

SUMMARY
Glia have been suggested to regulate sleep-like states in vertebrates and invertebrates alike. In the nematode Caenorhabditis elegans, sleep is associated with molting between larval stages. To understand if glia modulate neural circuits driving sleep in C. elegans larvae, we ablated the astrocyte-like CEPsh glia. We found that glia-ablated animals exhibit episodes of immobility preceding sleep, prolonged sleep, molting-independent short-duration locomotory pausing, and delayed development. CEPsh glia ensheath synapses between the sleep-associated ALA neuron and its postsynaptic partner AVE, a major locomotion interneuron. While AVE calcium transients normally correlate with head retraction, glia ablation results in prolonged calcium transients that are uncoupled from movement. Strikingly, all these glia ablation defects are suppressed by the ablation of ALA. Our results suggest that glia attenuate sleep-promoting inhibitory connections between ALA and AVE, uncovering specific roles for glia in sleep behavior. We propose that similar mechanisms may underlie glial roles in sleep in other animals.
In Brief
Sleep is characterized by reduced mobility. How neural circuits drive sleep-associated locomotion inhibition is not known. Here, Katz et al. demonstrate that neuron-associated glial cells prevent unintended sleep entry by inhibiting neuronal connections that promote sleep-related locomotion attenuation.
INTRODUCTION
Sleep is an ancient and conserved behavior, whose purpose remains largely unknown (Zimmerman et al., 2008). Sleeping animals exhibit reduced motility, decreased responsiveness to the environment, and increased arousal thresholds (Brown et al., 2012), making them highly vulnerable to dangers such as predators. The benefits of sleep must, therefore, outweigh these considerable risks. Because sleep can be detrimental, the transitions in and out of this behavioral state must be prompt and tightly regulated. A number of mechanisms driving entry into and exit from sleep have been described. The circadian clock can influence sleep entry and recovery by regulating the activities of specific brain centers (Saper et al., 2005) or by controlling local brain circuits that collectively interact (Krueger et al., 2008). Homeostatic drivers such as adenosine, which accumulates during wakefulness, have also been suggested to play roles in sleep/wakefulness transitions (Brown et al., 2012).
In 1895, Ramón y Cajal postulated in a theoretical paper that astrocytic glia might be global modulators of sleep (García-Marín et al., 2007; Ramón y Cajal, 1895). A number of recent studies experimentally support this view. Astrocytes appear to be required for switching cortical activities between awake and sleep states (Poskanzer and Yuste, 2016). Consistent with this, optogenetic activation of astrocytes enhances the sleep state (Pelluru et al., 2016), and this may occur through the release of adenosine (Halassa et al., 2009) or by regulation of Notch signaling (Seugnet et al., 2011). Moreover, a number of studies implicate astrocytes in circadian behavior (Barca-Mayo et al., 2017; Brancaccio et al., 2017; Ng et al., 2011; Tso et al., 2017). Nonetheless, how sleep-promoting circuits are modulated by astrocytes to induce behavioral quiescence and whether this function is conserved is not clear.
A developmentally timed sleep-like state in the nematode Caenorhabditis elegans coincides with molting between larval stages (Raizen et al., 2008), and it is termed lethargus. Molting onset, duration, and associated sleep are regulated by LIN-42/Period protein (Jeon et al., 1999; Monsalve et al., 2011), whose homologs control circadian rhythms in insects and vertebrates. Like sleep in other animals, lethargus is characterized by behavioral quiescence, reduced sensory responsiveness, reversibility, homeostatic regulation, and stereotypical body posture (Cho and Sternberg, 2014; Iwanir et al., 2013; Nagy et al., 2014; Raizen et al., 2008; Schwarz et al., 2011). Several neurons have been implicated in C. elegans sleep and wakefulness, including the RMG, RIS, ALA, ASK, and RIA neurons, whose sleep-related activities involve responses to or release of specific neuropeptides (Choi et al., 2013; Nath et al., 2016; Nelson et al., 2013; Turek et al., 2013, 2016; Van Buskirk and Sternberg, 2007). Epidermal growth factor (EGF) and Notch signaling are also implicated in sleep control (Singh et al., 2011; Van Buskirk and Sternberg, 2007). Simultaneous imaging of nearly all C. elegans head neurons, using the calcium sensor GCaMP, reveals a reduction in activity of most neurons during lethargus (Nichols et al., 2017).
The C. elegans nerve ring is a brain-like neuropil consisting of most axons in the animal and harboring the majority of neuron-neuron synapses. This structure is associated with four CEPsh glial cells. These glia share a number of features with vertebrate astrocytes. Like astrocytes, CEPsh glia tile around the brain neuropil to define non-overlapping domains (Ogata and Kosaka, 2002; White et al., 1986). Within the neuropil, CEPsh glia en-sheath a well-defined set of synapses (White et al., 1986), resembling vertebrate tripartite synapses that consist of neuronal synaptic pairs and astrocyte processes (Halassa et al., 2007). CEPsh glia also promote synaptogenesis (Colón-Ramos et al., 2007), an important function of vertebrate astrocytes (Eroglu and Barres, 2010). During development, CEPsh glia transform from simple bipolar cells to highly branched cells, reminiscent of the radial glia-to-astrocyte transition in vertebrates (Rapti et al., 2017). Importantly, CEPsh glia are enriched for gene transcripts also enriched in astrocytes, including the glial glutamate transporter glt-1 (M.K., unpublished data) (Zhang et al., 2014). Glia-specific loss of GLT-1 in both C. elegans and vertebrates results in repetitive behaviors (M.K., unpublished data) (Aida et al., 2015).
Here, we show that CEPsh glia are important regulators of sleep entry in C. elegans. Ablation of these glia results in precocious sleep bouts during lethargus, increased sleep duration, and transient locomotory pausing in adults. Furthermore, larval development in glia-ablated animals is considerably prolonged. Remarkably, inactivation of the ALA neuron, whose synapses with the postsynaptic AVE interneuron are ensheathed by CEPsh glia processes, is sufficient to reverse all of these defects. Epistasis studies reveal that ALA, AVE, and CEPsh glia act in the same circuit, and activity imaging in AVE suggests that loss of CEPsh glia decouples input and output signals in this neuron, in a manner dependent on ALA. Our data highlight a specific synaptic connection mediating locomotion inhibition during sleep and suggest an integral role for glia in modulating this connection.
RESULTS
CEPsh Glia Regulate Sleep Entry, Adult Locomotory Pausing, and the Rate of Development
To determine whether C. elegans CEPsh glia play roles in sleep, we ablated all four cells in first-stage (L1) larvae, after nerve ring formation, using a reconstituted Caspase-3 gene expressed specifically in CEPsh glia (Chelur and Chalfie, 2007) (M.K., unpublished data), and we assessed the effects on locomotion during the L4-to-adult molt. As previously reported (Raizen et al., 2008), wild-type animals are constantly active during the intermolt period (Figures 1A, 1C, and S1A) and exhibit a single consolidated episode of elevated locomotion quiescence during the molt (Figures 1A, 1D, and S1A). However, the 3/3 CEPsh glia-ablated lines we generated experience ectopic quiescence bouts of ~20 min in duration, 1–2 hr before the beginning of a prolonged consolidated period of inactivity (Figures 1B, 1C, and S1A). As in both wild-type and glia-ablated animals consolidated quiescence of >2 hr is apparent, we define this period as lethargus. The shorter duration quiescence episodes preceding lethargus in glia-ablated animals are defined as precocious episodes. These observations demonstrate that CEPsh glia are required for regulating the transition between awake and sleep states. Consistent with these observations, we found that, in 2/3 glia-ablated lines, the main quiescence period is also prolonged (Figures 1D and S1A), suggesting that ablated animals exhibit a general propensity for sleep.
Figure 1. Post-embryonic Ablation of CEPsh Glia Affects Sleep Entry, Locomotion, and Development.

(A and B) Traces of the quiescence fraction (Experimental Procedures) during the L4-adult transition of a wild-type (A) or a CEPsh glia-ablated (B) animal. Lethargus onset occurs at t = 0.
(C and D) Average quiescence fraction 4 hr before (C) and after (D) lethargus initiation, binned in 1-hr intervals. WT, wild-type. GA, glia ablated. Error bars indicate SE. Number of animals is indicated within or above each bar.
(E) Ablation of CEPsh glia results in enhanced pausing (n = 5 movies). Vertical axis represents the percentage of total time animals spent pausing out of the total duration of all recorded tracks.
(F and G) EGL-4 affects pre-lethargus quiescence (F) and ectopic adult pausing (G; n = 3 movies) of CEPsh glia-ablated animals (GA1). Loss-of-function mutation, −/lof; gain-of-function mutation, g/gof.
(H) Percentage of animals reaching a given developmental stage 55 hr after embryos are laid. Experiments were done in triplicate.
(C–H) Bar graphs, mean ± SEM; ANOVA Tukey’s HSD post hoc test, *compared to WT, ^compared to glia-ablated line 1; number of symbols represents (1) p < 0.05 and (3) p < 0.0005.
See also Figure S1.
We wondered, therefore, whether CEPsh glia-ablated animals might also exhibit locomotory quiescence during adulthood, when animals no longer molt. To test this, we tracked the foraging behavior of adult animals on agar plates in the absence of food and used a Hidden Markov Model to identify states of motion and quiescence based on animal velocity (Experimental Procedures; Figures S1B–S1D). We found that, while wild-type animals exhibit continuous movement on plates, CEPsh glia-ablated adults display periods of locomotory pausing, which are variable between individual animals and which can last from a few seconds to several minutes (Figures 1E, S1D, and S1E; Movies S1 and S2). Of note, while reduced pharyngeal pumping normally accompanies lethargus-associated sleep in C. elegans, pumping rates are not altered in CEPsh glia-ablated adults (Figure S1F). Thus, glia appear to specifically control locomotory aspects of sleep, consistent with previous studies reporting that locomotion and pharyngeal pumping quiescence are regulated by different pathways (Nath et al., 2016).
To determine whether the ectopic pre-lethargus quiescence and the adult locomotory pausing seen in CEPsh glia-ablated animals are functionally related to lethargus quiescence, we examined animals defective in EGL-4, a cyclic GMP-dependent protein kinase expressed in many head neurons and previously characterized as a general regulator of sleep (Raizen et al., 2008). We examined animals carrying either a loss-of-function mutation (ks62; −) or a gain-of-function mutation (ad450; g) in the egl-4 gene. We found that, in addition to reducing the quiescence fraction during lethargus (Figure S1G), loss of egl-4(−) also suppresses pre-lethargus quiescence and adult locomotory pausing in animals lacking CEPsh glia (Figures 1F and 1G). Consistent with these observations, gain of EGL-4 function increases quiescence during lethargus and adult locomotory pausing in glia-ablated animals (Figures 1G and S1G), although pre-lethargus quiescence was not affected in this mutant (Figure 1F).
Although the mechanism of EGL-4 function is not well understood, our genetic results using egl-4 mutants nonetheless suggest that the various forms of locomotion quiescence we examined are likely related.
Finally, in addition to ectopic sleep bouts, we also observed a considerable developmental delay in CEPsh glia-ablated animals, a defect shared with egl-4(g) mutants (Figure S1H). While freshly laid wild-type embryos develop to adulthood within ~55 hr at 20°C, more than 20% of CEPsh glia-ablated animals only reach the L4 or younger larval stages after a similar amount of time (Figure 1H). This observation raises the possibility that increased somnolence slows development of the animal (see below).
Taken together, our data suggest that CEPsh glia normally block locomotory quiescence and that their absence promotes ectopic quiescence during and outside the period of lethargus.
Ectopic Locomotory Quiescence and Developmental Delay Require the ALA Neuron
To understand how CEPsh glia block locomotory quiescence, we sought to identify neurons relevant to this glial function. In addition to ensheathing the nerve ring, CEPsh glia also send anterior processes that wrap around sensory endings of the dopaminergic CEP neurons. We found that ablation of CEP neurons in wild-type animals does not phenocopy CEPsh glia ablation and does not rescue ectopic locomotory pausing of CEPsh glia-ablated adults (Figure S1I). Thus, CEP neurons are unlikely to be the relevant targets of CEPsh glia in locomotion control.
Unlike most neurons, the ALA neuron remains active during sleep (Nichols et al., 2017), and its optogenetic activation suppresses motion (Fry et al., 2014; Nelson et al., 2014). Further-more, ALA has been suggested to promote sleep entry (Van Buskirk and Sternberg, 2007). To test if locomotion quiescence observed in glia-ablated animals involves ALA, we disrupted the cell by either laser ablation or by crossing to a ceh-17(np1) mutant, which perturbs ALA development and activity (Pujol et al., 2000; Van Buskirk and Sternberg, 2007). While disruption of ALA has no effect on locomotion in wild-type adults, it significantly rescues the ectopic pausing episodes of CEPsh glia-ablated adults (Figures 2A and 2B). Importantly, loss of ALA function also fully rescues the pre-lethargus quiescence episodes (Figure 2C), the extended duration of lethargus (Figure S2A), and, remarkably, also the developmental delay (Figure 2D) of CEPsh glia-ablated animals. That ceh-17 mutants alone do not exhibit defects in sleep control suggests that other parallel pathways likely also act to induce developmentally regulated sleep in C. elegans.
Figure 2. CEPsh Glia Ablation Defects Are Rescued by Disrupting ALA.

(A) Ectopic pausing of CEPsh glia-ablated adults is suppressed by ALA ablation (−) (n = 2 movies). Details are as in Figure 1E.
(B) A ceh-17 loss-of-function (−) mutation also suppresses ectopic pausing of CEPsh glia-ablated adults (n = 4 movies). Details are as in Figure 1E.
(C) A ceh-17 mutation (−) suppresses ectopic pre-lethargus quiescence in CEPsh glia-ablated animals. Details are as in Figure 1G.
(D) A ceh-17 mutation (−) restores normal development of CEPsh glia-ablated animals (experiments done in triplicate).
(A–D) Bar graphs, mean ± SEM; ANOVA Tukey’s HSD post hoc test, *compared to WT, ^compared to glia-ablated parent; number of symbols represents (1) p < 0.05, (2) p < 0.005, and (3) p < 0.0005.
See also Figure S2.
Together, these results suggest that all forms of ectopic quiescence episodes in CEPsh glia-ablated animals are a consequence of inappropriate activity of ALA. Thus, CEPsh glia may normally act to block ALA.
Activation of LET-23/EGF receptor (EGFR) in ALA controls sleep induced by heat and other stressors (Hill et al., 2014; Van Buskirk and Sternberg, 2007). To determine if glial effects on locomotion quiescence involve EGF signaling, we examined CEPsh glia-ablated animals also defective in let-23(sy12). We found that these animals show enhanced pre-lethargus quiescence, prolonged lethargus duration, and increased locomotion pausing in adults, compared with CEPsh glia-ablated animals alone (Figures S2B–S2D). These observations suggest that LET-23 normally inhibits locomotory quiescence and that it can do so independently of CEPsh glia. Supporting this notion, ALA disruption does not significantly alleviate ectopic adult pausing of CEPsh glia-ablated animals also carrying a let-23 mutation (Figure S2D). These results are consistent with previous reports suggesting that developmentally timed sleep and stress-induced sleep are regulated by different mechanisms (Trojanowski et al., 2015).
The AVE Neuron Functions Downstream of ALA and CEPsh Glia for Locomotory Quiescence
ALA has only five synaptic outputs, four of which target the backward locomotion command interneuron AVE, which synapses onto motor neurons (Chalfie et al., 1985; White et al., 1986). Three of the four ALA-AVE synapses are ensheathed by CEPsh glia processes, and they represent one of only two classes of such tripartite synapses known in C. elegans (White et al., 1986). These observations raise the possibility that AVE may be a relevant target for ALA during locomotion quiescence. To test this idea, we generated animals expressing tetanus toxin, which blocks synaptic neurotransmitter release, under the control of an AVE-specific promoter. These animals exhibit twitching movements primarily at the nose and tail tips (as opposed to locomotion) during lethargus, which prevented us from reliably assaying quiescence behavior during the sleep period (Figures S3A and S3B). However, we were able to follow ectopic locomotory pausing in adults. We found that, as with CEPsh glia ablation, AVE inhibition results in ectopic pausing (Figure 3A). By contrast, disruption of ALA does not rescue this pausing (Figure 3A). Importantly, ablation of both AVE and CEPsh glia does not enhance pausing beyond what is observed for CEPsh ablation alone (Figure 3B). Thus, AVE appears to function in the same circuit as and downstream of ALA and CEPsh glia in locomotion quiescence.
Figure 3. AVE Functions with ALA and CEPsh Glia to Control Locomotory Pausing.

(A) Expression of tetanus toxin (TeTx) in AVE promotes ectopic pausing in adults, which is not rescued by a ceh-17 mutation (−) (n = 3 movies). Details are as in Figure 1E.
(B) Ablation of AVE (−) does not enhance pausing of CEPsh-ablated worms (n = 2 movies). Details are as in Figure 1E.
(A and B) Bar graphs, mean ± SEM; (A) ANOVA Tukey’s HSD post hoc test, *compared to WT; number of symbols represents (1) p < 0.05 and (3) p < 0.0005.
See also Figure S3.
Since CEPsh glia ensheath ALA-AVE synapses, we wondered whether CEPsh loss may affect the structural integrity of these synapses. To test this, we generated animals expressing separate components of membrane-localized split-GFP (Feinberg et al., 2008) in ALA and AVE, using non-overlapping cell-specific promoters, and assessed membrane juxtaposition by GFP fluorescence. AVE postsynaptic sites were marked in these animals with mCherry-tagged neuroligin (Figure S3C). We found a comparable percentage of animals with intact synapses in wild-type and CEPsh glia-ablated animals (47% and 58%, respectively; n > 90). These results suggest that the effects of CEPsh glia removal are unlikely to be structural and may be a result of altered synaptic activity.
CEPsh Glia and ALA Control Coupling between AVE Calcium Elevation and Backward Locomotion
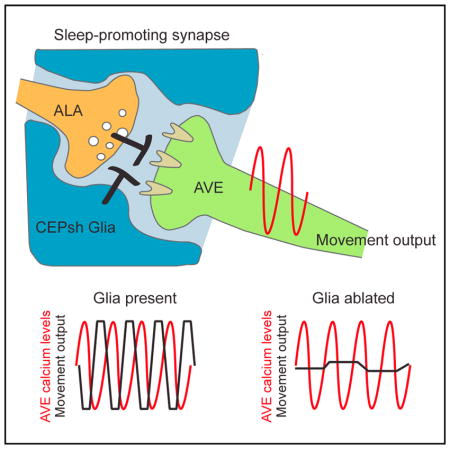
To study whether CEPsh glia affect AVE activity, we generated animals expressing the fluorescent calcium reporter GCaMP in AVE and confined them to a narrow trap in which they were able to move their head for short distances back and forth (Chronis et al., 2007). In this assay, frequent changes in AVE calcium levels and in head position are observed (Figures S4A and S4B; Movie S3). We classified AVE activity into four states, ramping up, ramping down, and high or low plateaus, and analyzed the transition between these states using a Hidden Markov Model (Figures S4C and S4D) (Kato et al., 2015). This analysis revealed a strong correlation between calcium increases in AVE and backward retraction of the head in wild-type animals (Figures 4A–4C). Calcium increases in AVE were previously shown to correlate with reversal movements (Kawano et al., 2011), suggesting that head retraction in our device likely indicates an intended reversal attempt. Importantly, the association between head movement and calcium activity is degraded in CEPsh glia-ablated animals (Figure 4B). Indeed, in CEPsh glia-ablated animals, >20% of calcium events are not accompanied by backward head retractions at all (error events; Figures 4C, S4E, and S4F). Moreover, while CEPsh glia-ablated and wild-type animals exhibit similar average AVE calcium amplitudes (Figure 4A) and frequencies (Figure S4G), the fraction of time AVE is in an active (on) state is significantly increased in CEPsh glia-ablated animals (Figure 4D).
Figure 4. CEPsh Glia and ALA Affect AVE Calcium Signal Duration and Behavioral Output.

(A) Average of AVE GCaMP signals, the number of total events analyzed for each strain is indicated on the graph. Dashed gray line, GCaMP signal rise initiation. All strains contain a GCaMP2.2 transgene.
(B) Head position relative to AVE GCaMP signal rise initiation (dashed gray line). Forward and backward head locations were determined by AVE cell body position.
(C) Percentage of events in which AVE GCaMP signal increase is not followed by backward head movement. Number of animals is indicated within or above each bar. (A–C) Error bars, mean ± SEM.
(D) The mean fraction of the time AVE calcium signal is at the high plateau “on” state (Experimental Procedures) is prolonged in CEPsh-ablated and AVE-TeTx animals. Number of animals is indicated within or above each bar. (C and D) *p < 0.01, random permutation test.
(E) Model for CEPsh glia function. CEPsh glia negatively regulate the inhibitory signaling between ALA and AVE.
See also Figure S4.
To determine whether changes in AVE calcium dynamics are related to the ALA-AVE synapses ensheathed by CEPsh glia, we assayed AVE calcium activity in CEPsh glia-ablated animals carrying the ceh-17 mutation. As shown in Figures 4A–4D, ALA inhibition is sufficient to restore normal AVE calcium peak duration and correlation with head movement to CEPsh glia-ablated animals. Thus, CEPsh glia control of AVE functional output requires ALA.
We wondered whether the prolonged calcium peaks and the decoupling of AVE activation from backward head movement are related defects. To test this, we examined AVE calcium levels in animals expressing tetanus toxin in AVE. We found that, by blocking AVE synaptic release, the mean duration of calcium signals is prolonged, as with CEPsh glia-ablated animals (Figure 4D). These results suggest that the prolonged AVE calcium peaks observed in CEPsh glia-ablated animals could be a result of inhibition of AVE synaptic output.
Our data suggest that the synapses between ALA and AVE are unusual. Activation of these inhibitory synapses blocks AVE output, but not by reducing AVE excitability, as the frequency and amplitude of AVE calcium peaks are not perturbed in CEPsh glia-ablated animals. We wondered, therefore, whether standard neurotransmitter signaling occurs at ALA-AVE synapses. We examined mutations in different neurotransmitter and neuropeptide pathways and assessed whether these can rescue the ectopic pausing episodes of adult CEPsh-ablated animals, as do mutations in ceh-17 or ALA ablation. Some mutants we tested confer pausing behavior independently of glia ablation. For these, we assessed the ability to rescue the developmental delay of CEPsh-ablated animals. As summarized in Table S1, mutations affecting dopamine (cat-2), serotonin (tph-1), and glutamate (eat-4) signaling fail to rescue pausing, as does a mutation in snf-11, the transporter required for GABA accumulation in ALA (Gendrel et al., 2016). Acetylcholine (ACh) mutants could not be tested, as they exhibit greatly reduced locomotion and strong developmental delays; however, ALA does not express canonical ACh synthesis or uptake proteins (Pereira et al., 2015). Thus, ACh is likely not involved.
To examine the involvement of neuropeptide signaling, we tested mutations in genes controlling dense-core vesicle fusion (unc-31 and ida-1) or neuropeptide maturation (egl-21/carboxy-peptidase E and aex-5, egl-3, kpc-1, and bli-4 peptide convertases). Only aex-5 mutants show moderate rescue, possibly suggesting the involvement of neuropeptide signaling. We therefore examined mutations in neuropeptide/receptor genes that are highly enriched in ALA (Nath et al., 2016) or that have been previously associated with sleep regulation (flp-7, flp-13, flp-24, nlp-8, Nath et al., 2016; sNPF/flp-27, Shang et al., 2013; and npr-22 implicated in the response of ALA to the RIS-released neuropeptide FLP-11, Turek et al., 2016). However, no suppression of CEPsh glia ablation defects was found. Pausing is markedly reduced by mutation in npr-1, a neuropeptide Y receptor implicated in lethargus (Choi et al., 2013). However, we found that NPR-1 expression in sensory neurons (Choi et al., 2013) is sufficient to restore ectopic pausing (Table S1), indicating that, at least in part, neuropeptide signaling can function upstream of the ALA-AVE synapse to control ectopic pausing.
Our results, therefore, eliminate most non-peptidergic neurotransmitter pathways as relevant for ALA-AVE synaptic function and suggest that either unknown aex-5-dependent neuropeptides or non-canonical signals mediate AVE synaptic inhibition by ALA.
DISCUSSION
Except for dwelling episodes associated with food (Fujiwara et al., 2002), stress (Hill et al., 2014), and molting (Raizen et al., 2008), the free-living nematode C. elegans continually moves. Here, we find that CEPsh glia of this animal control the transition into locomotion quiescence in the context of sleep. Importantly, glia are involved in sleep regulation in mice (Halassa et al., 2009; Pelluru et al., 2016) and flies (Seugnet et al., 2011), indicating an evolutionarily conserved function for this cell class.
Our results suggest a model (Figure 4E) in which CEPsh glia block an inhibitory synapse between the ALA and AVE neurons. Removal of CEPsh glia allows ALA to promote AVE inactivation, not by hyperpolarizing AVE but by uncoupling its synaptic input from its synaptic output.
Loss of CEPsh glia results in precocious episodes of quiescence immediately preceding lethargus in an ALA neuron-dependent manner. This unusual quiescence profile raises the possibility that sleep initiation may be governed by a gradual accumulation of sleep factors past a threshold, driving quiescence. Glia can serve to uptake a variety of neurotransmitters and secreted proteins from their surroundings (Conti et al., 2004; Rothstein et al., 1996), and it could therefore be that secreted moieties are relevant sleep factors. In this scenario, in CEPsh glia-ablated animals, the threshold for sleep entry is lowered by ectopic accumulation of sleep-inducing factors, resulting in premature sleep onset. Such a model may also explain the slower recovery from lethargus in CEPsh glia-ablated animals. A threshold model predicts that other sleep factors independent of CEPsh glia exist, as pre-lethargus quiescence episodes are transient and not consolidated as the main lethargus bout. Supporting this prediction, the ALA-dependent narcoleptic-like pausing behavior in CEPsh glia-ablated adults uncouples locomotion quiescence from other aspects of sleep, such as pharyngeal pumping. These findings not only suggest that sleep is a sum of different processes controlled by different pathways, but also implicate CEPsh glia and the ALA-AVE synapses they ensheath specifically in locomotion quiescence.
Only one other synaptic pairing is documented to be tightly ensheathed by CEPsh glia. This pairing, between the AIN and BAG presynaptic and postsynaptic neurons (White et al., 1986), is not known to have roles in sleep control. Nonetheless, the tight ensheathment may reflect an important synaptic function for glia that is not needed elsewhere. Indeed, our studies suggest that the ALA-AVE synapses may be atypical in other ways. Loss of CEPsh glia perturbs AVE functional output (reversal movement) in a manner dependent on ALA, but it affects neither the amplitude nor the frequency of AVE activation in our experimental setup. Such uncoupling of calcium induction and synaptic output has been previously reported in the ASER neuron of C. elegans in the context of a simple learning behavior. In that work, salt, which is usually attractive, becomes aversive when paired with starvation. Imaging of ASER reveals that salt cues usually induce calcium signals that drive synaptic vesicle release. Following starvation, however, neuronal outputs are inhibited even though calcium signals are enhanced (Oda et al., 2011). Thus, in addition to exhibiting uncoupling of synaptic input and output, inhibition of ASER by starvation and of AVE by CEPsh glia ablation are associated with increased calcium signal in these cells, suggesting the possibility of common, yet still unknown, underlying mechanisms.
It is of note that the ALA-AVE synapses are the most cell body-proximal synapses on AVE neurites (White et al., 1986). Whether this anatomical configuration reflects on the mechanism by which synaptic input and output are uncoupled in this neuron is unclear. Understanding the molecular nature of ALA-AVE neurotransmission should greatly aid in understanding how CEPsh glia inhibition of this synapse works. We have been able to eliminate most neurotransmitter classes and some ALA-expressed neuropeptides; however, peptidergic transmitters could still be relevant.
Finally, whether CEPsh glial activities in the context of sleep are dynamically modulated or whether these cells establish a fixed set point for neuronal activity that is not altered during the animal’s life remains unknown. Identification of mediators of glial function and assessment of temporal changes in their activities, or lack thereof, will allow this question to be addressed.
EXPERIMENTAL PROCEDURES
For strains and plasmids used in this study as well as detailed descriptions of GFP reconstitution across synaptic partners (GRASP) signal acquisition and scoring, cell ablation, and development scoring, see the Supplemental Experimental Procedures.
Locomotion Analysis
First-day adults were washed off food and transferred to a 6-cm nematode growth media (NGM)-agar plate with a high-osmolarity barrier (4M Fructose) at the plate periphery to prevent wandering of animals off the plate. Animals were allowed to adjust to the plate for 20 min, and locomotion was recorded using a camera for 30 min at 2 frame/s. Custom Java scripts identified and tracked moving animals and analyzed worm pausing behaviors (see the Supplemental Experimental Procedures for more details). The fraction of the total time animals were found pausing in all tracks relative to the overall duration of all the tracks in a movie was used to calculate the percentage of time adult animals spent pausing.
Lethargus Behavior
Intermolt and lethargus quiescence assays were performed in artificial dirt poly-dimethylsiloxane (PDMS) chips as previously described (Iwanir et al., 2013), except that mid-L4 larvae were picked from a population of mixed-stage animals according to their morphology, and behavior was recorded for 10 hr. Generally, the onset of lethargus was defined as the first time point from which the fraction of quiescence remained above 5% for 20 consecutive min; for animals displaying substantial pre-lethargic quiescence, the starting point for the quiescence threshold search was determined manually based on the quiescence profile (see the Supplemental Experimental Procedures for more details).
Analysis of AVE Activity and Response
Calcium imaging from the AVE neuron was performed using a microfluidic device as described (Chronis et al., 2007). Images were captured for 5 min at 4 frames/s and analyzed using a custom Java code for AVE activity and position within the microfluidic device (see the Supplemental Experimental Procedures for more details).
Statistics
ANOVA Tukey’s honest significant difference (HSD) post hoc test was used in the behavioral and developmental assays for comparison of multiple populations of animals using an R script with the null hypothesis that all samples are similar. Random permutation tests were used to evaluate the significance of differences in AVE activity and worm response between worm populations, using a Java code. Consider two sets of recordings from individual worms, n1 from population 1 and n2 from population 2. Each recording, which can comprise multiple AVE activation events, is treated as an independent sample. To test the null hypothesis that the two worm populations are equivalent, a large number (105) of random rearrangements of the two sets of samples into two groups of sizes n1 and n2 is generated. The stated significance figures are obtained as the fraction of the rearrangements for which the difference in the quantity of interest (fraction of error events or fraction of the time in the “on” state) between the rearranged groups is at least as large as the difference between the original groups. Unpaired two-sided Student’s t test was used to determine the statistical significance between glia-ablated and non-ablated animals that are mutated for the various neurotransmitter and neuropeptide pathways.
Supplementary Material
Highlights.
C. elegans CEPsh glia modulate locomotory quiescence during sleep
Glia attenuate sleep-promoting inhibitory synapses between ALA and AVE neurons
ALA promotes uncoupling of calcium transients and synaptic output in AVE
Acknowledgments
We thank Daniel Colón-Ramos, Kang Shen, Cori Bargmann, Cheryl Van Buskirk, and the Caenorhabditis Genetic Center (CGC) (NIH P40 OD010440) for strains and reagents and the Shaham lab for comments and discussions. This work was supported by EMBO fellowship ALTD 870-200 to M.K. and by NIH grants NS095795 and NS064273 to S.S.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, four figures, one table, and three movies and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.02.036.
AUTHOR CONTRIBUTIONS
M.K. and S.S. designed experiments, interpreted data, and wrote the manuscript. F.C. developed the worm tracking and locomotion analysis code and analyzed the calcium kinetics studies. S.I. and D.B. developed the code for analysis of lethargus behavior. S.I. analyzed the lethargus behavior. M.K. conducted all experiments.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Aida T, Yoshida J, Nomura M, Tanimura A, Iino Y, Soma M, Bai N, Ito Y, Cui W, Aizawa H, et al. Astroglial glutamate transporter deficiency increases synaptic excitability and leads to pathological repetitive behaviors in mice. Neuropsychopharmacology. 2015;40:1569–1579. doi: 10.1038/npp.2015.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barca-Mayo O, Pons-Espinal M, Follert P, Armirotti A, Berdondini L, De Pietri Tonelli D. Astrocyte deletion of Bmal1 alters daily locomotor activity and cognitive functions via GABA signalling. Nat Commun. 2017;8:14336. doi: 10.1038/ncomms14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancaccio M, Patton AP, Chesham JE, Maywood ES, Hastings MH. Astrocytes Control Circadian Timekeeping in the Suprachiasmatic Nucleus via Glutamatergic Signaling. Neuron. 2017;93:1420–1435. e5. doi: 10.1016/j.neuron.2017.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Basheer R, McKenna JT, Strecker RE, McCarley RW. Control of sleep and wakefulness. Physiol Rev. 2012;92:1087–1187. doi: 10.1152/physrev.00032.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfie M, Sulston JE, White JG, Southgate E, Thomson JN, Brenner S. The neural circuit for touch sensitivity in Caenorhabditis elegans. J Neurosci. 1985;5:956–964. doi: 10.1523/JNEUROSCI.05-04-00956.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelur DS, Chalfie M. Targeted cell killing by reconstituted caspases. Proc Natl Acad Sci USA. 2007;104:2283–2288. doi: 10.1073/pnas.0610877104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JY, Sternberg PW. Multilevel modulation of a sensory motor circuit during C. elegans sleep and arousal. Cell. 2014;156:249–260. doi: 10.1016/j.cell.2013.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Chatzigeorgiou M, Taylor KP, Schafer WR, Kaplan JM. Analysis of NPR-1 reveals a circuit mechanism for behavioral quiescence in C. elegans. Neuron. 2013;78:869–880. doi: 10.1016/j.neuron.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chronis N, Zimmer M, Bargmann CI. Microfluidics for in vivo imaging of neuronal and behavioral activity in Caenorhabditis elegans. Nat Methods. 2007;4:727–731. doi: 10.1038/nmeth1075. [DOI] [PubMed] [Google Scholar]
- Colón-Ramos DA, Margeta MA, Shen K. Glia promote local synaptogenesis through UNC-6 (netrin) signaling in C. elegans. Science. 2007;318:103–106. doi: 10.1126/science.1143762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti F, Minelli A, Melone M. GABA transporters in the mammalian cerebral cortex: localization, development and pathological implications. Brain Res Brain Res Rev. 2004;45:196–212. doi: 10.1016/j.brainresrev.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature. 2010;468:223–231. doi: 10.1038/nature09612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg EH, Vanhoven MK, Bendesky A, Wang G, Fetter RD, Shen K, Bargmann CI. GFP Reconstitution Across Synaptic Partners (GRASP) defines cell contacts and synapses in living nervous systems. Neuron. 2008;57:353–363. doi: 10.1016/j.neuron.2007.11.030. [DOI] [PubMed] [Google Scholar]
- Fry AL, Laboy JT, Norman KR. VAV-1 acts in a single inter-neuron to inhibit motor circuit activity in Caenorhabditis elegans. Nat Commun. 2014;5:5579. doi: 10.1038/ncomms6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara M, Sengupta P, McIntire SL. Regulation of body size and behavioral state of C. elegans by sensory perception and the EGL-4 cGMP-dependent protein kinase. Neuron. 2002;36:1091–1102. doi: 10.1016/s0896-6273(02)01093-0. [DOI] [PubMed] [Google Scholar]
- García-Marín V, García-López P, Freire M. Cajal’s contributions to glia research. Trends Neurosci. 2007;30:479–487. doi: 10.1016/j.tins.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Gendrel M, Atlas EG, Hobert O. A cellular and regulatory map of the GABAergic nervous system of C. elegans. eLife. 2016;5:e17686. doi: 10.7554/eLife.17686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Haydon PG. The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol Med. 2007;13:54–63. doi: 10.1016/j.molmed.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Florian C, Fellin T, Munoz JR, Lee SY, Abel T, Haydon PG, Frank MG. Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron. 2009;61:213–219. doi: 10.1016/j.neuron.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AJ, Mansfield R, Lopez JM, Raizen DM, Van Buskirk C. Cellular stress induces a protective sleep-like state in C. elegans. Curr Biol. 2014;24:2399–2405. doi: 10.1016/j.cub.2014.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwanir S, Tramm N, Nagy S, Wright C, Ish D, Biron D. The microarchitecture of C. elegans behavior during lethargus: homeostatic bout dynamics, a typical body posture, and regulation by a central neuron. Sleep (Basel) 2013;36:385–395. doi: 10.5665/sleep.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon M, Gardner HF, Miller EA, Deshler J, Rougvie AE. Similarity of the C. elegans developmental timing protein LIN-42 to circadian rhythm proteins. Science. 1999;286:1141–1146. doi: 10.1126/science.286.5442.1141. [DOI] [PubMed] [Google Scholar]
- Kato S, Kaplan HS, Schrödel T, Skora S, Lindsay TH, Yemini E, Lockery S, Zimmer M. Global brain dynamics embed the motor command sequence of Caenorhabditis elegans. Cell. 2015;163:656–669. doi: 10.1016/j.cell.2015.09.034. [DOI] [PubMed] [Google Scholar]
- Kawano T, Po MD, Gao S, Leung G, Ryu WS, Zhen M. An imbalancing act: gap junctions reduce the backward motor circuit activity to bias C. elegans for forward locomotion. Neuron. 2011;72:572–586. doi: 10.1016/j.neuron.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Krueger JM, Rector DM, Roy S, Van Dongen HP, Belenky G, Panksepp J. Sleep as a fundamental property of neuronal assemblies. Nat Rev Neurosci. 2008;9:910–919. doi: 10.1038/nrn2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsalve GC, Van Buskirk C, Frand AR. LIN-42/PERIOD controls cyclical and developmental progression of C. elegans molts. Curr Biol. 2011;21:2033–2045. doi: 10.1016/j.cub.2011.10.054. [DOI] [PubMed] [Google Scholar]
- Nagy S, Tramm N, Sanders J, Iwanir S, Shirley IA, Levine E, Biron D. Homeostasis in C. elegans sleep is characterized by two behaviorally and genetically distinct mechanisms. eLife. 2014;3:e04380. doi: 10.7554/eLife.04380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath RD, Chow ES, Wang H, Schwarz EM, Sternberg PW. C. elegans Stress-Induced Sleep Emerges from the Collective Action of Multiple Neuropeptides. Curr Biol. 2016;26:2446–2455. doi: 10.1016/j.cub.2016.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MD, Trojanowski NF, George-Raizen JB, Smith CJ, Yu CC, Fang-Yen C, Raizen DM. The neuropeptide NLP-22 regulates a sleep-like state in Caenorhabditis elegans. Nat Commun. 2013;4:2846. doi: 10.1038/ncomms3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MD, Lee KH, Churgin MA, Hill AJ, Van Buskirk C, Fang-Yen C, Raizen DM. FMRFamide-like FLP-13 neuropeptides promote quiescence following heat stress in Caenorhabditis elegans. Curr Biol. 2014;24:2406–2410. doi: 10.1016/j.cub.2014.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng FS, Tangredi MM, Jackson FR. Glial cells physiologically modulate clock neurons and circadian behavior in a calcium-dependent manner. Curr Biol. 2011;21:625–634. doi: 10.1016/j.cub.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols ALA, Eichler T, Latham R, Zimmer M. A global brain state underlies C. elegans sleep behavior. Science. 2017;356:eaam6851. doi: 10.1126/science.aam6851. [DOI] [PubMed] [Google Scholar]
- Oda S, Tomioka M, Iino Y. Neuronal plasticity regulated by the insulin-like signaling pathway underlies salt chemotaxis learning in Caenorhabditis elegans. J Neurophysiol. 2011;106:301–308. doi: 10.1152/jn.01029.2010. [DOI] [PubMed] [Google Scholar]
- Ogata K, Kosaka T. Structural and quantitative analysis of astrocytes in the mouse hippocampus. Neuroscience. 2002;113:221–233. doi: 10.1016/s0306-4522(02)00041-6. [DOI] [PubMed] [Google Scholar]
- Pelluru D, Konadhode RR, Bhat NR, Shiromani PJ. Optogenetic stimulation of astrocytes in the posterior hypothalamus increases sleep at night in C57BL/6J mice. Eur J Neurosci. 2016;43:1298–1306. doi: 10.1111/ejn.13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira L, Kratsios P, Serrano-Saiz E, Sheftel H, Mayo AE, Hall DH, White JG, LeBoeuf B, Garcia LR, Alon U, Hobert O. A cellular and regulatory map of the cholinergic nervous system of C. elegans. eLife. 2015;4:e12432. doi: 10.7554/eLife.12432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poskanzer KE, Yuste R. Astrocytes regulate cortical state switching in vivo. Proc Natl Acad Sci USA. 2016;113:E2675–E2684. doi: 10.1073/pnas.1520759113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujol N, Torregrossa P, Ewbank JJ, Brunet JF. The homeodomain protein CePHOX2/CEH-17 controls anteroposterior axonal growth in C. elegans. Development. 2000;127:3361–3371. doi: 10.1242/dev.127.15.3361. [DOI] [PubMed] [Google Scholar]
- Raizen DM, Zimmerman JE, Maycock MH, Ta UD, You YJ, Sundaram MV, Pack AI. Lethargus is a Caenorhabditis elegans sleep-like state. Nature. 2008;451:569–572. doi: 10.1038/nature06535. [DOI] [PubMed] [Google Scholar]
- Ramón y Cajal S. Algunas conjeturas sobre el mecanismo anatómico de la ideación, asociación y atención (Imprenta y Librería de Nicolás Moya) 1895. [Google Scholar]
- Rapti G, Li C, Shan A, Lu Y, Shaham S. Glia initiate brain assembly through noncanonical Chimaerin-Furin axon guidance in C. elegans. Nat Neurosci. 2017;20:1350–1360. doi: 10.1038/nn.4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–1263. doi: 10.1038/nature04284. [DOI] [PubMed] [Google Scholar]
- Schwarz J, Lewandrowski I, Bringmann H. Reduced activity of a sensory neuron during a sleep-like state in Caenorhabditis elegans. Curr Biol. 2011;21:R983–R984. doi: 10.1016/j.cub.2011.10.046. [DOI] [PubMed] [Google Scholar]
- Seugnet L, Suzuki Y, Merlin G, Gottschalk L, Duntley SP, Shaw PJ. Notch signaling modulates sleep homeostasis and learning after sleep deprivation in Drosophila. Curr Biol. 2011;21:835–840. doi: 10.1016/j.cub.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, Donelson NC, Vecsey CG, Guo F, Rosbash M, Griffith LC. Short neuropeptide F is a sleep-promoting inhibitory modulator. Neuron. 2013;80:171–183. doi: 10.1016/j.neuron.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K, Chao MY, Somers GA, Komatsu H, Corkins ME, Larkins-Ford J, Tucey T, Dionne HM, Walsh MB, Beaumont EK, et al. C. elegans Notch signaling regulates adult chemosensory response and larval molting quiescence. Curr Biol. 2011;21:825–834. doi: 10.1016/j.cub.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojanowski NF, Nelson MD, Flavell SW, Fang-Yen C, Raizen DM. Distinct Mechanisms Underlie Quiescence during Two Caenorhabditis elegans Sleep-Like States. J Neurosci. 2015;35:14571–14584. doi: 10.1523/JNEUROSCI.1369-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tso CF, Simon T, Greenlaw AC, Puri T, Mieda M, Herzog ED. Astrocytes Regulate Daily Rhythms in the Suprachiasmatic Nucleus and Behavior. Curr Biol. 2017;27:1055–1061. doi: 10.1016/j.cub.2017.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turek M, Lewandrowski I, Bringmann H. An AP2 transcription factor is required for a sleep-active neuron to induce sleep-like quiescence in C. elegans. Curr Biol. 2013;23:2215–2223. doi: 10.1016/j.cub.2013.09.028. [DOI] [PubMed] [Google Scholar]
- Turek M, Besseling J, Spies JP, König S, Bringmann H. Sleep-active neuron specification and sleep induction require FLP-11 neuropeptides to systemically induce sleep. eLife. 2016;5:e12499. doi: 10.7554/eLife.12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Buskirk C, Sternberg PW. Epidermal growth factor signaling induces behavioral quiescence in Caenorhabditis elegans. Nat Neurosci. 2007;10:1300–1307. doi: 10.1038/nn1981. [DOI] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci. 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman JE, Naidoo N, Raizen DM, Pack AI. Conservation of sleep: insights from non-mammalian model systems. Trends Neurosci. 2008;31:371–376. doi: 10.1016/j.tins.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.