

Abstract
The assembly of individual molecules into hierarchical structures is a promising strategy for developing three-dimensional materials with properties arising from interaction between the individual building blocks. Virus capsids are elegant examples of biomolecular nanostructures, which are themselves hierarchically assembled from a limited number of protein subunits. Here we demonstrate the bio-inspired modular construction of materials with two levels of hierarchy; the formation of catalytically active individual virus-like particles (VLPs) through directed self-assembly of capsid subunits with enzyme encapsulation, and the assembly of these VLP building blocks into three-dimensional arrays. The structure of the assembled arrays was successfully altered from an amorphous aggregate to an ordered structure, with a face-centered cubic lattice, by modifying the exterior surface of the VLP without changing its overall morphology, to modulate interparticle interactions. The assembly behavior and resultant lattice structure was a consequence of interparticle interaction between exterior surfaces of individual particles, and thus independent of the enzyme cargos encapsulated within the VLPs. These superlattice materials, composed of two populations of enzyme packaged VLP modules, retained the coupled catalytic activity in a two-step reaction for isobutanol synthesis. This study demonstrates a significant step toward the bottom-up fabrication of functional superlattice materials using a self-assembly process across multiple length scales, and exhibits properties and function that arise from the interaction between individual building blocks.
Keywords: virus-like particle (VLP), self-assembly, superlattice, hierarchical structure, enzyme encapsulation, nano-reactor, coupled catalysis
There is growing interest in bottom-up approaches for the fabrication of active, functional materials via hierarchical self-assembly of nanoscale building block components.1–9 Material scientists increasingly look to nature for design principles for materials and draw inspiration from the many examples of complex self-assembled biomolecular structures formed through multiple levels of hierarchical organization.10 Bacterial microcompartments such as the carboxysome are one such example in nature. They are cellular containers, self-assembled from protein subunits, which encapsulate a series of enzymes. These containers act as organelle-like compartments that segregate chemical reactions leading to improved catalytic efficiencies.11–12 Viruses, many of which self-assemble from protein subunits into regular spherical or rod-like structures, encapsulate and protect their viral genome and are another quintessential example of hierarchically self-assembled biomolecular structures. They have been used as templates for a wide range of nano-materials synthesis13 and are versatile platforms for both chemical and genetic modification. Bio-inspired approaches, which harness the elegant biomolecules and processes described above, hold the promise for the construction of functional materials.
The morphogenesis and directed cargo packaging observed in viruses has been particularly inspiring to material scientists, who have implemented virus-like particle (VLP) derivatives as building blocks for modular materials assembly.14–22 VLPs self-assemble from proteins that form the viral capsid in the absence of the viral genome, leading to non-infective protein cage shells with hollow interior compartments. VLPs exhibit near perfect icosahedral symmetry as a consequence of highly ordered assembly of coat proteins (CPs) forming the capsid. Since VLPs can be re-designed by genetic and chemical modification for synthetic cargo encapsulation, a wide variety of functional VLP nanomaterials have been developed.13, 23–25 The VLP derived from bacteriophage P22 has proven to be a powerful platform, as evidenced by its versatility in encapsulation of cargos including inorganic nanoparticles,26–27 polymers28–29 and proteins.30 The P22 VLP assembles from 420 copies of the CP and a variable number of scaffolding proteins (SP) (typically 100 – 300 copies), which direct the assembly of CPs into an approximately 56 nm diameter icosahedral capsid (Supplementary Figures S1 and S2).31–34 We have previously shown that the P22 VLP self-assembly can be co-opted to direct the encapsulation of multiple active enzymatic cargos through heterologous expression of two gene products (CP and truncated SP-cargo fusion) (Figure 1a).35–39 The capsid shell is highly porous to small molecules allowing diffusion of substrates and products into and out of the VLPs. Enzyme encapsulated VLPs function as nanoreactors, where the enzymes confined within the nanoscale cavity are protected from the external environment by the protein shell37–38 (Figure 1b). The assembly of the P22 VLP is dependent on the presence of scaffolding proteins but almost independent of the nature of the cargo. This convergent assembly of multiple copies of individual proteins results in the formation of a homogeneous population of stable nanoparticles and represents one level of hierarchical assembly.
Figure 1.
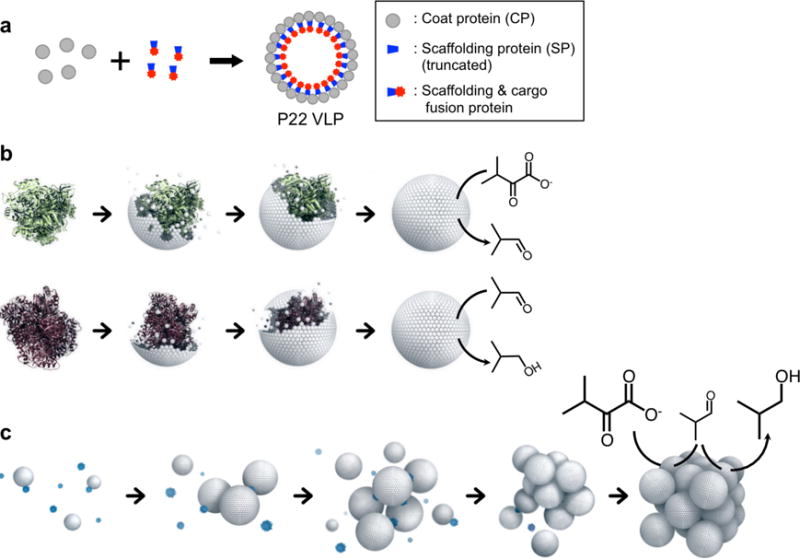
Schematic illustration of hierarchical self-assembly from protein subunit building blocks to a superlattice of catalytically active virus-like particles (VLPs). (a) Illustration of directed assembly of a P22 VLP with encapsulated cargo. Scaffolding proteins (SPs) direct the assembly of coat proteins (CPs) into the capsid structure. Cargos are guided towards co-encapsulation inside of the VLP via SP fusion. (b) Individual CPs self-assemble to form a VLP, templated by a SP-cargo fusion protein. The green protein represents a SP fusion protein with KivD, which catalyzes the conversion of α-ketoisovalerate to isobutyraldehyde, and the purple protein represents a SP fusion protein with AdhA, which catalyzes the conversion of isobutyraldehyde to isobutanol. (c) VLPs self-assemble into higher ordered superlattice materials, mediated by positively charged PAMAM dendrimers (blue). The array material, which is comprised of the two separately encapsulated enzymes, retains catalytic communication after assembly and performs a two-step reaction to produce isobutanol.
Further levels of hierarchical assembly, such as the controlled assembly of the individual VLP modules into bulk materials, could lead to the fabrication of sophisticated three-dimensional materials exhibiting properties arising from interaction between the individual modules. Here we demonstrate a second level of hierarchical materials construction to form active superlattice materials through the assembly of two populations of enzyme encapsulated P22 VLP nanoreactor modules. In the superlattices, the two separately encapsulated enzymes retained their catalytic communication (i.e. the product of the first reaction is the substrate for the second enzyme) and together catalyze a coupled two-step reaction resulting in the production of isobutanol (Figure 1c). Because the superlattices are bulk materials, they are readily condensed or recovered via gentle centrifugation, which leads to simple means to accelerate catalytic conversion or enable the reuse of the materials.
RESULTS AND DISCUSSION
Directed assembly of P22 VLP superlattices
We examined the higher order assembly of individual P22 VLPs into arrayed materials mediated through electrostatic interactions. Wild type P22 VLP (wtP22), and a surface modified P22 VLP variant were utilized for this study (Supporting Information Section 3). The variant displays a small peptide repeat on the C-terminus of each P22 subunit, exposed on the exterior surface of the VLP, and is thus expected to provide sterically repulsive interactions between the P22 VLPs.40 The introduced peptide was net negatively charged (VAALEKE)2 and the VLP construct is referred to as P22-E2.
We investigated the assembly behavior of the VLPs at varying ionic strengths (I) to modulate the magnitude of the electrostatic interactions (Supplementary Table S3). Initially, wtP22 was mixed with either an amine-terminated generation 6 PAMAM dendrimer (G6, positively charged) or a carboxyl-terminated generation 6.5 dendrimer (G6.5, negatively charged) in buffer solutions (pH 7.0) with a range of ionic strengths (I). The turbidity measurements shown in Figure 2a reveal that solutions of wtP22 capsids assembled to larger aggregates, upon addition of G6 (1000 G6 per VLP), at ionic strengths up to I = 247 mM whereas the solutions remained clear upon addition of G6.5 over the entire range of ionic strengths tested (Supplementary Figure S3). This suggests that the assembly is mediated by electrostatic interactions between positively charged G6 and the negatively charged P22 VLP (zeta potential for wtP22 of −30.7 ± 2.3 mV; Figure 2b). This is also consistent with the hierarchical assembly of other VLPs reported previously.14–17 We define the ionic strength above which assembly does not occur as the threshold ionic strength (IT). The zeta potential of P22-E2 (−30.4 ± 2.8 mV) is almost identical to that of wtP22, and formed assemblies with G6 up to I = 247 mM, the same as observed for wtP22. A critical factor that contributes to the ionic strength threshold is charge screening, described by the Debye screening length κ−1, the distance over which electrostatic interactions between particles become attenuated in solution (Supporting Information Section 4). With increasing ionic strength, κ−1 decreases, and electrostatic interactions between P22 VLPs and G6 become weaker. Above the IT, electrostatic interactions are too weak to mediate the assembly of particles. It is interesting that the higher order assembly of P22 VLP/G6 shows a sharp ionic strength dependence, similar to reported studies of array formation using other virus capsids.15–16 This very sharp transition is likely due to multivalent interactions between VLP and dendrimer. When the interaction between VLP and G6 is initiated, it quickly propagates the assembly of multiple VLPs and G6 dendrimers.
Figure 2.
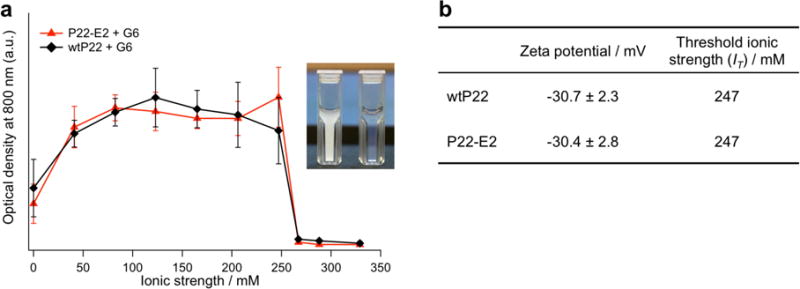
Assembly of P22 VLPs mediated by G6 PAMAM dendrimer. (a) Assembly of wtP22 and P22-E2 variant in solution under a range of ionic strengths (I) was assessed by monitoring light scattering at 800 nm. A rapid increase in optical density was observed upon addition of G6 PAMAM dendrimer to both wtP22 and P22-E2 under I = 0 – 247 mM due to the formation of large aggregates. In the case of P22-E2 + G6, an increase in optical density was observed upon addition of G6 under the same I range as wtP22 + G6. (Inset) A solution of P22-E2 mixed with G6 in I = 206 mM became turbid (left), whereas when mixed in I = 329 mM remained clear (right). (b) The zeta potential of each P22 VLP at pH 7.0 and the IT for assembly with G6.
The hierarchically assembled structures of each P22 VLP construct with G6 were analyzed by small angle X-ray scattering (SAXS) (Figure 3, Supplementary Figures S4 – S8, and Supplementary Table S4). The broad peaks observed in the SAXS data of wtP22-derived samples (Figure 3a) indicate that they do not possess significant long-range order. The result suggests that strong electrostatic attraction alone leads to kinetically trapped amorphous aggregates and might not allow rearrangement of particles within the assembly. In contrast, the samples constructed with P22-E2 exhibited sharp SAXS peaks when they were assembled near their IT (Figure 3b, c), suggesting that P22-E2 VLPs assemble into ordered superlattice structures. This result demonstrates that the structure of the VLP assembly can be converted from an amorphous aggregate to an ordered superlattice by tuning interparticle interactions. This is achieved by genetically modifying the exterior surface of the VLPs to provide sterically repulsive interactions between the P22 VLPs and allow the assembly to adopt an energetically more favored ordered structure. This is consistent with other colloidal systems, where the combination of repulsive and attractive interactions allows for particle reorganization and minimizes kinetically trapped aggregation.1, 4, 41 At lower ionic strengths, the SAXS peaks of the P22-E2 are as broad as those observed for the wtP22 assembly. This indicates that theP22-E2 can form kinetically trapped amorphous structures when the ionic strength is far below the IT where the attractive electrostatic interactions are too strong. The domain sizes of the crystalline P22-E2/G6 assemblies are estimated to be at least ~ 400 nm, much larger than that of the wtP22 and G6 assembly (Supporting Information Section 7). Optical microscopy of the P22-E2 superlattices revealed the presence of particulates with sizes in the range of 1 – 10 μm (Supplementary Figure S9), i.e. larger than the estimated average crystalline domain size indicating that the prepared VLP arrays are likely best thought of as polycrystalline materials.
Figure 3.
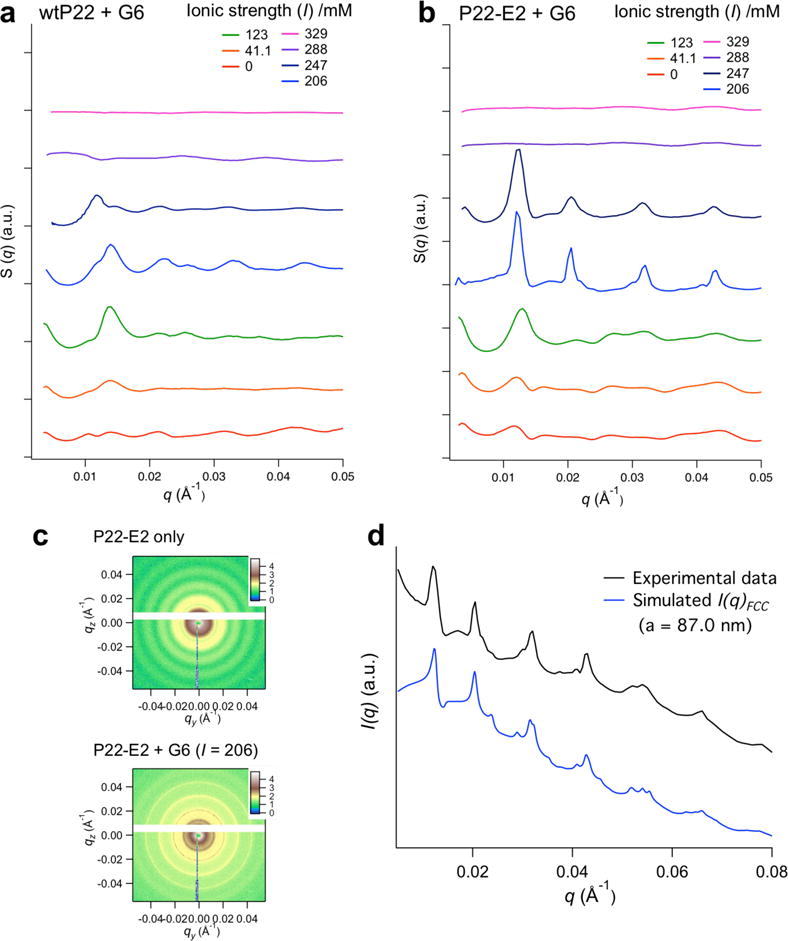
SAXS analysis of P22 VLP assemblies mediated by G6 PAMAM dendrimers. (a, b) The structure of P22 assemblies formed under various ionic strengths (I) was investigated by SAXS. SAXS data are plots of structure factor S(q) versus scattering vector q (plots are vertically offset for clarity). In the case of P22-E2 (b) new peaks attributed to a structure factor of the assembly became more prominent with increasing ionic strength up to the threshold ionic strength (IT) for each construct. This indicates that the array materials with longer-range order are achieved near the IT. Conversely, the structure of the wtP22 assembly (a) shows broad peaks, suggesting some short-range order of wtP22 VLPs in the assembly, however no prominent peaks due to long-range order were observed. (c) 2D scattering patterns of individual P22-E2 VLPs and P22-E2 + G6 assembled at I = 206 mM. (d) Comparison of the experimental scattering profile of P22-E2 + G6 assembled at I = 206 mM (black) matched with the simulated scattering pattern of an FCC structure with lattice parameter a = 87.0 nm and a nearest neighbor distance between VLPs of 61.5 nm (blue).
Based on the observed SAXS data and simulated SAXS profiles, the structure of the superlattice assembly is best modeled as a face centered cubic (FCC) structure with lattice parameter a = 87.0 nm, which corresponds to a nearest neighbor distance between P22 VLPs of 61.5 nm (Figure 3d, and Supplementary Figures S7 and S8). Because the P22 VLP is approximately 56 nm in diameter (Supplementary Figure S2)32 and G6 is 6.7 nm in diameter, the nearest neighbor distance of the simulated structures are reasonable if the G6 dendrimers were positioned between the VLPs in the assembly. We have measured several different batches of P22-E2 and G6 assemblies, prepared under the same conditions (I = 206 mM), and the prominent peaks assigned to FCC with a = 87 nm were consistently observed in all samples. While most of the samples showed a single FCC phase, in some samples, the structure could be interpreted as a mixture of a predominant FCC phase with a minor component of a hexagonal closed packed (HCP) phase with a = 61.5 nm and c = 101.1 nm (see Supplementary Figure S8). This has precedent in the assembly of spherical colloids where mixtures of FCC and HCP lattices are known to form due to the small energy difference between these phases.42–44
To further confirm the structure of the superlattice, assembled samples of P22-E2 and G6 prepared at I = 206 mM were imaged by transmission electron microscopy (TEM) after Pt-Pd shadowing of the sample grid. VLP superlattices showing both hexagonal close packed and square lattices, consistent with the {111} and {100} lattice planes of an FCC structure with a nearest neighbor distance of 62 nm, were observed (Figure 4a and Supplementary Figure S10). We also performed cryo-EM of the sample in vitreous ice and the Fourier Transform of the superlattice also indicated hexagonal packing of VLPs with an interparticle distance of 63nm (Supplementary Figure S11). Importantly, the TEM and cryo-EM observations agree well with the results from the SAXS measurements, which were performed in-situ where the assembled sample was dispersed in solution. Therefore the ordered lattice structures observed by TEM (Figure 4a and Supplementary Figure S10) are unlikely to be a consequence of artefacts due to drying during TEM grid preparation. Together the results from SAXS, TEM and cryo-EM suggest that the structure of the ordered VLP superlattice corresponds to an FCC lattice with a lattice constant a = 87 nm.
Figure 4.
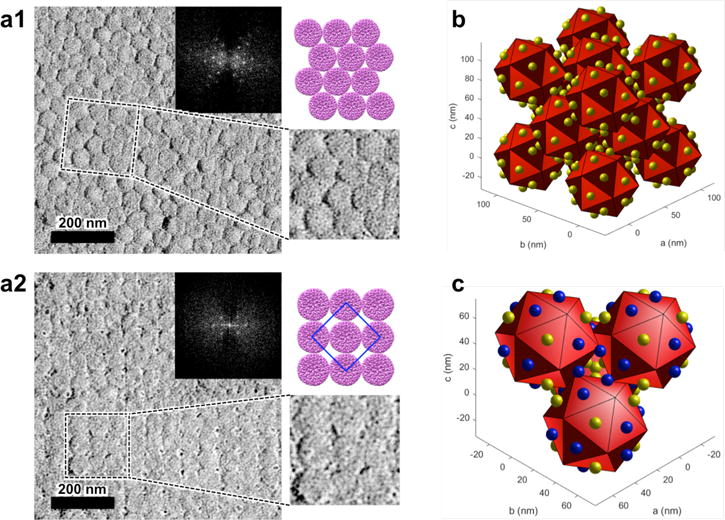
TEM characterization and structural model of P22-E2 superlattice. (a) The structure of the P22-E2 + G6 superlattice assembled at I = 206 mM was observed by TEM following Pt-Pd shadowing of the sample. An expanded image (bottom right) and an image of VLP arrangement in the corresponding area (top right) are also presented. (a1) An image of the assembly showing hexagonal close packing of P22-E2 VLPs. The FFT image (inset) indicates the inter-row spacing of about 54 nm, which corresponds to a nearest neighbor distance of 62 nm, which matches well with the SAXS data. (a2) An image of the assembly showing a square lattice arrangement of P22-E2 VLPs. The FFT image (inset) indicates the period that corresponds to the center-to-center distance between the nearest neighbor VLPs, is about 61 nm. The blue-line square in the top right illustration corresponds to a unit cell on the {100} face. (b) Structural model of an FCC unit cell, consisting of 14 icosahedrons. The yellow spheres associated with each face of the icosahedron represent the dendrimer molecules. Because a sphere at each lattice point in an FCC structure is surrounded by 12 nearest neighbor spheres and eight tetrahedral coordination sites, 12 faces of an icosahedron are in close proximity to the nearest neighbors and the other 8 faces are exposed to tetrahedral coordination sites. (c) Structural model of the tetrahedral coordination site in the FCC lattice. Here, 12 dendrimers on each icosahedron, which bridge to the 12 nearest neighbor icosahedra are shown in blue. The other eight dendrimers are shown in yellow. These eight dendrimers are located at the tetrahedral coordination sites in the FCC lattice.
The amount of G6 dendrimer incorporated into the superlattice of P22-E2 with G6 (prepared at I = 206 mM) was analyzed and approximately twenty G6 per P22-E2 VLP were found, using two independent analytical methods (Supporting Information Section 9). Additionally, DLS measurements of the assembly of P22-E2 VLP, with various ratios of G6, suggest that about 20 – 30 G6 dendrimers per VLP are needed to induce the higher order assembly (Supplementary Figure S14). These results were consistent and suggest that there is approximately one G6 dendrimer per triangular face of the icosahedral VLP (Figure 4b). We propose a model where icosahedral particles are packed into an FCC lattice and their 12 vertices are located near the face centers of their neighbors. The 12 G6 dendrimers can potentially bridge each vertex to one neighboring face of each nearest neighbor P22 VLP (Figure 4c). The 8 G6 dendrimers that do not play a role in bridging may be located at the tetrahedral sites of the FCC (Wyckoff position 8c) (Figure 4c). When too many G6 are bound to a VLP steric repulsion between G6 dendrimers at this site may prevent FCC packing of VLPs and result in a disordered amorphous assembly. In fact the P22-E2 assembly prepared at I = 41.1 mM, which shows an amorphous structure, incorporates twice as many G6 dendrimers as the ordered superlattice P22-E2 assembly (Supplementary Table S5), likely due to stronger electrostatic interactions as a consequence of lower ionic strength conditions.
Catalytic activity of enzyme encapsulated VLP superlattices
We examined the functionality of the P22 VLP superlattices with an incorporated catalytic reaction network comprising two different enzymes encapsulated within the P22 VLPs; ketoisovalerate decarboxylase (KivD) and alcohol dehydrogenase A (AdhA). These enzymes perform a consecutive two-step reaction in which α-ketoisovalerate is first converted to isobutyraldehyde by KivD and then to isobutanol by AdhA45–46 (Figure 5a). KivD and AdhA were encapsulated in P22-E2 VLPs separately as fusions to the SP (referred to as P22-E2-KivD and P22-E2-AdhA). The P22-E2-KivD and P22-E2-AdhA form capsids with morphologies indistinguishable from empty P22-E2 capsids (Supplementary Figure S15). Both KivD and AdhA encapsulated inside of the capsid maintain their catalytic activity (Supplementary Figure S16).
Figure 5.
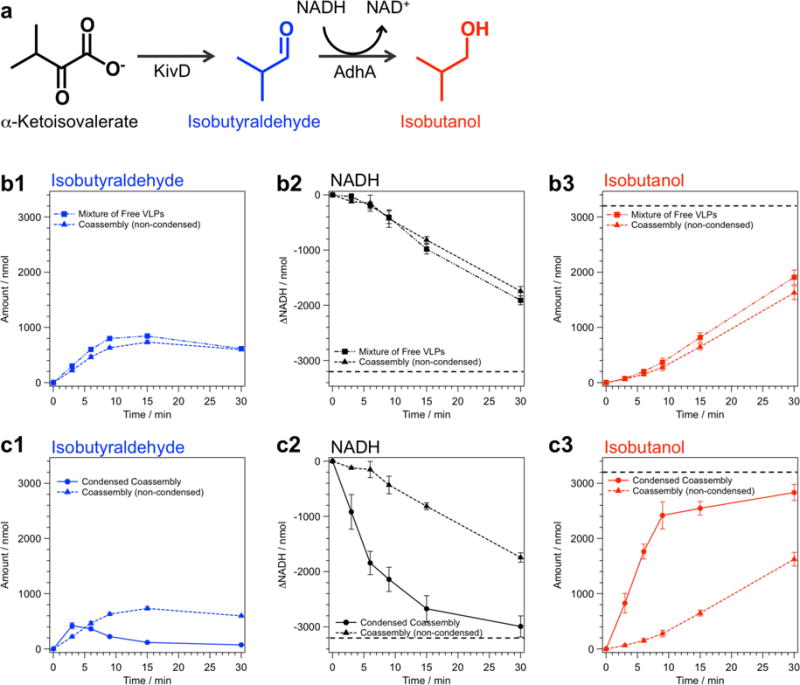
Catalytic activity of co-assembled array composed of P22-E2-KivD and P22-E2-AdhA and corresponding non-assembled mixture. (a) Reaction scheme of the sequential conversion of α-ketoisovalerate to isobutyraldehyde by Ketoisovalerate Decarboxylase (KivD) followed by the reduction of isobutyraldehyde to isobutanol by Alcohol Dehydrogenase A (AdhA) with the concomitant oxidation of NADH. (b, c) The progress of the two-step reaction catalyzed by the mixture of free nanoreactors, co-assembled nanoreactor superlattices, or condensed superlattices was monitored with gas chromatography-mass spectrometry (GC-MS) (production of isobutyraldehyde and isobutanol) and UV-Vis spectroscopy (conversion of NADH to NAD+). Catalytic activity of the co-assembled superlattices and the condensed material, in which the superlattice was resuspended in one-tenth of initial buffer volume, was compared with mixture of free P22-E2-KivD and P22-E2-AdhA under the same conditions of enzyme and substrate concentration. The net conversion profiles were similar between the free nanoreactors and the non-condensed superlattices (b), whereas the conversion was significantly accelerated in the condensed superlattice sample (c). The dotted line in the NADH depletion and isobutanol production indicates 100% conversion of supplied substrate to the final product.
The ionic strength dependence of the G6 mediated assembly of enzyme encapsulated P22-E2 VLPs was the same as that of P22-E2 without enzyme cargos (Supplementary Figure S17). Confocal microscopy observations of the VLP lattices co-assembled from P22-E2-KivD and P22-E2-AdhA revealed that the two types of VLPs are homogeneously distributed over the bulk (Supplementary Figure S18). Furthermore, G6 mediated assembly of either P22-E2-KivD or P22-E2-AdhA individually as well as mixtures of P22-E2-KivD and P22-E2-AdhA were analyzed by SAXS (Supplementary Figure S19). These samples showed the same FCC structure regardless of the presence or absence of cargo enzymes inside the P22-E2 VLP when they were assembled at ionic strengths near IT. These data indicate that higher order assembly of P22 VLPs is independent of cargo proteins encapsulated inside the VLP. This is most likely because the cargo is partitioned away from exterior surface of the VLP by the capsid shell and higher order assembly is a consequence of interparticle interactions (electrostatic and steric) between the exterior surfaces of the VLPs and the G6 dendrimer. Because higher order assembly with a random distribution of VLPs is possible regardless of the interior cargos, this modular assembly approach could be applied to a wide range of functional VLPs. Based on the molecular weight measurements of the VLPs using MALS (Supporting Information Section 11) and the structural information of the superlattices from SAXS, the maximum enzyme concentration within the superlattices is estimated to be ~ 100 mg/cm3 (Supporting Information Section 13), which is significantly higher that the enzyme concentration in the free P22 VLP solution (~ 0.3 mg/cm3) from which the superlattices were formed.
Superlattices, composed of two populations of VLP nanoreactor modules, retain their individual and coupled activities resulting in production of isobutanol via a two-step reaction. The co-assembled nanoreactor superlattices and mixtures of free nanoreactors followed similar patterns of isobutyraldehyde (intermediate) build-up, NADH depletion, and production of isobutanol (Figure 5b, Supplementary Figure S21a, band S22 ), indicating that the superlattices retain comparable catalytic activity to the free VLPs despite the fact that they are bulk materials in which enzymes are immobilized. The use of enzymes as synthetic biocatalysts often requires their immobilization on a support material as a means to separate enzymes from substrates and products thus facilitating their recovery and reuse.47 However, immobilization of enzymes into bulk materials often results in significant loss of activity and mitigating the loss is a critical challenge in biotechnology,48 which is exacerbated for multi-enzyme systems.49 The retained activity of enzymes in nanoreactor superlattices is likely due to the nature of the hierarchically assembled structure of the material. Individual enzymes are confined inside the VLP and are protected from the external environment.37–38 Additionally, the P22 VLPs have 2-3 nanometer pores between CP subunits (full length SPs can escape from intact P22 VLPs50), hence diffusion of small molecule substrates across the capsid shell are not hindered. Molecular diffusion through the lattice also does not appear to be a rate-limiting step in the overall reaction.
The overall coupled catalytic conversion could be readily accelerated by taking advantage of the properties of the superlattice material as a condensed phase, which allows it to settle over time (or be pelleted via gentle centrifugation) into a highly condensed material (Supplementary Figure S20). The co-assembled superlattices were condensed to one-tenth of the initial volume via gentle centrifugation followed by removal of supernatant. Addition of the same amount (3200 nmol) of substrate (α-ketoisovalerate) to the condensed and non-condensed nanoreactor superlattices demonstrated that the condensed sample processed the two-step reaction significantly faster because of the higher substrate and catalyst concentration (Figure 5c and Supplementary Figure S21c). Achieving comparable enzyme concentrations using the free nanoreactors (or the free enzymes) would require significant effort to recover and concentrate the catalyst and could be beyond the solubility limit of some enzymes. This ease of handling of the nanoreactor superlattices also allows for the catalyst material to be easily recovered and recycled. We measured catalytic activity of the superlattice three times after their isolation from the product by gentle centrifugation followed by re-suspension and incubation with new substrate. These re-usability studies indicate the activity was maintained over multiple catalytic cycles (Supplementary Figure S23) and highlight the utility of using the enzyme encapsulated VLP superlattices for catalytic transformations.
CONCLUSION
The construction of ordered three-dimensional lattices from nano-size building blocks has been studied over the past decade and a range of ordered structures have been realized.1–9 However, it is still challenging to design and fabricate functional superlattices with properties arising from the interaction between individual building blocks. Here, VLP based nanoreactor superlattices that perform a coupled two-step reaction for isobutanol synthesis were successfully developed. This study demonstrates a significant step toward the bottom-up fabrication of functional superlattice materials using self-assembly processes across multiple length scales, which exhibit properties and function that arise from the interaction between individual building blocks. The interactions that govern the assembly of VLPs into higher order structures depend on the exterior surface of the VLPs and are independent of cargo molecules encapsulated on the VLP interior. A large diversity of functional cargo molecules have been encapsulated inside of VLPs via this self-assembly processes13, 23–26, 28, 30, 35–39, 51 and thus the approach to construct higher order assemblies presented here could be widely applicable not only in catalysis but also for the design of a wide range of functional materials.
METHODS
General
All water was deionized using a Milli-Q Advantage A10 water purification system (EMD Millipore). DNA modifying enzymes were purchased from New England Biolabs (Ipswich, MA) and Promega (Madison, Wisconsin). DNA primers were purchased from Eurofins MWG Operon (Huntsville, AL) and Integrated DNA Technologies (Coralville, IA). E. coli BL21(DE3) strain and XL1 strain were purchased from Novagen (Billerica, MA) and Agilent Technologies (Santa Clara, CA), respectively. L. lactis subsp. lactis DNA was purchased from ATCC (Manassas, VA). QIAquick gel extraction kit and QIAprep Spin Miniprep kit were purchased from Qiagen (Valencia, CA). DNase, RNase, Lysozyme, thiamine pyrophosphate (TPP), α-ketoisovalerate, Alcohol Dehydrogenase and PAMAM dendrimers were purchased from Sigma Aldrich (St. Louis, MO). All other chemical reagents were purchased from Fisher Scientific (Pittsburgh, PA). DNA sequences of the proteins used in this study are summarized in the Supporting Information Section 1.
Genetic engineering of P22 VLP mutant
A truncated form of P22 Scaffolding Protein (amino acids 142 to 303, hereafter referred to as SP141) and P22 Coat Protein (CP) were cloned into the first and the second multi cloning sites of the pRSFDuet-1 vector (Novagen), respectively. Two repeats of a net negatively charged peptide with sequence VAALEKE (E2 peptide) was introduced at the C-terminus of the CP (hereafter referred to as CP-E2). Because the P22 VLP is constituted with 420 copies of CP and the C-terminus of CP is known to be exposed to the exterior of the capsid,40 420 copies of the E2 peptide is displayed on the outside of the P22 VLP for the mutant. The P22 VLPs with the E2 peptide is referred to as P22-E2. The amino acids sequences of CP and CP-E2 are shown in Supplementary Table S1.
Expression of wtP22 and P22-E2 (without enzymes)
Each pRSFDuet-1 vector containing either wtCP or CP-E2, and SP genes, was transformed into BL21 (DE3) E. coli strain. The cells were seeded on an LB-agarose plate supplemented with 30 μg/mL of kanamycin, and the plate was incubated at 37°C overnight for screening. A colony of each vector was picked and grown in LB medium at 37 °C in the presence of 30 μg/mL kanamycin to maintain selection pressure. Expression of the proteins was induced by isopropyl β-D-thiogalactopyranoside (IPTG) to a final concentration of 0.3 mM when O.D. at 600 nm of the culture medium reached 0.6 a.u., and the medium was continuously cultured for 4 h. After the incubation, cells were harvested by centrifugation and cell pellets were stored at −80 ° C until subjected to purification of the protein.
Engineering of KivD-SP141 and AdhA-SP141
The genes for Ketoisovalerate Decarboxylase (KivD)52 and Alcohol Dehydrogenase A (AdhA)53 were amplified from L. lactis subsp. lactis genomic DNA. Primers used to amplify and clone genes are listed in Supplementary Table S2. Many variants of α-keto acid decarboxylases with similar gene sequences have been isolated from different subspecies of L. lactis. Some of these are reported as indole-3-pyruvate decarboxylase (I3PD) based on an automated computational analysis using gene prediction methods. However, biochemical characterization of an α-keto acid decarboxylase (NCBI Accession#: CAG34226), which has 99% amino acid identity with the KivD cloned for this study, revealed that the enzyme exhibited over 1000 times higher decarboxylation activity toward α-ketoisovalerate than indole-3-pyruvate.52 Therefore, we denote the enzyme we have cloned as KivD rather than I3PD.
It has been shown that a truncated version of SP, maintaining the C-terminal, CP binding region, is sufficient to promote VLP assembly.33, 54 We have demonstrated that genetic fusion of a cargo protein of interest to the truncated SP results in directed encapsulation of the cargo in the VLP.35–38 Therefore, each enzyme was genetically fused to the N-terminus of the truncated SP (SP141) spaced by a nine amino acid linker (GSLVPRGSC). Restriction sites NcoI, BamHI and SacI were added upstream and downstream (upstream of SP141) of each cargo gene, and after the stop codon, respectively. Each cargo-SP141 was inserted into a pBAD vector containing an ampicillin resistance gene. Bacterial colonies resulting from transformation of the ligation were screened by colony PCR, restriction enzyme digestion and the DNA from all positive colonies was sequenced for complete verification.
Expression of enzyme encapsulated P22-E2
The enzymes KivD and AdhA, which carry out a two-step cascade reaction (Figure 5a), were separately encapsulated in the P22-E2 capsid (hereafter referred to as P22-E2-KivD and P22-E2-AdhA, respectively). A combination of the two vectors, pRSFDuet-1 (containing a gene for kanamycin resistance) with CP-E2 (but no SP) and either pBAD (containing a gene for ampicillin resistance) with KivD-SP141 or pBAD with AdhA-SP141 were co-transformed into BL21 (DE3) E. coli cells. The cells were plated on an LB-agarose plate supplemented with 50 μg/mL of ampicillin and 30 μg/mL of kanamycin to select for E. coli colonies with both vectors, and the plate was incubated at 37°C overnight. One colony from each plate was picked and grown in LB medium at 37 °C in the presence of ampicillin and kanamycin to maintain selection for both plasmids. Expression of enzyme-SP141 was induced by the addition of L-arabinose to a final concentration of 13.3 mM once the cells reached mid-log phase (O.D.600=0.6). After enzyme-SP141 was expresses for 4 hours, expression of the CP-E2 was induced with IPTG to a final concentration of 0.3 mM for 2 additional hours. Cells were harvested by centrifugation and cell pellets were stored at −80 ° C.
Purification of P22 VLPs
P22 VLPs used in this study were purified by the following procedure, which was a minor modification of previously described method.28–29, 55 Cell pellets were resuspended in phosphate buffer (sodium phosphate 50 mM, sodium chloride 100 mM pH 7.0, hereafter refer as standard phosphate buffer) and lysozyme, DNase, and RNase were added to final concentrations of 50, 60, and 100 μg/mL, respectively. For the P22-E2-KivD construct, 5 mM MgCl2 and 2.5 mM TPP were also supplemented to the suspension, as they are necessary co-factors. The cell suspensions were incubated at room temperature with gentle shaking for 30 minutes and lysed by sonication for 3 minutes at 50% amplitude on ice. The sonication process was repeated three times. Cell debris was removed by centrifugation at 12,000 g for 45 min at 4 °C and then passed through a 0.45μm filter. Samples were purified by ultracentrifugation over a 35% (w/v) sucrose cushion at 45,000 rpm (F50L-8×39 rotor, Piramoon Technologies) for 50 min. The resulting viral pellets were resuspended in the standard phosphate buffer and further purified over a Sephacryl S-500 HR size exclusion column (SEC: GE Healthcare Life Sciences) using fast protein liquid chromatography (BioLogic DuoFlow, Bio-Rad, Hercules, CA). The flow rate for SEC purification was 1 mL/min of phosphate buffer. Fractions containing P22 VLP were concentrated by ultracentrifugation at 45,000 rpm (F50L-8×39 rotor) for 50 min and the resulting pellets were re-suspended in an adequate volume of phosphate buffer.
Characterization of P22 VLPs
The molecular weights of P22 VLPs were measured by multi-angle light scattering (MALS: DAWN8+, Wyatt Technology, Santa Barbara, CA) equipped with a He-Ne laser source, quasi-elastic light scattering detector, and refractive index (RI) detector (Optilab T-rEX, Wyatt Technology). Samples were separated over a WTC-200S5 (Wyatt Technologies) size exclusion column (SEC) utilizing an Agilent 1200 HPLC to apply and maintain a 0.7 mL/min flow rate of phosphate buffer at pH 7.2 containing 50 mM sodium phosphate, 100 mM sodium chloride and 200 ppm sodium azide. Samples of 25 μL were injected onto the column and total run time was 30 minutes. The number average molecular weight, Mn was calculated with Astra software ver. 6.0.3.16 (Wyatt Technology). A refractive index increment (dn/dc) of protein (0.185) was used to calculate the molecular weight of all samples. Hydrodynamic diameters of P22 VLPs were measured by dynamic light scattering (DLS: Zetasizer Nano ZS, Malvern Instruments, Worcestershire, UK). Zeta-potentials of P22 VLPs in buffer solution (sodium phosphate 10 mM, sodium chloride 20 mM, pH 7.0) were measured using Zetasizer Nano ZS (Malvern Instruments). The Smoluchowski approximation was used to convert the electrophoretic mobility to zeta potential.
Protein concentration was determined by UV absorption of guanidine hydrochloride denatured samples measured at 280 nm. Absorbance values were divided by calculated molar extinction coefficients using the ExPASy ProtParam tool (Swiss Institute of Bioinformatics) yielding values of ε280 = 44,920 M−1cm−1 for CP and CP-E2, ε280 = 12,090 M−1cm−1 for SP141, ε280 = 57,425 M−1cm−1 for KivD- SP141, and ε280 = 41,995 M−1cm−1 for AdhA- SP141. The ratio of enzyme-SP fusion protein to CP was determined from molecular weight data ascertained from MALS as described previously.35 The molecular weight of 420 CP-E2 (20.28 MDa) was subtracted from the average molecular weight of either P22-E2-KivD (30.41 MDa) or P22-E2-AdhA (33.20 MDa) obtained by SEC-MALS analysis to determine the average mass of total enzyme-SP141 encapsulated in P22. These values were divided by the molecular weight of the enzyme-SP141 (79,693.8 Da and 54,685.7 Da for KivD-SP141 and AdhA-SP141, respectively) to obtain the number of enzyme subunits packaged per capsid. The analysis revealed that 127.1±1.4 KivD and 236.2±1.3 AdhA are encapsulated per P22-E2 on average (Supplementary Table S6). This number was further used in kinetic assays to determine proper ratios of the two enzymes.
Preparation and monitoring of P22 VLP assembly mediated by dendrimer
P22 VLP was buffer exchanged to various ionic strength buffers as summarized in Supplementary Table S3. PAMAM dendrimer G6 or G6.5 in methanol (as purchased) was diluted with the same buffer solution at a ratio of 1:4 (vol/vol). Typically, 150 μL of P22 VLP (50 nM) was placed in an eppendorf tube followed by addition of the PAMAM dendrimer solution at a defined ratio of 1000 dendrimer/P22 VLP. Optical density of the samples at 800 nm was measured (UV-Vis; Agilent 8453) 30 min after the addition of the dendrimer. In the Figure 2a and S17, the data was normalized to the highest optical density of each P22 VLP mutant data set.
Small-angle X-ray scattering (SAXS) measurement
SAXS experiments were conducted at the X9 beamline at the National Synchrotron Light Source (NSLS) and the 12ID-B beamline at the Advanced Photon Source (APS). All the samples were measured in the buffer solutions as prepared. The measurements were carried out at 13.5 keV (X9) or at 14 keV (12ID-B) and the scattering data were collected with a Pilatus 1M detector at the X9 and a Pilatus 2M detector at the 12ID-B. Scattering angle was calibrated using silver behenate as a standard. One-dimensional scattering profiles were obtained by averaging two-dimensional scattering patterns. The data were represented as scattering intensity as a function of scattering vector q:
where θ is half of the scattering angle 2θ and λ is the x-ray wavelength used for the measurements.
Transmission electron microscopy
All samples for TEM were diluted 10 times with water prior to the observation. Samples of individual P22 VLPs (5 μL, 0.1 mg/mL protein) were applied to formvar coated copper grids and incubated for 30 seconds and excess liquid was blotted with filter paper. Grids were then stained with 5 μL 2.0% uranyl acetate and excess stain was wicked away with filter paper. Images were taken on a JEM 1010 transmission electron microscope (JEOL Ltd., Akishima, Japan) at accelerating voltage of 80 keV. Hierarchical assemblies of P22-E2 and G6 dendrimer were imaged by placing 4 μL of solution onto an electron microscope grid with carbon support film and excess liquid was blotted with filter paper. Platinum-Palladium wire was evaporated at an angle of 40° over the grid for shadowing and specimens were examined by JEM 1010 at an acceleration voltage of 80 keV.
Samples for cryo-electron microscopy (cryo-EM) of the assembly of P22-E2 and G6 were embedded in a thin layer of vitreous ice on holey carbon grids with 2 μm holes (C-flat holey carbon grid, Electron Microscopy Sciences, Hatfield, PA) and plunge-frozen using a Vitrobot (FEI, Hillsboro, OR). The cryo-EM data was collected on a JEM 3200FS (JEOL Ltd.) with the cryo-transfer holder model 626 (Gatan Inc., Pleasonton, CA) at liquid nitrogen temperature. Zero loss images, through a 30 eV energy filter, were recorded on a CCD camera (UltraScan4000, Gatan Inc.).
Quantification of G6/P22-E2 ratio in the superlattice
The amount of G6 dendrimers incorporated in the superlattice per P22-E2 VLP was quantified by two different approaches: SAXS and quantitative amino acid analysis (AAA). For SAXS, the assembly of P22-E2 and G6 dendrimer prepared in phosphate buffer (I = 206 mM) was pelleted down in a table-top centrifuge (21,100 g) followed by resuspension in buffer twice to remove excess free dendrimer and P22-E2. The second resuspension was done in a high salt buffer at I = 329 mM (80 mM sodium phosphate, 160 mM sodium chloride, pH7.0). Under the high salt condition, the sample disassembles into the individual components P22-E2 and G6. The SAXS profile of the disassembled sample was fit with a combination of separately measured P22-E2 and G6 experimental SAXS profiles to determine the ratio of G6/P22-E2 VLP in the superlattice (Supplementary Figure S12). For AAA, the assembled samples (prepared at I = 206 mM and I = 41.1 mM) were washed as described above and then disassembled into a high salt buffer (80 mM sodium phosphate, 160 mM sodium chloride, pH7.0). These samples were acid hydrolyzed at 145 °C for 2 hours, followed by derivatization of amino acids with a fluorescence tag (AccQ-Fluor reagent kit, Waters, Milford, MA), and then analyzed with HPLC to quantify each component. The elution peak attributed to the dendrimer was observed directly after the phenylalanine peak. The concentrations of G6 in the samples were determined in comparison with a standard curve (Supplementary Figure S13). P22 VLP concentrations were also quantified through amino acid analysis and the ratio of G6/P22 VLP was determined (Supplementary Table S5).
Confocal fluorescence microscopy
P22-E2-KivD and P22-E2-AdhA were labeled with DyLight 405 maleimide (Thermo Fisher Scientific, Waltham, MA, USA) and Texas Red C2 maleimide (Thermo Fisher Scientific), respectively. The CP of P22 does not possess reactive cysteines55 whereas SP-KivD and SP-AdhA have two and ten cysteine residues, respectively, hence the dyes were conjugated with the SP-enzyme fusion proteins encapsulated inside of VLPs. For P22-E2-KivD, the VLP was treated with 1 mM DTT at room temperature for 4h prior to the labeling with DyLight 405. P22-E2-KivD in standard phosphate buffer (I = 206 mM) was reacted with the dye at a concentration of four molar equivalents per SP-KivD subunit at room temperature for 4 h. Unreacted free dye was removed using Micro Bio-Spin Columns P-30 (Bio-Rad). Purified P22-E2-KivD was measured with UV-Vis spectroscopy and confirmed that approximately one dye was conjugated per SP-KivD subunit. For P22-E2-AdhA, the VLP was mixed with a 0.25 molar equivalent of Texas Red dye per SP-AdhA and reacted at room temperature for 4 h. After purification, using Micro Bio-Spin Columns P-30, nearly all dye (i.e. 0.25 dye per subunit) was confirmed to be attached. The fluorescently labeled P22-E2-KivD and P22-E2-AdhA were mixed at a 1:1 molar ratio followed by G6 dendrimer addition to form higher order assemblies as described above. Fluorescence images of assembled VLPs were acquired using a Leica TCS SP8 confocal scanning microscope and Leica LAS-X software. Images were modified only in terms of brightness and contrast.
Enzyme activity assay of individual P22 VLPs
Turnover rates for P22-E2-AdhA in standard phosphate buffer (I = 206 mM) were monitored in a range of isobutyraldehyde concentrations (1 mM - 90 mM). Activity was monitored by recording the decrease in absorbance at 340 nm, indicative of conversion of NADH to NAD+. P22-E2-KivD turnover rate was measured in standard phosphate buffer in a range of α-ketoisovalerate concentrations (2.5 mM - 70 mM) with excess commercially available Alcohol Dehydrogenase, which acted as a reporter enzyme. All KivD activity measurements were done in the presence of 2.5 mM TPP. Activity was monitored by recording the decrease in absorbance at 340 nm (NADH consumption), as well as the decrease at 314 nm, indicative of the conversion of α-ketoisovalerate to isobutyraldehyde. Extinction coefficients were experimentally determined for NADH and α-ketoisovalerate at 340 nm to be 5.73 mM−1 cm−1 and 1.59 × 10−2 mM−1 cm−1, respectively, and at 314 nm to be 3.57 mM−1 cm−1 and 3.0 × 10−2 mM−1 cm−1, respectively. Initial turnover rates were analyzed for each substrate concentration at 314 nm and at 340 nm, and then compared to verify precision of the assay. Kinetic parameters of the enzyme encapsulated P22 VLPs were measured on an Agilent 8453 UV-Vis Spectrophotometer with an 8 position multicell transport. All assays were conducted with a final assay volume of 100 μL at room temperature and performed in triplicate. NADH was added to each reaction so that final absorbance at 340 nm = 1.6 - 2.0. NADH thermal degradation was monitored for 100 - 200 seconds then 1 μL enzyme was thoroughly mixed in the solution. Initial rates were applied to a Michaelis-Menten fit (Igor Pro 6.37) after subtraction of non-enzymatic activity to determine kcat and KM of each type of P22-enzyme VLP (Supplementary Figure S16).
Pathway activity of nanoreactor superlattices and free P22 nanoreactors
Experimentally derived kcat values of P22-E2-KivD and P22-E2-AdhA were used to determine molar ratios of enzymes such that their maximum potential rates (Vmax) were at a 1:1 ratio; i.e., kcat of P22-E2-KivD is 3.5 times higher than that of P22-E2-AdhA (Supplementary Figure S16), hence samples were mixed at a 1:3.5 KivD:AdhA molar ratio in standard phosphate buffer (50 mM sodium phosphate, 100 mM sodium chloride, pH 7.0). The G6 dendrimer in methanol (as purchased) was diluted with standard phosphate buffer at a ratio of 1:4 (vol/vol) and 585 μL of the P22 VLP solution was mixed with 215 μL of G6 solution to co-assemble P22-E2-KivD and P22-E2-AdhA nanoreactors into the superlattices. As a control, the same volume of the P22 VLP solution was mixed with standard phosphate buffer with 20% v/v methanol (such that the final methanol concentrations matched the quantity in the superlattice samples). The samples that do not contain G6 dendrimer are referred to as free nanoreactors. The co-assembled superlattices were condensed to one-tenth of their original volume. Eight hundred μL of superlattice solution was allowed to settle or centrifuged at 5000 g for 3 min to gently pellet the superlattices, then 720 μL of supernatant was removed to reduce the buffer volume to 10% and the pellet was gently resuspended in buffer.
The coupled activities of nanoreactor superlattices in the condensed phase, non-condensed condition and free nanoreactors were investigated using exactly the same amount of substrate and enzymes. For condensed phase, the two-step reaction was conducted in a 400 μL solution, which contained 80 μL of condensed suspension of superlattice, 3,200 nmol (8 mM) of α-ketoisovalerate, 3,200 nmol (8 mM) of NADH, and 1,000 nmol (2.5 mM) of TPP. Stock solutions of 160 mM α-ketoisovalerate, 60 mM NADH, and 100 mM TPP were prepared with water and mixed to phosphate buffer to achieve the final concentrations. At 3, 6, 9, 15 and 30 minutes, 70 μL of the reaction solution was mixed with 630 μL of buffer and 70 μL of 1 N HCl to quench enzyme activity, flash frozen in liquid nitrogen, and stored at −80°C for GC-MS analysis. For the non-condensed superlattice and free nanoreactors, the two-step reaction was conducted in a 4000 μL solution, which contained 800 μL of either superlattice or free nanoreactor solution, 3200 nmol (0.8 mM) of α-ketoisovalerate, 3,200 nmol (0.8 mM) of NADH, and 10,000 nmol (2.5 mM) of TPP. At 3, 6, 9, 15 and 30 minutes, 700 μL of reaction solution was mixed with 70 μL of 1 N HCl to quench enzyme activity, flash frozen in liquid nitrogen, and stored at −80°C for GC-MS analysis.
Parallel measurements were conducted under the same assay conditions outlined above but sample activity was monitored by measuring the decrease in absorbance at 340 nm, indicative of conversion of NADH to NAD+. Instead of acid quenching, a fraction of reaction solution was removed from each sample at the time points listed above and added to high ionic strength buffer (100 mM sodium phosphate 200 mM sodium chloride pH 7.0). This step ensured disassembly of G6 mediated superlattices (reducing scattering contributions), significantly slowed the reaction for accurate measurement, and diluted NADH to a measurable absorbance. For the condensed superlattices sample, 5 μL of the reaction solution was added to 195 μL of phosphate buffer. For the non-condensed superlattices sample and free nanoreactor sample, 50 μL of the reaction solutions were added to 150 μL of phosphate buffer. Absorbances were converted to concentrations using the experimentally determined extinction coefficient for NADH of 5.73 mM−1 cm−1. The total amount of oxidized NADH during the duration of the experiment was determined and compared to GC-MS measurements.
Gas chromatography-mass spectrometry detection of isobutyraldehyde and isobutanol
Acid quenched, coupled enzyme reactions were further analyzed by gas chromatography-mass spectrometry (GC-MS) to verify production of isobutyraldehyde (shared intermediate between KivD and AdhA) and isobutanol (final product). Time point measurements from both condensed and non-condensed nanoreactor superlattice samples composed of P22-E2-KivD, P22-E2-AdhA and G6 as well as free nanoreactor mixtures of P22-E2-KivD and P22-E2-AdhA were analyzed. Samples that were acid quenched and flash frozen previously were thawed to room temperature. Samples were analyzed on an Agilent 6890 gas chromatograph by static headspace GC with an auto sampler (Gerstel MPS 2) and heated syringe (constant 75 °C). One hundred μL of sample from each time point was aliquoted into a 10 mL headspace vial (Gerstel) in triplicate. Samples were heated to 80 °C for 1.5 minutes with agitation (250 rpm 15 sec on and 2 sec off), then 500 μL of headspace was injected over 8 sec with a 2:1 split ratio into a 60 meter long, 250 μm inner diameter capillary GC column (1.4 μm thick DB-624, Agilent) with He carrier gas at 1.2 mL/min. The temperature program for the column was hold 50°C for 3 min, ramp 8 °C/min to 100 °C, hold 100 °C 1 min, ramp 25 °C/min to 150 °C. The volatile reaction products were detected by electron ionization mass spectrometry (EI-MS) (Agilent 5973inert), which monitored m/z = 43, 72, and 74 in select ion monitoring (SIM) mode. The sample syringe and needle were flushed with 30 psi nitrogen for 6 min between injections. The elution times of isobutyraldehyde and isobutanol were confirmed to be approximately 8.2 min and 10.4 min, respectively, by measuring isobutyraldehyde and isobutanol standards individually; the intensity ratios (vs m/z 43) of qualifier ions @ 72 and 74, respectively, were monitored to ensure that no other co-eluting compounds were present. An external standard curve for quantitative analysis was generated using mixtures of pure isobutyraldehyde (intermediate) and isobutanol (final product) in phosphate buffer from 20 to 2000 μmol/L. Extracted ion chromatograms of m/z = 43, which correspond to isopropyl fragment ions of both isobutyraldehyde and isobutanol, were used to quantify the amounts of isobutyraldehyde and isobutanol in each sample.
Reusability of nanoreactor superlattices
P22-E2-KivD and P22-E2-AdhA were co-assembled into the superlattices and then condensed as described above. Protein concentration of the condensed sample was measured spectroscopically at absorbance 280 nm following disassembly of the superlattices into free VLPs. Disassembly was achieved by mixing 10 μL of the suspended superlattice with 90 μL of a high salt buffer (100 mM sodium phosphate 200 mM sodium chloride pH 7.0). The absorbance at 280 nm was 0.6 and this value was used to adjust the superlattices concentrations after their recovery in subsequent trials. Because TPP shows strong absorbance at 280 nm and therefore interferes with protein concentration measurements, it was supplemented to the superlattice sample after adjusting protein concentration. The two-step enzyme reaction was conducted by mixing the condensed suspension of superlattice and phosphate buffer with α-ketoisovalerate, NADH, and TPP at a 1:4 ratio (vol/vol). The final concentrations of α-ketoisovalerate, NADH, and TPP were 8 mM, 8 mM, and 2.5 mM, respectively. Progress of the two-step reaction was evaluated by monitoring depletion of NADH for 30 min as described above. After each period of evaluation, the reaction solution was subjected to centrifugation (5000 g for 3 min) to pellet down the superlattices. The recovered superlattices were resuspended in phosphate buffer supplemented with G6 dendrimer followed by centrifugation (5000 g for 3 min) twice to remove product and other remnants. Approximately 80 % of the total superlattices were recovered at the end of each evaluation cycle. The superlattices were subjected to the activity assay three times in total.
Supplementary Material
Acknowledgments
The enzyme encapsulation studies were supported by a grant from the National Science Foundation (NSF-BMAT DMR-1507282). Characterization of P22 assembly into superlattices was supported by a grant from US Department of Energy, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering (DE-SC0016155). BL was supported by MICCoM as part of the Computational Materials Sciences Program funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Materials Sciences and Engineering Division. The BNL contribution to this work was supported by the US Department of Energy, Basic Energy Sciences, by the Materials Sciences and Engineering Division (MF) and through the National Synchrotron Light Source II (MF and LY), under contract No. DE-AC02-98CH10886. LY is also supported by DOE-BER under contract No. DE-SC0012704 and by NIH-NIGMS under grant P41GM111244. This research used Beamline X9 of the National Synchrotron Light Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-AC02-98CH10886 with additional support from NIH through grant S10 OD012331. This research also used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We thank the IU Nanoscale Characterization Facility and the IU Mass Spectrometry Facility for access to their instrumentation. We also thank J. Powers of the IU Light Microscopy Imaging Center (LMIC) for help and advice.
Footnotes
Supporting Information
The supporting information is available free of charge on the ACS Publications website where DNA and amino acid sequences of constructs as well as additional experimental details and data (PDF) are provided.
ORCID
Masaki Uchida: 0000-0003-0710-8834
Dustin P. Patterson: 0000-0001-5291-2966
Byeongdu Lee: 0000-0003-2514-8805
Trevor Douglas: 0000-0002-7882-2704
Notes
The authors declare no competing financial interest.
References
- 1.Park SY, Lytton-Jean AK, Lee B, Weigand S, Schatz GC, Mirkin CA. DNA-Programmable Nanoparticle Crystallization. Nature. 2008;451:553–556. doi: 10.1038/nature06508. [DOI] [PubMed] [Google Scholar]
- 2.Macfarlane RJ, Lee B, Jones MR, Harris N, Schatz GC, Mirkin CA. Nanoparticle Superlattice Engineering with DNA. Science. 2011;334:204–208. doi: 10.1126/science.1210493. [DOI] [PubMed] [Google Scholar]
- 3.Brodin JD, Auyeung E, Mirkin CA. DNA-Mediated Engineering of Multicomponent Enzyme Crystals. Proc Natl Acad Sci USA. 2015;112:4564–4569. doi: 10.1073/pnas.1503533112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nykypanchuk D, Maye MM, van der Lelie D, Gang O. DNA-Guided Crystallization of Colloidal Nanoparticles. Nature. 2008;451:549–552. doi: 10.1038/nature06560. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Y, Lu F, Yager KG, van der Lelie D, Gang O. A General Strategy for the DNA-Mediated Self-Assembly of Functional Nanoparticles into Heterogeneous Systems. Nat Nanotechnol. 2013;8:865–872. doi: 10.1038/nnano.2013.209. [DOI] [PubMed] [Google Scholar]
- 6.Shevchenko EV, Talapin DV, Kotov NA, O’Brien S, Murray CB. Structural Diversity in Binary Nanoparticle Superlattices. Nature. 2006;439:55–59. doi: 10.1038/nature04414. [DOI] [PubMed] [Google Scholar]
- 7.Kalsin AM. Electrostatic Self-Assembly of Binary Nanoparticle Crystals with a Diamond-Like Lattice. Science. 2006;312:420–424. doi: 10.1126/science.1125124. [DOI] [PubMed] [Google Scholar]
- 8.Feng L, Laderman B, Sacanna S, Chaikin P. Re-Entrant Solidification in Polymer-Colloid Mixtures as a Consequence of Competing Entropic and Enthalpic Attractions. Nat Mater. 2015;14:61–65. doi: 10.1038/nmat4109. [DOI] [PubMed] [Google Scholar]
- 9.Kitagawa S, Kitaura R, Noro S. Functional Porous Coordination Polymers. Angew Chem Int Ed. 2004;43:2334–2375. doi: 10.1002/anie.200300610. [DOI] [PubMed] [Google Scholar]
- 10.Whitesides GM, Grzybowski B. Self-Assembly at All Scales. Science. 2002;295:2418–2421. doi: 10.1126/science.1070821. [DOI] [PubMed] [Google Scholar]
- 11.Chowdhury C, Sinha S, Chun S, Yeates TO, Bobik TA. Diverse Bacterial Microcompartment Organelles. Microbiol Mol Biol Rev. 2014;78:438–468. doi: 10.1128/MMBR.00009-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanaka S, Kerfeld CA, Sawaya MR, Cai F, Heinhorst S, Cannon GC, Yeates TO. Atomic-Level Models of the Bacterial Carboxysome Shell. Science. 2008;319:1083–1086. doi: 10.1126/science.1151458. [DOI] [PubMed] [Google Scholar]
- 13.Douglas T, Young M. Viruses: Making Friends with Old Foes. Science. 2006;312:873–875. doi: 10.1126/science.1123223. [DOI] [PubMed] [Google Scholar]
- 14.Kostiainen MA, Kasyutich O, Cornelissen JJLM, Nolte RJM. Self-Assembly and Optically Triggered Disassembly of Hierarchical Dendron-Virus Complexes. Nat Chem. 2010;2:394–399. doi: 10.1038/nchem.592. [DOI] [PubMed] [Google Scholar]
- 15.Kostiainen MA, Hiekkataipale P, Laiho A, Lemieux V, Seitsonen J, Ruokolainen J, Ceci P. Electrostatic Assembly of Binary Nanoparticle Superlattices Using Protein Cages. Nat Nanotechnol. 2012;8:52–56. doi: 10.1038/nnano.2012.220. [DOI] [PubMed] [Google Scholar]
- 16.Liljestrom V, Mikkila J, Kostiainen MA. Self-Assembly and Modular Functionalization of Three-Dimensional Crystals from Oppositely Charged Proteins. Nat Commun. 2014;5:4445. doi: 10.1038/ncomms5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liljestrom V, Seitsonen J, Kostiainen MA. Electrostatic Self-Assembly of Soft Matter Nanoparticle Cocrystals with Tunable Lattice Parameters. ACS Nano. 2015;9:11278–11285. doi: 10.1021/acsnano.5b04912. [DOI] [PubMed] [Google Scholar]
- 18.Cigler P, Lytton-Jean AKR, Anderson DG, Finn MG, Park SY. DNA-Controlled Assembly of a NaTl Lattice Structure from Gold Nanoparticles and Protein nanoparticles. Nat Mater. 2010;9:918–922. doi: 10.1038/nmat2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sontz PA, Bailey JB, Ahn S, Tezcan FA. A Metal Organic Framework with Spherical Protein Nodes: Rational Chemical Design of 3D Protein Crystals. J Am Chem Soc. 2015;137:11598–11601. doi: 10.1021/jacs.5b07463. [DOI] [PubMed] [Google Scholar]
- 20.Uchida M, LaFrance B, Broomell CC, Prevelige PEJr, Douglas T. Higher Order Assembly of Virus-Like Particles (VLPs) Mediated by Multi-Valent Protein Linkers. Small. 2015;11:1562–1570. doi: 10.1002/smll.201402067. [DOI] [PubMed] [Google Scholar]
- 21.Chung W-J, Oh J-W, Kwak K, Lee BY, Meyer J, Wang E, Hexemer A, Lee S-W. Biomimetic Self-Templating Supramolecular Structures. Nature. 2011;478:364–368. doi: 10.1038/nature10513. [DOI] [PubMed] [Google Scholar]
- 22.Liu Z, Qiao J, Niu Z, Wang Q. Natural Supramolecular Building Blocks: From Virus Coat Proteins to Viral Nanoparticles. Chem Soc Rev. 2012;41:6178–6117. doi: 10.1039/c2cs35108k. [DOI] [PubMed] [Google Scholar]
- 23.Witus LS, Francis MB. Using Synthetically Modified Proteins to Make New Materials. Acc Chem Res. 2011;44:774–783. doi: 10.1021/ar2001292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Worsdorfer B, Woycechowsky KJ, Hilvert D. Directed Evolution of a Protein Container. Science. 2011;331:589–592. doi: 10.1126/science.1199081. [DOI] [PubMed] [Google Scholar]
- 25.Comellas-Aragonès M, Engelkamp H, Claessen VI, Sommerdijk NAJM, Rowan AE, Christianen PCM, Maan JC, Verduin BJM, Cornelissen JJLM, Nolte RJM. A Virus-Based Single-Enzyme Nanoreactor. Nat Nanotechnol. 2007;2:635–639. doi: 10.1038/nnano.2007.299. [DOI] [PubMed] [Google Scholar]
- 26.Reichhardt C, Uchida M, O’Neil A, Li R, Prevelige PE, Douglas T. Templated Assembly of Organic–Inorganic Materials Using the Core Shell Structure of the P22 Bacteriophage. Chem Commun. 2011;47:6326. doi: 10.1039/c1cc11215e. [DOI] [PubMed] [Google Scholar]
- 27.Bedwell GJ, Zhou Z, Uchida M, Douglas T, Gupta A, Prevelige PE. Selective Biotemplated Synthesis of TiO2 inside a Protein Cage. Biomacromolecules. 2015;16:214–218. doi: 10.1021/bm501443e. [DOI] [PubMed] [Google Scholar]
- 28.Lucon J, Qazi S, Uchida M, Bedwell GJ, LaFrance B, Prevelige PEJr, Douglas T. Use of the Interior Cavity of the P22 Capsid for Site-Specific Initiation of Atom-Transfer Radical Polymerization with High-Density Cargo Loading. Nat Chem. 2012;4:781–788. doi: 10.1038/nchem.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uchida M, Morris DS, Kang S, Jolley CC, Lucon J, Liepold LO, LaFrance B, Prevelige PE., Jr Douglas, TSite-Directed Coordination Chemistry with P22 Virus-Like Particles. Langmuir. 2012;28:1998–2006. doi: 10.1021/la203866c. [DOI] [PubMed] [Google Scholar]
- 30.O’Neil A, Reichhardt C, Johnson B, Prevelige PE, Douglas T. Genetically Programmed in Vivo Packaging of Protein Cargo and Its Controlled Release from Bacteriophage P22. Angew Chem Int Ed. 2011;50:7425–7428. doi: 10.1002/anie.201102036. [DOI] [PubMed] [Google Scholar]
- 31.Prevelige PE, Thomas D, King J. Scaffolding Protein Regulates the Polymerization of P22 Coat Subunits into Icosahedral Shells in Vitro. J Mol Biol. 1988;202:743–757. doi: 10.1016/0022-2836(88)90555-4. [DOI] [PubMed] [Google Scholar]
- 32.Thuman-Commike PA, Greene B, Malinski JA, Burbea M, McGough A, Chiu W, Prevelige PE., Jr Mechanism of Scaffolding-Directed Virus Assembly Suggested by Comparison of Scaffolding-Containing and Scaffolding-Lacking P22 Procapsids. Biophys J. 1999;76:3267–3277. doi: 10.1016/S0006-3495(99)77479-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parker MH, Casjens S, Prevelige PE. Functional Domains of Bacteriophage P22 Scaffolding Protein. J Mol Biol. 1998;281:69–79. doi: 10.1006/jmbi.1998.1917. [DOI] [PubMed] [Google Scholar]
- 34.Lander GC, Tang L, Casjens SR, Gilcrease EB, Prevelige P, Poliakov A, Potter CS, Carragher B, Johnson JE. The Structure of an Infectious P22 Virion Shows the Signal for Headful DNA Packaging. Science. 2006;312:1791–1795. doi: 10.1126/science.1127981. [DOI] [PubMed] [Google Scholar]
- 35.Patterson DP, Prevelige PE, Douglas T. Nanoreactors by Programmed Enzyme Encapsulation inside the Capsid of the Bacteriophage P22. ACS Nano. 2012;6:5000–5009. doi: 10.1021/nn300545z. [DOI] [PubMed] [Google Scholar]
- 36.Patterson DP, Schwarz B, Waters RS, Gedeon T, Douglas T. Encapsulation of an Enzyme Cascade within the Bacteriophage P22 Virus-Like Particle. ACS Chem Biol. 2014;9:359–365. doi: 10.1021/cb4006529. [DOI] [PubMed] [Google Scholar]
- 37.Jordan PC, Patterson DP, Saboda KN, Edwards EJ, Miettinen HM, Basu G, Thielges MC, Douglas T. Self-Assembling Biomolecular Catalysts for Hydrogen Production. Nat Chem. 2016;8:179–185. doi: 10.1038/nchem.2416. [DOI] [PubMed] [Google Scholar]
- 38.O’Neil A, Prevelige PE, Douglas T. Stabilizing Viral Nano-Reactors for Nerve-Agent Degradation. Biometer Sci. 2013;1:881. doi: 10.1039/c3bm60063g. [DOI] [PubMed] [Google Scholar]
- 39.Patterson DP, McCoy K, Fijen C, Douglas T. Constructing Catalytic Antimicrobial Nanoparticles by Encapsulation of Hydrogen Peroxide Producing Enzyme inside the P22 VLP. J Mater Chem B. 2014;2:5948. doi: 10.1039/c4tb00983e. [DOI] [PubMed] [Google Scholar]
- 40.Servid A, Jordan P, O’Neil A, Prevelige P, Douglas T. Location of the Bacteriophage P22 Coat Protein C-Terminus Provides Opportunities for the Design of Capsid-Based Materials. Biomacromolecules. 2013;14:2989–2995. doi: 10.1021/bm400796c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bartlett P, Campbell AI. Three-Dimensional Binary Superlattices of Oppositely Charged Colloids. Phys Rev Lett. 2005;95:128302. doi: 10.1103/PhysRevLett.95.128302. [DOI] [PubMed] [Google Scholar]
- 42.Pusey PN, van Megen W, Bartlett P, Ackerson BJ, Rarity JG, Underweed SM. Structure of Crystals of Hard Colloidal Spheres. Phys Rev Lett. 1989;63:2753–2756. doi: 10.1103/PhysRevLett.63.2753. [DOI] [PubMed] [Google Scholar]
- 43.Gasser U, Weeks ER, Schofield A, Pusey PN, Weitz DA. Real-Space Imaging of Nucleation and Growth in Colloidal Crystallization. Science. 2001;292:258–262. doi: 10.1126/science.1058457. [DOI] [PubMed] [Google Scholar]
- 44.Mahynski NA, Panagiotopoulos AZ, Meng D, Kumar SK. Stabilizing Colloidal Crystals by Leveraging Void Distributions. Nat Commun. 2014;5:1–8. doi: 10.1038/ncomms5472. [DOI] [PubMed] [Google Scholar]
- 45.Atsumi S, Hanai T, Liao JC. Non-Fermentative Pathways for Synthesis of Branched-Chain Higher Alcohols as Biofuels. Nature. 2008;451:86–89. doi: 10.1038/nature06450. [DOI] [PubMed] [Google Scholar]
- 46.Peralta-Yahya PP, Zhang F, del Cardayre SB, Keasling JD. Microbial Engineering for the Production of Advanced Biofuels. Nature. 2012;488:320–328. doi: 10.1038/nature11478. [DOI] [PubMed] [Google Scholar]
- 47.Sheldon RA. Enzyme Immobilization: The Quest for Optimum Performance. Adv Synth Catal. 2007;349:1289–1307. [Google Scholar]
- 48.Rodrigues RC, Ortiz C, Berenguer-Murcia A, Torres R, Fernández-Lafuente R. Modifying Enzyme Activity and Selectivity by Immobilization. Chem Soc Rev. 2013;42:6290–6307. doi: 10.1039/c2cs35231a. [DOI] [PubMed] [Google Scholar]
- 49.Jia F, Narasimhan B, Mallapragada S. Materials-Based Strategies for Multi-Enzyme Immobilization and Co-Localization: A Review. Biotechnol Bioeng. 2014;111:209–222. doi: 10.1002/bit.25136. [DOI] [PubMed] [Google Scholar]
- 50.Fuller MT, King J. Purification of the Coat and Scaffolding Proteins from Procapsids of Bacteriophage P22. Virology. 1981;112:529–547. doi: 10.1016/0042-6822(81)90300-7. [DOI] [PubMed] [Google Scholar]
- 51.Huang X, Bronstein LM, Dragnea B. Self-Assembled Virus-Like Particles with Magnetic Cores. Nano Lett. 2007;7:2407–2416. doi: 10.1021/nl071083l. [DOI] [PubMed] [Google Scholar]
- 52.de la Plaza M, de Palencia PF, Pelaez C, Requena T. Biochemical and Molecular Characterization of Alpha-Ketoisovalerate Decarboxylase, an Enzyme Involved in the Formation of Aldehydes from Amino Acids by Lactococcus Lactis. FEMS Microbiol Lett. 2004;238:367–374. doi: 10.1016/j.femsle.2004.07.057. [DOI] [PubMed] [Google Scholar]
- 53.Liu X, Bastian S, Snow CD, Brustad EM, Saleski TE, Xu J-H, Meinhold P, Arnold FH. Structure-Guided Engineering of Lactococcus Lactis Alcohol Dehydrogenase LlAdhA for Improved Conversion of Isobutyraldehyde to Isobutanol. J Biotechnol. 2012;164:188–195. doi: 10.1016/j.jbiotec.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weigele PR, Sampson L, Winn-Stapley D, Casjens SR. Molecular Genetics of Bacteriophage P22 Scaffolding Protein’s Functional Domains. J Mol Biol. 2005;348:831–844. doi: 10.1016/j.jmb.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 55.Kang S, Uchida M, O’Neil A, Li R, Prevelige PE, Douglas T. Implementation of P22 Viral Capsids as Nanoplatforms. Biomacromolecules. 2010;11:2804–2809. doi: 10.1021/bm100877q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.