequency in ALNs, followed by MP+ macrophages, while in ILNs the frequency of MP+ DCs and MP+ macrophages was equivalent (Fig. 2K). The frequency of MP+ neutrophils was low both in ALNs and ILNs. In addition, subcutaneously injection of MPs, did not result in serum elevation of TGF-β1 and GM-CSF at 2, 4, and 7 days compared to no treatment (Fig. 2L-M). These results demonstrate that this dMP system does not cause serum elevation of tolerogenic factors enclosed in MPs and subcutaneously delivered, suggesting that systemic immunosuppression is unlikely. In sum, these proof-of-concept studies emphasize the feasibility of this platform to modulate DC recruitment and phenotype, as well as the distribution of the MP-loaded cells proximally, but not into the distal lymphoid organs or systemically.
3.3 dMP-MOG35-55 formulation blocks experimental autoimmune encephalomyelitis in a semi-therapeutic treatment setting
We examined whether the dMP system formulated with the antigenic MOG35-55 peptide (dMP-MOG35-55) can be used to treat EAE, the mouse model for MS. Specifically, the dMP-MOG35-55 formulation, consisting of non-phagocytosable TGF-β1 and GM-CSF MPs and phagocytosable vitamin D3 and MOG35-55 MPs (dMP-MOG35-55), was used. The dMP formulation without MOG35-55 consisting of soluble TGF-β1, GM-CSF, vitamin D3, and unloaded phagocytosable MPs was used as control (dMP No MOG). The dMP-MOG35-55 treatment and the corresponding control was administered subcutaneously on days 4, 7, and 10, following EAE induction in C57BL/6 mice. Our results show that EAE mice treated with the dMP-MOG35-55 developed minimal EAE scores in significant contrast to EAE mice treated with the dMP No MOG (Fig. 3A). Linear regression analysis of EAE disease score development revealed no disease development in EAE mice treated with dMP-MOG35-55 compared to positive disease progression in EAE mice treated with dMP without MOG35-55 (Fig. 3B). Thus, these results demonstrate that early administration dMP-MOG35-55, after disease induction, is highly efficacious in the prevention of EAE disease.
3.4 EAE mice treated with dMP-MOG35-55 have reduced leukocytes and CD4+ T cells in filtrating into the CNS
The hallmark of active MS and EAE is mononuclear immune infiltration into the CNS [4, 9]. Histopathological examination of spinal cord sections from EAE mice revealed perivascular cuffing with mononuclear inflammatory cells as well as extension of mononuclear inflammatory cell infiltrate into parenchyma in mice treated with the control of soluble factors (equivalent doses of TGF-β1, GM-CSF, vitamin D3 and MOG35-55 peptide) co-administered with empty MPs (S+U MPs) (Fig. 4A). However, spinal cord sections of EAE mice treated with dMP-MOG35-55 revealed an intact parenchyma with the absence of perivascular mononuclear inflammatory cells, indicating that the dMP-MOG35-55 treatment prevented EAE disease development through successfully blocking inflammatory cell infiltrating into the CNS. Lymphocyte infiltration into the CNS is observed in early and active MS and EAE and is considered the cause of autoimmune pathogenesis [4]. We further evaluated by flow cytometry the percentages and absolute numbers of T cells in the CNS of dMP-MOG35-55 versus S+U MPs-treated EAE mice and naïve mice and found that the percentages and absolute numbers of CD4+ T cells were drastically reduced in the EAE mice treated with dMP-MOG35-55, but still slightly higher than that of naïve healthy mice (Fig. 4B, C). Thus, the dMP-MOG35-55 treatment reduced the total mononuclear inflammatory cell infiltrating into CNS (Fig. 4A) in EAE mice, and also significantly reduced the total number of CD4+ T cells infiltrating into CNS (Fig. 4C), which suggests that dMP-MOG35-55 prevents EAE disease development through impeding CD4+ T cell infiltration in the CNS.
Figure 4. EAE mice treated with dMP-MOG35-55 have reduced leukocytes and CD4+ T cells infiltrating into the CNS.

(A) Representative Hematoxylin and Eosin staining of spinal cord section from B6 EAE mice treated either with dMP MOG35-55 or unloaded MPs or S+U MPs; white bar represents 200 μm; n=10 per group. (B) Representative flow cytometry analysis performed on day 21 following EAE induction of live CD4+ and CD8+ T cell frequencies from CNS of mice treated on day 4 following EAE induction either with dMP MOG35-55 or S+U MPs; n=5 per group. (C) Absolute numbers of CD4+ T cells in CNS of healthy naïve mice, or on day 21 following EAE induction of mice treated on day 4 following EAE induction either with dMP MOG35-55 or S+U MPs. p value was obtained from Student's t test.
3.5 EAE mice treated with dMP-MOG35-55 have reduced CD4+ T cells producing IL-17A, GM-CSF and IFNγ in the CNS
Given that production of proinflammatory cytokines IL-17A, GM-CSF, and IFNγ by pathogenic autoreactive CD4+ T cells is critical in the EAE disease pathogenesis [14, 58], we examined whether dMP-MOG35-55 treatment suppresses production of these proinflammatory cytokines. Not only was the number of CNS-infiltrating CD4+ T cells reduced in EAE mice treated with dMP-MOG35-55, but the production of IL-17A, GM-CSF, IFNγ, and co-production of these cytokines by the few CNS-infiltrating CD4+ T cells was also severely reduced in EAE mice treated with dMP-MOG35-55 (Fig. 5). Both the frequencies (Fig. 5A) and absolute numbers (Fig. 5B) of IL-17A, GM-CSF, IFNγ, and dual cytokine-producing CD4+ T cells in the CNS were significantly reduced in EAE mice treated with dMP-MOG35-55, suggesting that in addition to preventing the infiltration of CD4+ T cells in the CNS, dMP-MOG35-55 also suppress the production of IL-17A, GM-CSF, and IFNγ by pathogenic CD4+ T cells.
Figure 5. EAE mice treated with dMP-MOG35-55 have reduced CD4+ T cells producing IL-17A, GM-CSF and IFNγ in the CNS.
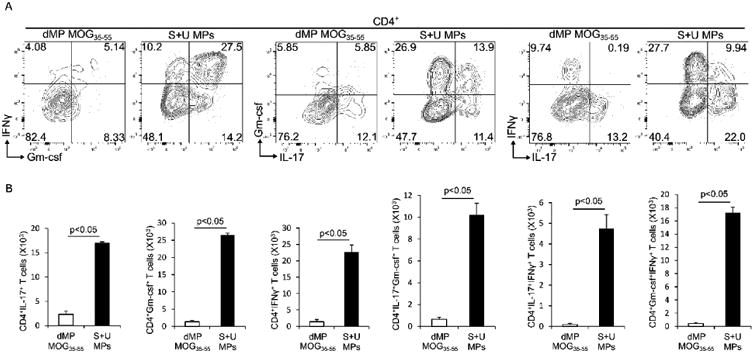
(A) Representative frequencies of CD4+ T cells positive for IL-17A, GM-CSF, IFNγ, and dual cytokines analyzed by intracellular cytokine staining and flow cytometry on day 21 following EAE induction in the brain of EAE mice treated on day 4 following EAE induction either with dMP MOG35-55 or S+U MPs; n=5 per group. (B) Absolute numbers of the CNS-infiltrating CD4+ T cells producing the indicated cytokines on day 21 following EAE induction in the brain of EAE mice treated on day 4 following EAE induction either with dMP MOG35-55 or S+U MPs; n=5 per group. p value was obtained from Student's t test.
3.6 EAE mice treated with dMP-MOG35-55 have decreased pathogenic CD4+ T cellsexpressing the transcription factors Rorγt and T-bet in the CNS
The Th17 transcription factor, Rorγt, and the Th1 transcription factor, T-bet, have been demonstrated to be crucial for GM-CSF production in pathogenic CD4+ T cells and EAE disease pathogenesis [13, 15, 59-61]. We thus examined whether dMP-MOG35-55 treatment suppresses Rorγt and T-bet expression in CD4+ T cells in the CNS. EAE mice treated with dMP-MOG35-55 showed significant reduction in frequencies and absolute numbers of Rorγt+, T-bet+, and dual Rorγt and T-bet- expressing CD4+ T cells in the CNS (Fig. 6), as well as diminished Rorγt and T-bet MFIs (data not shown). These results suggest that dMP-MOG35-55 treatment may block the entire transcriptional program of pathogenic CD4+ T cells in EAE mice. Thus, taken together, these results suggest that the reduced EAE disease scores in the dMP-MOG35-55-treated EAE mice are linked to reduced pathogenic CD4+ T cells in the CNS.
Figure 6. EAE mice treated with dMP-MOG35-55 have decreased pathogenic CD4+ T cells expressing the transcription factors Rorγt and T-bet in the CNS.
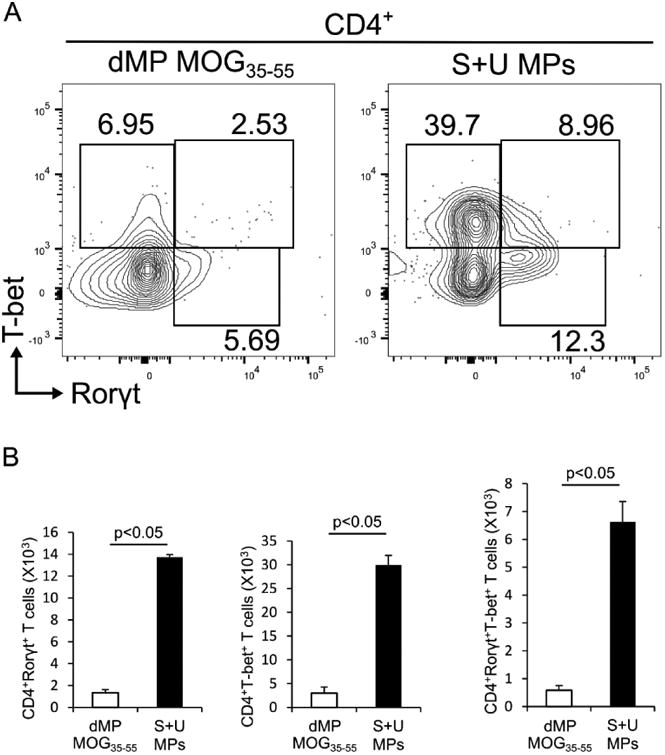
(A) Representative intranuclear flow cytometry analysis of frequencies of Rorγt+, T-bet+, and Rorγt+T-bet+ CD4+ T cells on day 21 following EAE induction in the brain of EAE mice treated on day 4 following EAE induction either with dMP MOG35-55 or S+U MPs; n=5 per group. (B) Absolute numbers of the CNS-infiltrating Rorγt+, T-bet+, and Rorγt+T-bet+ CD4+ T cells on day 21 following EAE induction of EAE mice treated on day 4 following EAE induction either with dMP MOG35-55 or S+U MPs; n=5 per group. p value was obtained from Student's t test.
3.7 Activated macrophages/microglial cells are reduced in the CNS of mice treated with dMP-MOG35-55
In EAE and MS, pathogenic effector CD4+ T cells trigger activation of CNS resident microglia and the recruitment of macrophages, which are essential for inflammatory demyelinating lesions (reviewed in [62]). We therefore examined whether dMP-MOG35-55 treatment affected activated microglia/macrophage populations in the CNS of EAE mice. Both microglia and macrophages are CD11b+F4/80+CD68+ and upregulate MHCII and CD80 following activation [63] (reviewed in [64]). We demonstrate that the frequency (Fig. 7A) and absolute number (Fig. 7B) of CD11b+CD68+F4/80+CD80+ cells, which include both activated macrophages and microglia, were significantly reduced in the CNS of EAE mice treated with dMP-MOG35-55. This is likely in relation to the decreased IL-17A, GM-CSF, and IFNγ production by CNS-infiltrating CD4+ T cells (Fig. 4). Thus, overall, the reduced EAE disease severity in mice treated with dMP-MOG35-55 can be explained by the decreased pathogenic CD4+ T cells and reduced activated macrophages/microglia in the CNS.
Figure 7. Activated macrophages/microglial cells are reduced in the CNS of mice treated with dMP-MOG35-55.
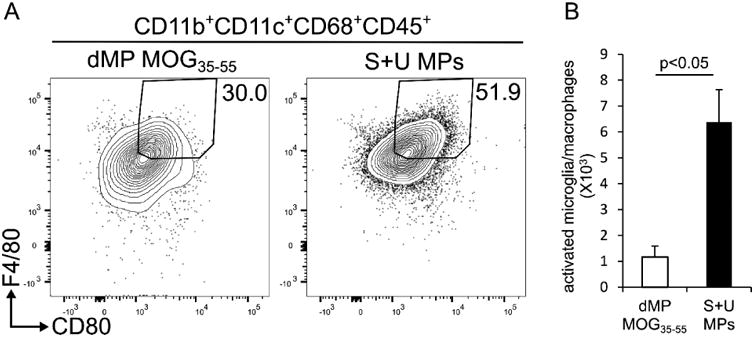
(A) Representative flow cytometry analysis performed on day 21 following EAE induction of leukocytes isolated from CNS of mice treated on day 4 following EAE induction either with dMP MOG35-55 or S+U MPs; n=5 per group. (B) Absolute numbers of activated microglia/macrophages on day 21 following EAE induction in CNS of mice treated on day 4 following EAE induction either with dMP MOG35-55 or S+U MPs; n=5 per group. Live leukocytes were gated on CD11b+CD11c+CD68+CD45+ and further on F4/80 and CD80 high. p value was obtained from Student's t test.
3.8 Efficacy of dMP treatment is dependent on antigen specificity
Because MS and EAE are established with a major autoimmune component (reviewed in [4]), we examined whether this dMP semi-therapeutic treatment was antigen-specific. We thus treated EAE mice with either dMP-MOG35-55, which includes the MOG35-55 antigenic peptide-loaded MPs, or dMP-Ova323-339, which includes MPs loaded with an irrelevant antigenic peptide, Ova323-339, derived from ovalbumin. Treatment with dMP-MOG35-55 prevented EAE disease development, but treatment with dMP-Ova323-339 did not (Fig. 8A). Linear regression analysis of EAE disease score development revealed no disease development in EAE mice treated with dMP-MOG35-55 compared to positive disease progression in EAE mice treated with dMP-Ova323-339 (Fig. 8B), thus demonstrating that the success of the treatment is dependent on antigen-specificity.
3.9 T cells from EAE mice treated with dMP-MOG35-55, but not with dMP-Ova323-339, fail to expand in response to MOG35-55-dependent stimulation
Based on the demonstration that dMP-MOG35-55 treatment of EAE is antigen-specific, we further asked the question whether T cells isolated from dMP-MOG35-55 treated EAE mice can proliferate as efficiently as T cells derived from EAE mice treated with dMP-Ova323-339, following exogenous stimulation with splenocytes loaded with MOG35-55, in an in vitro antigen re-stimulation assay. Our results show that CD4+ T cells isolated from dMP-MOG35-55 treated EAE mice failed to proliferate, while CD4+ T cells dMP-OVA323-339 treated EAE mice expanded robustly in response to MOG35-55-loaded splenocyte co-culture (Fig. 9). These results indicate these T cells from dMP-MOG35-55-treated EAE mice are anergic, unable to respond to this EAE-related antigen.
Figure 9. T cells from EAE mice treated with dMP-MOG35-55, but not with dMP-Ova323-339, failed to expand in response to MOG35-55-dependent stimulation.
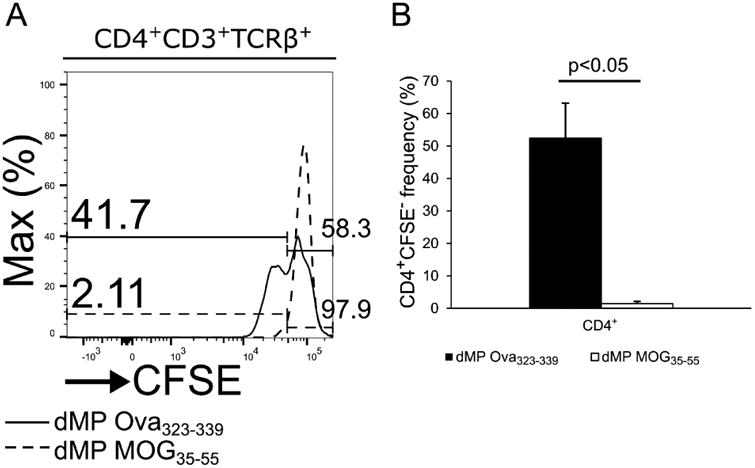
(A) Representative flow cytometry analysis of CFSE level in CD4+ T cells isolated on day 21 following EAE induction from draining lymph nodes of B6 mice treated with either dMP MOG35-55 (dash line) or dMP Ova323-339 (solid line) on days 4 and 7 (filled arrows) following EAE induction after 72 co-culture with T-cell depleted splenocytes loaded with MOG35-55. (B) Frequencies of CFSE negative CD4+ T cells isolated on day 21 following EAE induction from draining lymph nodes of B6 mice treated with either dMP MOG35-55 (dash line) or dMP Ova323-339 (solid line) on days 4 and 7 (filled arrows) following EAE induction after 72h co-culture with T-cell depleted splenocytes loaded with MOG35-55. p value was obtained from Student's t test.
3.10. Dendritic cells from draining lymph nodes of EAE mice treated with dMP-MOG35-55 display a tolerized phenotype
We finally investigated whether the dMP-MOG35-55 treatment of EAE mice induced a suppressive DC phenotype, as indicated by reduced expression of CD86 and MHCII. The frequency of CD11b+CD11c+ DCs that highly co-express CD86 and MHCII in draining lymph nodes is significantly reduced in EAE mice treated with dMP-MOG35-55 compared with those treated with irrelevant antigen-loaded dMP-Ova323-339 (Fig. 10A-B). Similarly, the mean fluorescence intensity (MFI) of CD86 expression in dMP-MOG35-55 was reduced (Fig. 10C). Thus, not only are the CD4+ T cells anergic in the dMP-MOG35-55-treated group (Fig. 9), but the DCs are also suppressive in an antigen-specific manner (Fig. 10).
Figure 10. Dendritic cells from draining lymph nodes of EAE mice treated with dMP-MOG35-55 displayed a tolerized phenotype.
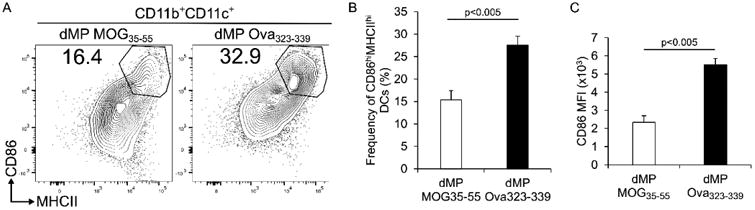
(A) Representative flow cytometry analysis of CD86 and MHCII expression on CD11b+CD11b+ dendritic cells isolated from draining lymph nodes of mice treated on day 4 and 7 following EAE induction either with dMP MOG35-55 or dMP Ova323-339; n=4 per group. (B) Frequency of CD86hiMHCIIhi CD11b+CD11c+ dendritic cells isolated from draining lymph nodes of mice treated on day 4 and 7 following EAE induction either with dMP MOG35-55 or dMP Ova323-339; n=4 per group. (C) Mean fluorescence intensity of CD86 in CD11b+CD11c+ dendritic cells isolated from draining lymph nodes of mice treated on day 4 and 7 following EAE induction either with dMP MOG35-55 or dMP Ova323-339; n=4 per group. p value was obtained from Student's t test.
4. Discussion
Therapeutic interventions to mitigate autoimmune-mediated demyelination in the CNS are limited for patients suffering from MS. While multiple immunosuppressant approaches (e.g., corticosteroids, anti-CD25, anti-CD20, anti-CD52) are either FDA approved or in clinical trials for progressive and relapsing MS, systemic immunosuppression leaves patients at risk for opportunistic infections and tumor development [16]. The advent of DC-based, antigen-specific immunotherapy is an exciting therapeutic avenue to restore homeostatic immunity in a disease-relevant manner [65, 66]. The first DC-based vaccine, Sipuleucel-T, was approved by the FDA in 2010 as a cancer immunotherapy approach [67]. Along this line, adoptive transfer of tolerogenic DCs to restore homeostatic immunity has been explored in models of graft survival and type 1 diabetes [68, 69], and a recent phase I clinical trial for type 1 diabetics has as a goal to modulate autologous DCs toward an immunosuppressive state via a combination of antisense oligonucleotides against co-stimulatory molecules CD40, CD80, and CD86 before being re-administered [70]. Results from this approach demonstrated tolerable safety profiles. Similarly, MS-specific DC-based immunotherapy has been investigated through delivery of exogenously-loaded DCs with MOG40-55 tolerized with vitamin D3, and was shown to ameliorate EAE [71]. Unfortunately, adoptive DC therapy is limited by high cost, inadequate cell sources, and safety concerns [66, 72]. Biomaterial approaches to modulate DC behavior have the potential to circumvent several limitations associated with exogenous delivery [31-35, 73-75]. In the present study, we have improved upon a dual-sized microparticle platform that previously demonstrated efficacy in the prevention of type 1 diabetes in NOD mice, and apply the therapy in the treatment of EAE, a mouse model of MS.
Notably, dMP-MOG35-55 treatment showed high efficacy and specificity in the treatment of EAE. We found that early subcutaneous administration of dMP-MOG35-55 treatment at 4, 7, and 10 days following EAE induction completely suppressed EAE manifestation in all mice treated. The dMP-MOG35-55 approach is based on delivery of specific antigens and tolerogenic factors encapsulated in PLGA MPs for controlled release in a local microenvironment. Similar approaches using PLGA nanoparticles and EAE-specific antigens with or without tolerogenic factors have been investigated [51, 76-79]. However, the present dMP-MOG35-55 treatment, administered subcutaneously, suppressed EAE, whereas several previously reported PLGA-based nanoparticle formulations relied on intravenous administration [51, 76] or required intra-lymph node injection [78]. Nanoparticles administered intravenously are typically cleared within hours by hepatic filtration and renal clearance [80]. Thus, a low percentage of drug is expected to be delivered to disease-relevant lymphoid organs and immune cell targets. By using a MP system delivered subcutaneously, we see significant effects on DC recruitment, modulation toward a tolerogenic phenotype with upregulation of PD-L1 and downregulation of CD86, and trafficking of MP+ DCs to draining lymph nodes, even after eight days post injection. Additionally, this delivery strategy did not result in systemic distribution of MPs, as fluorescently-labelled MPs were isolated only from proximal draining lymph nodes and not from the spleen or mesenteric lymph nodes. This is highly desirable to safeguard against systemic immunosuppression. In contrast, nanoparticles can enter directly through lymphatics without the aid of trafficking phagocytes [81]. Nanoparticle formulations that have prevented EAE have included highly immunosuppressive drugs such as rapamycin [77], which has demonstrated toxicity for other cell types including pancreatic β-cells [82]. The present dMP-MOG35-55 therapy mitigates EAE without highly immunosuppressive drugs, simultaneously providing more efficacious therapeutic aid compared to other PLGA vaccines, such as IL-10 nanoparticles [79]. IL-10 nanoparticles in combination with MOG35-55 peptide were previously used in prophylactic as well as therapeutic regimens and showed some decrease in EAE severity [79], however more modest than in our treatments. IL10 is known to limit adaptive immunity mediated by CD4+ T cell IFNγ response to infections such as Leismania donovani, Yersinia pestis, and Yersinia enterocolitica, and its elimination leads to enhanced survival after bacterial and viral infection (reviewed in [83]). Thus therapies involving IL-10 may not be optimal given the enhanced immune suppressive effects for bacterial and viral infections. In the current study, TGF-β1, GM-CSF, and vitamin D3 were chosen as immunomodulatory factors designed to suppress autoimmunity but not cause overwhelming immunosuppression [41, 42]. The regulation of immunity by TGF-β1 is highly intricate and context-dependent where it maintains a delicate balance of tolerogenic versus immunogenic pathways of the immune system while restricting bystander inflammatory tissue damage (reviewed in [84-86]). The multifaceted signaling pathways, including PI3K, STAT, MAPK, and NF-κB signaling, downstream of the GM-CSF receptor attest to the complex role that GM-CSF plays in the immune system (reviewed in [87]). Low concentration of GM-CSF was shown to only activate the PI3K pathway for DC survival, decrease MHCII/CD80/CD86 expression on DCs, and increase PD-L1 expression, which is appropriate for tolerogenic DC induction [49, 50]. Recently, through metabolic and signaling pathway analysis, vitamin D3 was also shown to induce tolerogenic DCs through glucose metabolism, glycolysis, and PI3K/Akt/mTOR pathway [44], which seem to act on the same pathways as low GM-CSF dose [87]. Thus, the choice of TGF-β1, GM-CSF, and vitamin D3 as tolerogenic factors for the current dMP system were based on their immunomodulatory effects that avoiding the broadly overwhelming immunosuppression of cytokines such as IL-10. The present subcutaneously administered dMP-MOG35-55 treatment suppressed EAE through reduction of total leukocytes, CD4+ T cells, including those expressing inflammatory cytokine, and activated macrophages/microglia in the CNS, which is similar to the reduction in CNS immune infiltration and cytokine secretion induced by the previously reported intravenous PLGA-based nanoparticle encapsulating EAE-specific antigen formulation [51]. The relative strength of tolerance achieved in the present study using a dMP system and semi-therapeutic regimen administered subcutaneously after EAE induction is comparable, if not superior, to the preventative/prophylactic and therapeutic regimen utilized for nanoparticle platforms described previously [51, 52] in that the current study achieved total suppression of EAE clinical disease and drastic reduction of CD4+ T cell infiltration into CNS. The quality and timing of tolerance achieved herein with the dMP administered in semi-therapeutic regimen is superior compared to the preventative/prophylactic and therapeutic regimen reported previously for nanoparticle platforms, as it can be used after disease initiation, which has clinical implication for further development into a therapeutic to be administered after disease clinical sign onset.
Controlled-release platforms for immunomodulatory applications are advantageous versus soluble administration. In addition to mitigating the risk for systemic immunosuppression, soluble drugs are rapidly cleared from the body. Thus, pharmacokinetics may prevent soluble administration to effectively restore homeostatic immunity or require more frequent or higher dosing. The impact of biomaterial encapsulation in our system was demonstrated by the requirement for encapsulation of drugs in MPs, as soluble injections of the factors along with unloaded MPs did not prevent T cell infiltration in the CNS. Importantly, the encapsulated drugs could not be detected in the blood, thus being unlikely that the dMP encapsulation will cause immune suppression.
Induction of antigen-specific tolerance is crucial to developing a safe, translatable therapy for EAE/MS, with the goal to achieve therapeutic efficacy without inducing broad immunosuppression. By utilizing multiple control groups, we demonstrated that the dMP-MOG35-55 treatment specifically suppressed EAE in an antigen-dependent manner. Comparing the dMP-MOG35-55 to a similar formulation without antigen-loaded MPs, we saw that omission of antigen resulted in mice developing disease. Similarly, disease was only blocked with dMP-MOG35-55 treatment, but not by treatment with an irrelevant antigen, dMP-Ova323-339. These are hallmarks of successful antigen-specific immunotherapy approaches, and correspond to such demonstrations by previous reports utilizing nanoparticles encapsulating EAE-specific antigens and tolerogenic factors [51, 52, 88]. This antigen-specificity in a newly developed microparticle-based immunotherapy is especially important since the safety profile of the MS antigen-specific tolerogenic drug glatiramer acetate, an FDA approved therapy for MS, is significantly superior to that of other approved immunotherapies, such as Natalizumab and Fingolimod, which do not rely on antigen-specificity [19, 89-91]. Our observation that CD4+ T cells isolated from EAE mice treated with dMP-MOG35-55 were not responsive to stimulation by MOG35-55-loaded splenocytes whereas EAE mice treated with dMP Ova323-339 proliferated, showed that T cell anergy is effectively induced by the dMP-MOG35-55 therapy in an antigen-specific manner, which is similar to other PLGA-based nanoparticle encapsulated EAE therapies [51, 52, 88].
Together, these results highlight an exciting combinatorial controlled release and immunologically-driven approach to restore immune homeostasis in a mouse model of a MS. Through a dMP system that delivers local sustained release of multiple immunomodulatory factors, and targets both intra- and extracellular tolerogenic receptors, we demonstrate robust and durable antigen-specific autoimmune protection, more so than soluble factors or irrelevant antigen formulations. Additionally, the significance of these studies is underscored in the versatility of the dMP platform, as substitution of antigen and/or factors has the potential to elicit tolerogenic or immunogenic responses in a tailored, disease-specific fashion
Acknowledgments
Support is gratefully acknowledged by R01 AI133623 to B.G.K and D.A, the University of Florida Gatorade Trust to D.A. and J.J.C., the United States Navy, United States Armed Forces Health Professions Scholarship Program, funds medical education of J.J.C., and grants from the National Institute of Diabetes and Digestive and Kidney Diseases, R01 DK091658, and R01 DK098589 (to B.G.K.). We acknowledge Xiaoping Luo for care of mice. We acknowledge Upasana Parthasarathy, Tammy Dowie, Regina Oshins, and laboratory of Mark L. Brantly for support in ELISA. The content is solely the responsibility of the authors and does not necessarily represent the official views, policy or position of the Department of the Navy, Department of Defense, the U.S. Government, nor National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372(9648):1502–17. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 2.Kobelt G, Berg J, Atherly D, Hadjimichael O. Costs and quality of life in multiple sclerosis: a cross-sectional study in the United States. Neurology. 2006;66(11):1696–702. doi: 10.1212/01.wnl.0000218309.01322.5c. [DOI] [PubMed] [Google Scholar]
- 3.Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, Thompson AJ, Wolinsky JS, Balcer LJ, Banwell B, Barkhof F, Bebo B, Calabresi PA, Clanet M, Comi G, Fox RJ, Freedman MS, Goodman AD, Inglese M, Kappos L, Kieseier BC, Lincoln JA, Lubetzki C, Miller AE, Montalban X, O'Connor PW, Petkau J, Pozzilli C, Rudick RA, Sormani MP, Stüve O, Waubant E, Polman CH. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology. 2014;83(3):278–86. doi: 10.1212/WNL.0000000000000560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15(9):545–58. doi: 10.1038/nri3871. [DOI] [PubMed] [Google Scholar]
- 5.Nylander A, Hafler DA. Multiple sclerosis. J Clin Invest. 2012;122(4):1180–8. doi: 10.1172/JCI58649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiner HL. Multiple sclerosis is an inflammatory T-cell-mediated autoimmune disease. Arch Neurol. 2004;61(10):1613–5. doi: 10.1001/archneur.61.10.1613. [DOI] [PubMed] [Google Scholar]
- 7.Bielekova B, Sung MH, Kadom N, Simon R, McFarland H, Martin R. Expansion and functional relevance of high-avidity myelin-specific CD4+ T cells in multiple sclerosis. J Immunol. 2004;172(6):3893–904. doi: 10.4049/jimmunol.172.6.3893. [DOI] [PubMed] [Google Scholar]
- 8.Hellings N, Barée M, Verhoeven C, D'hooghe MB, Medaer R, Bernard CC, Raus J, Stinissen P. T-cell reactivity to multiple myelin antigens in multiple sclerosis patients and healthy controls. J Neurosci Res. 2001;63(3):290–302. doi: 10.1002/1097-4547(20010201)63:3<290::AID-JNR1023>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 9.Lavi E, Constantinescu CS. Experimental models of multiple sclerosis. Springer; New York: 2005. [Google Scholar]
- 10.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201(2):233–40. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6(11):1133–41. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126(6):1121–33. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 13.Codarri L, Gyülvészi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12(6):560–7. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- 14.El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang GX, Dittel BN, Rostami A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12(6):568–75. doi: 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK. Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med. 2004;200(1):79–87. doi: 10.1084/jem.20031819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wingerchuk DM, Carter JL. Multiple sclerosis: current and emerging disease-modifying therapies and treatment strategies. Mayo Clin Proc. 2014;89(2):225–40. doi: 10.1016/j.mayocp.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 17.Goodin DS, Frohman EM, Garmany GP, Halper J, Likosky WH, Lublin FD, Silberberg DH, Stuart WH, van den Noort S T.a.T.A.S.o.t.A.A.o.N.a.t.M.C.f.C.P. Guidelines. Disease modifying therapies in multiple sclerosis: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and the MS Council for Clinical Practice Guidelines. Neurology. 2002;58(2):169–78. doi: 10.1212/wnl.58.2.169. [DOI] [PubMed] [Google Scholar]
- 18.Goodin DS, Reder AT, Ebers GC, Cutter G, Kremenchutzky M, Oger J, Langdon D, Rametta M, Beckmann K, DeSimone TM, Knappertz V. Survival in MS: a randomized cohort study 21 years after the start of the pivotal IFNβ-1b trial. Neurology. 2012;78(17):1315–22. doi: 10.1212/WNL.0b013e3182535cf6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goodin DS, Cohen BA, O'Connor P, Kappos L, Stevens JC T.a.T.A.S.o.t.A.A.o. Neurology. Assessment: the use of natalizumab (Tysabri) for the treatment of multiple sclerosis (an evidence-based review): report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2008;71(10):766–73. doi: 10.1212/01.wnl.0000320512.21919.d2. [DOI] [PubMed] [Google Scholar]
- 20.Gasperini C, Ruggieri S. Development of oral agent in the treatment of multiple sclerosis: how the first available oral therapy, fingolimod will change therapeutic paradigm approach. Drug Des Devel Ther. 2012;6:175–86. doi: 10.2147/DDDT.S8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feinstein A, Freeman J, Lo AC. Treatment of progressive multiple sclerosis: what works, what does not, and what is needed. Lancet Neurol. 2015;14(2):194–207. doi: 10.1016/S1474-4422(14)70231-5. [DOI] [PubMed] [Google Scholar]
- 22.Hilas O, Patel PN, Lam S. Disease modifying agents for multiple sclerosis. Open Neurol J. 2010;4:15–24. doi: 10.2174/1874205X01004010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morelli AE, Thomson AW. Dendritic cells: regulators of alloimmunity and opportunities for tolerance induction. Immunol Rev. 2003;196:125–46. doi: 10.1046/j.1600-065x.2003.00079.x. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Ma B, Zhou Y, Zhang M, Qiu X, Sui Y, Zhang X, Fan Q. Dendritic cells fused with allogeneic breast cancer cell line induce tumor antigen-specific CTL responses against autologous breast cancer cells. Breast Cancer Res Treat. 2007;105(3):277–86. doi: 10.1007/s10549-006-9457-8. [DOI] [PubMed] [Google Scholar]
- 25.Zhou J, Weng D, Zhou F, Pan K, Song H, Wang Q, Wang H, Li Y, Huang L, Zhang H, Huang W, Xia J. Patient-derived renal cell carcinoma cells fused with allogeneic dendritic cells elicit anti-tumor activity: in vitro results and clinical responses. Cancer Immunol Immunother. 2009;58(10):1587–97. doi: 10.1007/s00262-009-0668-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim JH, Lee Y, Bae YS, Kim WS, Kim K, Im HY, Kang WK, Park K, Choi HY, Lee HM, Baek SY, Lee H, Doh H, Kim BM, Kim CY, Jeon C, Jung CW. Phase I/II study of immunotherapy using autologous tumor lysate-pulsed dendritic cells in patients with metastatic renal cell carcinoma. Clin Immunol. 2007;125(3):257–67. doi: 10.1016/j.clim.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 27.Höltl L, Zelle-Rieser C, Gander H, Papesh C, Ramoner R, Bartsch G, Rogatsch H, Barsoum AL, Coggin JH, Thurnher M. Immunotherapy of metastatic renal cell carcinoma with tumor lysate-pulsed autologous dendritic cells. Clin Cancer Res. 2002;8(11):3369–76. [PubMed] [Google Scholar]
- 28.Reddy ST, Swartz MA, Hubbell JA. Targeting dendritic cells with biomaterials: developing the next generation of vaccines. Trends Immunol. 2006;27(12):573–9. doi: 10.1016/j.it.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 29.Bracho-Sanchez E, Xia CQ, Clare-Salzler MJ, Keselowsky BG. Micro and Nano Material Carriers for Immunomodulation. Am J Transplant. 2016 doi: 10.1111/ajt.13878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lewis JS, Zaveri TD, Crooks CP, 2nd, Keselowsky BG. Microparticle surface modifications targeting dendritic cells for non-activating applications. Biomaterials. 2012;33(29):7221–32. doi: 10.1016/j.biomaterials.2012.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phillips B, Nylander K, Harnaha J, Machen J, Lakomy R, Styche A, Gillis K, Brown L, Lafreniere D, Gallo M, Knox J, Hogeland K, Trucco M, Giannoukakis N. A microsphere-based vaccine prevents and reverses new-onset autoimmune diabetes. Diabetes. 2008;57(6):1544–55. doi: 10.2337/db07-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schlosser E, Mueller M, Fischer S, Basta S, Busch DH, Gander B, Groettrup M. TLR ligands and antigen need to be coencapsulated into the same biodegradable microsphere for the generation of potent cytotoxic T lymphocyte responses. Vaccine. 2008;26(13):1626–37. doi: 10.1016/j.vaccine.2008.01.030. [DOI] [PubMed] [Google Scholar]
- 33.Elamanchili P, Lutsiak CM, Hamdy S, Diwan M, Samuel J. “Pathogen-mimicking” nanoparticles for vaccine delivery to dendritic cells. J Immunother. 2007;30(4):378–95. doi: 10.1097/CJI.0b013e31802cf3e3. [DOI] [PubMed] [Google Scholar]
- 34.Singh A, Nie H, Ghosn B, Qin H, Kwak LW, Roy K. Efficient modulation of T-cell response by dual-mode, single-carrier delivery of cytokine-targeted siRNA and DNA vaccine to antigen-presenting cells. Mol Ther. 2008;16(12):2011–21. doi: 10.1038/mt.2008.206. [DOI] [PubMed] [Google Scholar]
- 35.Jhunjhunwala S, Raimondi G, Thomson AW, Little SR. Delivery of rapamycin to dendritic cells using degradable microparticles. J Control Release. 2009;133(3):191–7. doi: 10.1016/j.jconrel.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoon YM, Lewis JS, Carstens MR, Campbell-Thompson M, Wasserfall CH, Atkinson MA, Keselowsky BG. A combination hydrogel microparticle-based vaccine prevents type 1 diabetes in non-obese diabetic mice. Sci Rep. 2015;5:13155. doi: 10.1038/srep13155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Champion JA, Walker A, Mitragotri S. Role of particle size in phagocytosis of polymeric microspheres. Pharm Res. 2008;25(8):1815–21. doi: 10.1007/s11095-008-9562-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Waeckerle-Men Y, Gander B, Groettrup M. Delivery of tumor antigens to dendritic cells using biodegradable microspheres. Methods Mol Med. 2005;109:35–46. doi: 10.1385/1-59259-862-5:035. [DOI] [PubMed] [Google Scholar]
- 39.Gerelchuluun T, Lee YH, Lee YR, Im SA, Song S, Park JS, Han K, Kim K, Lee CK. Dendritic cells process antigens encapsulated in a biodegradable polymer, poly(D,L-lactide-co-glycolide), via an alternate class I MHC processing pathway. Arch Pharm Res. 2007;30(11):1440–6. doi: 10.1007/BF02977369. [DOI] [PubMed] [Google Scholar]
- 40.Shen H, Ackerman AL, Cody V, Giodini A, Hinson ER, Cresswell P, Edelson RL, Saltzman WM, Hanlon DJ. Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology. 2006;117(1):78–88. doi: 10.1111/j.1365-2567.2005.02268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lewis JS, Dolgova NV, Zhang Y, Xia CQ, Wasserfall CH, Atkinson MA, Clare-Salzler MJ, Keselowsky BG. A combination dual-sized microparticle system modulates dendritic cells and prevents type 1 diabetes in prediabetic NOD mice. Clin Immunol. 2015;160(1):90–102. doi: 10.1016/j.clim.2015.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lewis JS, Roche C, Zhang Y, Brusko TM, Wasserfall CH, Atkinson M, Clare-Salzler MJ, Keselowsky BG. Combinatorial delivery of immunosuppressive factors to dendritic cells using dual-sized microspheres. Journal of materials chemistry B, Materials for biology and medicine. 2014;2(17):2562–2574. doi: 10.1039/C3TB21460E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nikolic T, Roep BO. Regulatory multitasking of tolerogenic dendritic cells - lessons taken from vitamin d3-treated tolerogenic dendritic cells. Front Immunol. 2013;4:113. doi: 10.3389/fimmu.2013.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferreira GB, Vanherwegen AS, Eelen G, Gutiérrez AC, Van Lommel L, Marchal K, Verlinden L, Verstuyf A, Nogueira T, Georgiadou M, Schuit F, Eizirik DL, Gysemans C, Carmeliet P, Overbergh L, Mathieu C. Vitamin D3 Induces Tolerance in Human Dendritic Cells by Activation of Intracellular Metabolic Pathways. Cell Rep. 2015 doi: 10.1016/j.celrep.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 45.Belladonna ML, Volpi C, Bianchi R, Vacca C, Orabona C, Pallotta MT, Boon L, Gizzi S, Fioretti MC, Grohmann U, Puccetti P. Cutting edge: Autocrine TGF-beta sustains default tolerogenesis by IDO-competent dendritic cells. J Immunol. 2008;181(8):5194–8. doi: 10.4049/jimmunol.181.8.5194. [DOI] [PubMed] [Google Scholar]
- 46.Luo X, Tarbell KV, Yang H, Pothoven K, Bailey SL, Ding R, Steinman RM, Suthanthiran M. Dendritic cells with TGF-beta1 differentiate naive CD4+CD25- T cells into islet-protective Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A. 2007;104(8):2821–6. doi: 10.1073/pnas.0611646104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ali OA, Huebsch N, Cao L, Dranoff G, Mooney DJ. Infection-mimicking materials to program dendritic cells in situ. Nature materials. 2009;8(2):151–8. doi: 10.1038/nmat2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van de Laar L, Coffer PJ, Woltman AM. Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood. 2012;119(15):3383–93. doi: 10.1182/blood-2011-11-370130. [DOI] [PubMed] [Google Scholar]
- 49.Lutz MB, Suri RM, Niimi M, Ogilvie AL, Kukutsch NA, Rössner S, Schuler G, Austyn JM. Immature dendritic cells generated with low doses of GM-CSF in the absence of IL-4 are maturation resistant and prolong allograft survival in vivo. Eur J Immunol. 2000;30(7):1813–22. doi: 10.1002/1521-4141(200007)30:7<1813::AID-IMMU1813>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 50.Mayuzumi N, Matsushima H, Takashima A. IL-33 promotes DC development in BM culture by triggering GM-CSF production. Eur J Immunol. 2009;39(12):3331–42. doi: 10.1002/eji.200939472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hunter Z, McCarthy DP, Yap WT, Harp CT, Getts DR, Shea LD, Miller SD. A biodegradable nanoparticle platform for the induction of antigen-specific immune tolerance for treatment of autoimmune disease. ACS nano. 2014;8(3):2148–60. doi: 10.1021/nn405033r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yeste A, Nadeau M, Burns EJ, Weiner HL, Quintana FJ. Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2012;109(28):11270–5. doi: 10.1073/pnas.1120611109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmitz K, de Bruin N, Bishay P, Männich J, Häussler A, Altmann C, Ferreirós N, Lötsch J, Ultsch A, Parnham MJ, Geisslinger G, Tegeder I. R-flurbiprofen attenuates experimental autoimmune encephalomyelitis in mice. EMBO Mol Med. 2014;6(11):1398–422. doi: 10.15252/emmm.201404168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pino PA, Cardona AE. Isolation of brain and spinal cord mononuclear cells using percoll gradients. J Vis Exp. 2011;(48) doi: 10.3791/2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Califano D, Sweeney KJ, Le H, VanValkenburgh J, Yager E, O'Connor W, Kennedy JS, Jones DM, Avram D. Diverting T helper cell trafficking through increased plasticity attenuates autoimmune encephalomyelitis. J Clin Invest. 2014;124(1):174–87. doi: 10.1172/JCI70103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vanvalkenburgh J, Albu DI, Bapanpally C, Casanova S, Califano D, Jones DM, Ignatowicz L, Kawamoto S, Fagarasan S, Jenkins NA, Copeland NG, Liu P, Avram D. Critical role of Bcl11b in suppressor function of T regulatory cells and prevention of inflammatory bowel disease. J Exp Med. 2011;208(10):2069–81. doi: 10.1084/jem.20102683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Califano D, Cho JJ, Uddin MN, Lorentsen KJ, Yang Q, Bhandoola A, Li H, Avram D. Transcription Factor Bcl11b Controls Identity and Function of Mature Type 2 Innate Lymphoid Cells. Immunity. 2015;43(2):354–68. doi: 10.1016/j.immuni.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jäger A, Dardalhon V, Sobel RA, Bettelli E, Kuchroo VK. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol. 2009;183(11):7169–77. doi: 10.4049/jimmunol.0901906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, Garefalaki A, Potocnik AJ, Stockinger B. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol. 2011;12(3):255–63. doi: 10.1038/ni.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang Y, Weiner J, Liu Y, Smith AJ, Huss DJ, Winger R, Peng H, Cravens PD, Racke MK, Lovett-Racke AE. T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. J Exp Med. 2009;206(7):1549–64. doi: 10.1084/jem.20082584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, Sobel RA, Regev A, Kuchroo VK. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. 2012;13(10):991–9. doi: 10.1038/ni.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Codarri L, Greter M, Becher B. Communication between pathogenic T cells and myeloid cells in neuroinflammatory disease. Trends Immunol. 2013;34(3):114–9. doi: 10.1016/j.it.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 63.Xiao Y, Jin J, Chang M, Chang JH, Hu H, Zhou X, Brittain GC, Stansberg C, Torkildsen Ø, Wang X, Brink R, Cheng X, Sun SC. Peli1 promotes microglia-mediated CNS inflammation by regulating Traf3 degradation. Nat Med. 2013;19(5):595–602. doi: 10.1038/nm.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Northrup L, Christopher MA, Sullivan BP, Berkland C. Combining antigen and immunomodulators: Emerging trends in antigen-specific immunotherapy for autoimmunity. Adv Drug Deliv Rev. 2015 doi: 10.1016/j.addr.2015.10.020. [DOI] [PubMed] [Google Scholar]
- 66.Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol. 2007;7(10):790–802. doi: 10.1038/nri2173. [DOI] [PubMed] [Google Scholar]
- 67.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, Xu Y, Frohlich MW, Schellhammer PF, Investigators IS. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–22. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 68.Taner T, Hackstein H, Wang Z, Morelli AE, Thomson AW. Rapamycin-treated, alloantigen-pulsed host dendritic cells induce ag-specific T cell regulation and prolong graft survival. Am J Transplant. 2005;5(2):228–36. doi: 10.1046/j.1600-6143.2004.00673.x. [DOI] [PubMed] [Google Scholar]
- 69.Harrison LC. Vaccination against self to prevent autoimmune disease: the type 1 diabetes model. Immunol Cell Biol. 2008;86(2):139–45. doi: 10.1038/sj.icb.7100151. [DOI] [PubMed] [Google Scholar]
- 70.Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care. 2011;34(9):2026–32. doi: 10.2337/dc11-0472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mansilla MJ, Selles-Moreno C, Fabregas-Puig S, Amoedo J, Navarro-Barriuso J, Teniente-Serra A, Grau-Lopez L, Ramo-Tello C, Martinez-Caceres EM. Beneficial effect of tolerogenic dendritic cells pulsed with MOG autoantigen in experimental autoimmune encephalomyelitis. CNS Neurosci Ther. 2015;21(3):222–30. doi: 10.1111/cns.12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mart In-Fontecha A, Sebastiani S, Hopken UE, Uguccioni M, Lipp M, Lanzavecchia A, Sallusto F. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med. 2003;198(4):615–21. doi: 10.1084/jem.20030448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu Q, Jia J, Yang T, Fan Q, Wang L, Ma G. Pathogen-Mimicking Polymeric Nanoparticles based on Dopamine Polymerization as Vaccines Adjuvants Induce Robust Humoral and Cellular Immune Responses. Small. 2016;12(13):1744–57. doi: 10.1002/smll.201503662. [DOI] [PubMed] [Google Scholar]
- 74.Petersen LK, Ramer-Tait AE, Broderick SR, Kong CS, Ulery BD, Rajan K, Wannemuehler MJ, Narasimhan B. Activation of innate immune responses in a pathogen-mimicking manner by amphiphilic polyanhydride nanoparticle adjuvants. Biomaterials. 2011;32(28):6815–22. doi: 10.1016/j.biomaterials.2011.05.063. [DOI] [PubMed] [Google Scholar]
- 75.Jilek S, Merkle HP, Walter E. DNA-loaded biodegradable microparticles as vaccine delivery systems and their interaction with dendritic cells. Adv Drug Deliv Rev. 2005;57(3):377–90. doi: 10.1016/j.addr.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 76.Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZN, Yap WT, Getts MT, Pleiss M, Luo X, King NJ, Shea LD, Miller SD. Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat Biotechnol. 2012;30(12):1217–24. doi: 10.1038/nbt.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maldonado RA, LaMothe RA, Ferrari JD, Zhang AH, Rossi RJ, Kolte PN, Griset AP, O'Neil C, Altreuter DH, Browning E, Johnston L, Farokhzad OC, Langer R, Scott DW, von Andrian UH, Kishimoto TK. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc Natl Acad Sci U S A. 2015;112(2):E156–65. doi: 10.1073/pnas.1408686111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tostanoski LH, Chiu YC, Gammon JM, Simon T, Andorko JI, Bromberg JS, Jewell CM. Reprogramming the Local Lymph Node Microenvironment Promotes Tolerance that Is Systemic and Antigen Specific. Cell reports. 2016;16(11):2940–52. doi: 10.1016/j.celrep.2016.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cappellano G, Woldetsadik AD, Orilieri E, Shivakumar Y, Rizzi M, Carniato F, Gigliotti CL, Boggio E, Clemente N, Comi C, Dianzani C, Boldorini R, Chiocchetti A, Reno F, Dianzani U. Subcutaneous inverse vaccination with PLGA particles loaded with a MOG peptide and IL-10 decreases the severity of experimental autoimmune encephalomyelitis. Vaccine. 2014;32(43):5681–9. doi: 10.1016/j.vaccine.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 80.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–15. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O'Neil CP, Lee LK, Swartz MA, Hubbell JA. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat Biotechnol. 2007;25(10):1159–64. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- 82.Barlow AD, Nicholson ML, Herbert TP. Evidence for rapamycin toxicity in pancreatic beta-cells and a review of the underlying molecular mechanisms. Diabetes. 2013;62(8):2674–82. doi: 10.2337/db13-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Iyer SS, Cheng G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit Rev Immunol. 2012;32(1):23–63. doi: 10.1615/critrevimmunol.v32.i1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov. 2012;11(10):790–811. doi: 10.1038/nrd3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rubtsov YP, Rudensky AY. TGFbeta signalling in control of T-cell-mediated self-reactivity. Nat Rev Immunol. 2007;7(6):443–53. doi: 10.1038/nri2095. [DOI] [PubMed] [Google Scholar]
- 86.Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 2010;10(8):554–67. doi: 10.1038/nri2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hercus TR, Thomas D, Guthridge MA, Ekert PG, King-Scott J, Parker MW, Lopez AF. The granulocyte-macrophage colony-stimulating factor receptor: linking its structure to cell signaling and its role in disease. Blood. 2009;114(7):1289–98. doi: 10.1182/blood-2008-12-164004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.McCarthy DP, Yap JW, Harp CT, Song WK, Chen J, Pearson RM, Miller SD, Shea LD. An antigen-encapsulating nanoparticle platform for TH1/17 immune tolerance therapy. Nanomedicine. 2017;13(1):191–200. doi: 10.1016/j.nano.2016.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Arnon R, Aharoni R. Mechanism of action of glatiramer acetate in multiple sclerosis and its potential for the development of new applications. Proc Natl Acad Sci U S A. 2004;101(2):14593–8. doi: 10.1073/pnas.0404887101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M, Yang M, Raghupathi K, Novas M, Sweetser MT, Viglietta V, Dawson KT, Investigators CS. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367(12):1087–97. doi: 10.1056/NEJMoa1206328. [DOI] [PubMed] [Google Scholar]
- 91.Cohen JA, Chun J. Mechanisms of fingolimod's efficacy and adverse effects in multiple sclerosis. Ann Neurol. 2011;69(5):759–77. doi: 10.1002/ana.22426. [DOI] [PubMed] [Google Scholar]