

Abstract
Here we report the synthesis and in vitro evaluation of 25 new quinolinyl analogues for α-synuclein aggregates. Three lead compounds were subsequently labeled with carbon-11 or fluorine-18 to directly assess their potency in a direct radioactive competitive binding assay ng both α-synuclein fibrils and tissue homogenates from Alzheimer’s disease (AD) cases. The modest binding affinities of these three radioligands toward α-synuclein were comparable with results from the Thioflavin T fluorescence assay. However, all three ligand also showed modest binding affinity to the AD homogenates and lack selectivity for α-synuclein. The structure-activity relationship data from these 25 analogues will provide useful information for design and synthesis of new compounds for imaging α-synuclein aggregation.
Keywords: α-Synuclein fibrils, quinolinyl analogue, radiosynthesis, PET radiotracer, Thioflavin T fluorescence assay, Parkinson’s disease
Graphical abstract
Parkinson’s disease (PD) is a chronic and progressive neurodegenerative disease. The classical motor symptoms of PD include bradykinesia, resting tremor, rigidity, while non-motor symptoms involve depression, olfactory malfunction, and cognitive impairment, all of which significantly compromise the patient’s daily life.1 PD is characterized by the loss of dopamine neurons within the substantia nigra pars compacta along with the formation of intracellular fibrillar Lewy bodies (LBs) and Lewy neuritis inclusions (LNs). Aggregated α-synuclein is the predominant component of LBs and LNs.1 α-Synuclein is a highly soluble when unfolded presynaptic protein that consists of 140 amino acids which is ubiquitously expressed in the brain.2 It plays an important role in recycling synaptic vesicles and in regulating the synthesis, storage, and release of neurotransmitters.3, 4 The accumulation of misfolded α-synuclein protein is widely recognized as the pathological hallmark of PD and other synucleinopathies including Dementia with Lewy bodies and Multiple System Atrophy (MSA).5 Furthermore, Braak staging of brain tissue from PD cases suggests that the appearance of α-synuclein inclusions precedes dopaminergic loss6 and that α-synuclein deposition may occur years prior to the appearance of motor symptoms.7, 8 An additional concern is that treatment of PD with dopaminergic drugs alters the striatal uptake of dopaminergic imaging biomarkers used for monitoring disease progression;9 poor correlation has been reported between PET measures with these tracers and clinical status in therapeutic trials of PD progression.10, 11 Therefore, the strategy of imaging deposited α-synuclein aggregates rather than dopaminergic changes could provide a more accurate definition of early-stage premotor PD. Identifying a suitable positron emission tomography (PET) radioligand that is able to map the α-synuclein aggregation in the brain at early stage of PD is imperative for development of disease-modifying therapeutics aimed at slowing or terminating the progression of PD. The use of PET in assessing α-synuclein aggregation in the brain has been hampered by the lack of agents having high binding potency and high selectivity for α-synuclein over other proteins associated with neurodegenerative disease, particularly, amyloid beta (Aβ) and tau protein. Investigators have put tremendous effort into developing highly potent and highly selective α-synuclein PET radiotracers.12 To date, two classes of small molecular probes have been reported: fluorescent probes, and radiolabeled probes.12 Fluorescent probes, such as Thioflavin T, Thioflavin S, N-arylaminonaphthalene sulfonate analogues, cyanine dyes, Evans blue, LDS798, quinoxalin analogues, and Nile red analogues, have not shown selectivity for Lewy bodies versus Aβ plaques.12 For the radiolabeled probes, the benzoxazole derivative, [2-[2-(2-dimethylaminothiazol-5-yl-)ethenyl]-6-[2-[18F]fluoroethoxy]-benzoxazole ([18F]BF227), 11C-Pittsburgh compound-B ([11C]PIB), and chalcone analogs [125I]IDP-4 have been evaluated for imaging α-synuclein (Figure 1).13 [18F]BF227 failed to bind to Aβ-free Dementia with Lewy Bodies or age-matched control samples while it bound to AD brain homogenates.13 [18F]BF227 did not show promise in imaging α-synuclein aggregation in an accelerated mouse model of synucleinopathy which develops α-synuclein deposits in brainstem and thalamus,.14 Preclinical evaluation of PIB as an α-synuclein specific PET tracer has been inconsistent.15 The higher affinity of PIB for Aβ proteins over α-synuclein poses another clear concern. The single-photon emission computed tomography (SPECT) tracer [125I]IDP-4 reportedly has high binding affinity for α-synuclein fibrils (Kd = 5.4 nM); however its high molecular weight and high lipophilicity caused low brain uptake in vivo.16
Figure 1.
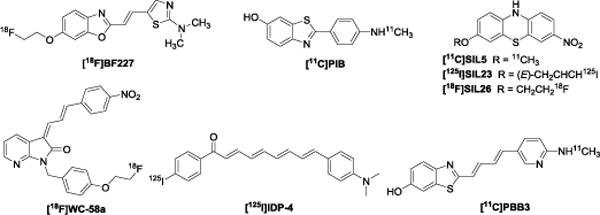
Reported radiotracers for α-synuclein.
Recently, we reported several radiolabeled tricyclic ligands [11C]SIL5, [125I]SIL23, and [18F]SIL26 which showed reasonable in vitro binding affinity and selectivity for recombinant α-synuclein fibrils compared to Aβ and tau fibrils (Figure 1). However, in vitro evaluation of these radioligands in postmortem PD cases showed only modest affinity for tissue homogenates from PD cases, this lack of selectivity discouraged further evaluation of these radiotracers for PET imaging of α-synuclein aggregates in human brain.17–19 We also reported a series of indolinone and indolinone-diene analogues which bind to α-synuclein fibrils.20 Although the indolinone-diene analogues had a higher binding affinity for α-synuclein fibrils than the related indolinone congeners, most indolinone-diene analogues exist as two inseparable regioisomers, which can make identification of clear structure-activity relationships challenging. This issue was overcome by introduction of distinct substituents into the benzene ring of the -ene and -diene moieties (i.e., [18F]WC-58a, Figure 1).20 Thioflavin T indirect competetive binding and direct homologous radioligand competitive binding assays with the single isomer clearly indicated favorable binding affinity of these ligands towards α- synuclein fibrils and good selectivity (> 50-fold) towards Aβ and tau fibrils. However, the high log P value of the most potent compound limited the in vivo application of [18F]WC-58a.20 More recently, researchers used the tau PET ligand 2-((1E,3E)-4-(6-([11C]methylamino)pyridin-3-yl)buta-1,3-dienyl)benzo[d]thiazol-6-ol ([11C]PBB3) (Figure 1) to assess α-synuclein pathology by in vitro fluorescence and autoradiographic studies; although α-synuclein pathology in Lewy body disease was not detected by [11C]PBB3 PET, the tracer was able to image MSA cases having high densities of glial cytoplasmic inclusions.21
Here we report our continuing efforts on the development of PET tracers for imaging α-synuclein aggregation.12, 17–20 New analogues containing a quinolinyl moiety and six-membered aromatic rings linked by either a double bond or an oxadiazole bridge were synthesized and their in vitro binding properties were characterized (Figure 2). The rationales behind the design of these new compounds include: 1) LDS798 contains multiple double bounds and is able to label Lewy bodies and penetrate the blood brain barrier, therefore we introduced a quinolinyl group that has a double bond side chain;12 2) N-arylaminonaphthalene sulfonate analogue 2,6-ANS shows binding to α-synuclein fibrils, therefore, we introduced heteroatoms on both aromatic rings of 2,6-ANS and modified the secondary amine bridge of our new analogues; 3) an oxadiazole fragment was used to replace the bridged double bond in order to: optimize the lipophilicity of the target compounds, restrict the flexibility of the molecular structure, and overcome the potential isomerization of the double bound. This strategy was based on the observation that oxadiazole moieties have been employed for radiotracers that target Aβ in Alzheimer’s disease, neurotransmitter α7 nicotinic acetylcholine receptors in neuropsychiatric diseases, and type 2 cannabinoid receptor for neuroinflammation.22–25 As described in detail below, the in vitro binding affinity of the 25 newly synthesized quinolinyl analogues was measured using a Thioflavin T fluorescence assay (Table 1); this screening identified six compounds with binding affinity <60 nM towards recombinant α-synuclein fibrils. Subsequently, we carbon-11 or fluorine-18 radiolabeled three compounds to perform direct homologous radioactive competitive binding assays, results showed modest binding potency for α-synuclein fibrils; further characterization using AD tissue homogenates also revealed modest binding with AD tissue (Figure 3).
Figure 2.
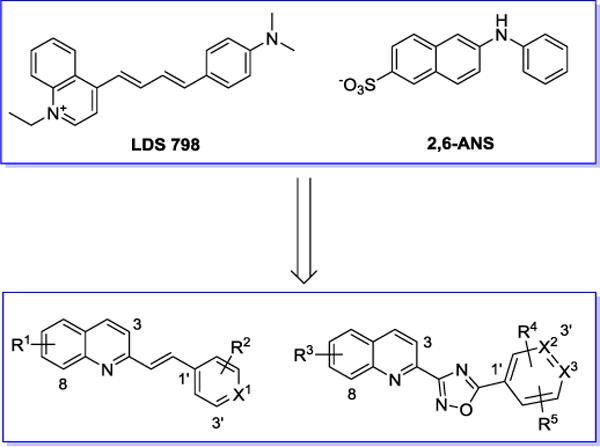
Rationale of newly designed α-synuclein compounds.
Table 1.
Binding affinities (Ki) of new compounds determined by Thioflavin T indirect competitive binding assay.
| Entry | Compound | Thioflavin T assay Ki (nM) a |
Log Pb |
|---|---|---|---|
| 1 | 1 | NB | 4.61 |
| 2 | 2 | 192 ± 1.0 | 4.61 |
| 3 | 3 | 680 ± 139 | 4.6 |
| 4 | 4 | NB | 4.61 |
| 5 | 5 | NB | 3.27 |
| 6 | 6 | NB | 3.27 |
| 7 | 7 | 56 ± 40 | 3.27 |
| 8 | 8 | 52 ± 38 | 3.27 |
| 9 | 9 | NB | 3.27 |
| 10 | 10 | NB | 3.27 |
| 11 | 11 | NB | 3.89 |
| 12 | 12 | 54 ± 2 | 4.17 |
| 13 | 13 | 52 ± 21 | 3.86 |
| 14 | 14 | 18 ±1 | 4.05 |
| 15 | 15 | NB | 3.83 |
| 16 | 16 | 167 ± 28 | 4.10 |
| 17 | 26 | NB | 3.42 |
| 18 | 27 | 630 | 4.01 |
| 19 | 28 | 15 ± 3 | 4.20 |
| 20 | 29 | NB | 4.13 |
| 21 | 30 | NB | 4.67 |
| 22 | 31 | NB | 3.62 |
| 23 | 32 | NB | 4.28 |
| 24 | 33 | NB | 3.81 |
| 25 | 34 | NB | 3.74 |
Ki values (mean ± SD nM) were determined in at least three experiments, NB; no binding was measurable in the Thioflavin T assay;
Calculated by ChemBioDraw Ultra 16.0.
Figure 3.
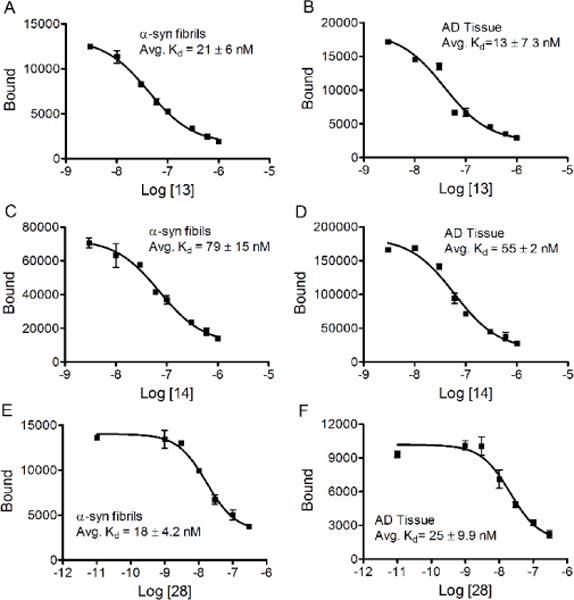
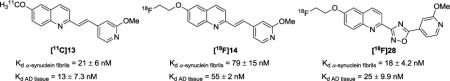
Kd values for [11C]13, [18F]14, and [18F]28 determined by the direct radioligand competitive binding assay.
The synthesis of quinolinyl analogues with a double bond bridge is outlined in Scheme 1. Condensation of 2-methylqinoline derivatives with aldehyde in the presence of p-toluenesulfonamide at 130 °C afforded compounds 1 – 16 in high yields.26 The final compounds include diverse substituents with a fluoroethoxy group in the 6-position, or a methoxy group in the 3–8-position of the quinolinyl fragment. Methoxy and halogen substituents were also introduced to the six-membered aromatic ring (pyridyl fragment); these substituents facilitate convenient carbon-11 or fluorine-18 radiolabeling.
Scheme 1.
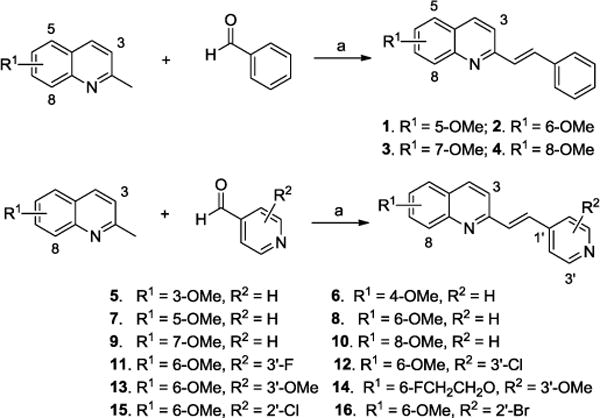
Synthesis of quinolinyl analogues with a double bond bridge. Reagents and conditions: (a) p-toluenesulfonamide, toluene, 130 °C.
Next, we synthesized additional analogues in which the bridged double bond in compounds 1 – 16 was replaced with an oxadiazole moiety. Lower lipophilicity could be expected by this substitution which has been used successfully for other CNS radiotracers.22–25 Reaction of commercially available 6-methoxyquinoline-2-carbonitrile 17 with hydroxylamine hydrochloride afforded carboximidamide 18 in near quantitative yield, which was used for next condensation reaction without further purification. Removal of the methyl group from compound 17 gave phenol 19, which was alkylated to produce the 6-position modified compounds 20 – 22. Compounds 20 – 22 were converted to corresponding carboximidamides 23 – 25 using similar strategy as used to produce 18. Condensation of compounds 18, and 23 – 25 with nicotinic acid or isonicotinic acid afforded compounds 26 – 30. By similar strategy, the four imidazole pyridine analogues 31 – 34 were obtained in moderate yield (Scheme 2).
Scheme 2.
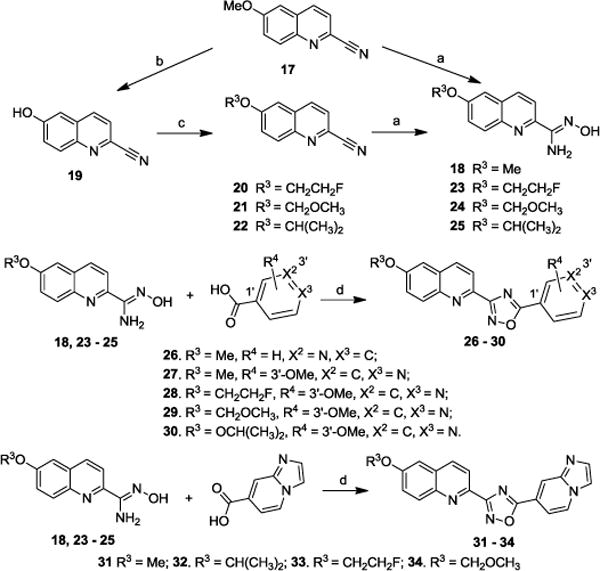
Synthesis of oxadiazole containing α-synuclein compounds. Reagents and conditons: (a) NH2OH HCl, EtOH/H2O (2/1, v/v), NaHCO3; (b) AlCl3, ClCH2CH2Cl; (c) Cs2CO3, THF, 70 °C, TsOCH2CH2F for 20, MeOCH2Cl for 21, CHBr(CH3)2 for 22; (d) EDCl·HCl, HOBt, DIPEA, DMF, 130 °C.
The binding affinities of the quinolinyl compounds for recombinant α-synuclein fibrils were determined using a Thioflavin T assay as previously reported (Table 1).17 Ten of the 25 compounds produced measurable displacement of Thioflavin T in the competitive binding assay, However the maximum displacement of ThioT binding was only ~ 50%, indicating that the binding interaction of quinolinyl compounds with α-synuclein fibrils did not completely overlap with that of Thioflavin T. Fifteen of the compounds did not produce sufficient displacement of Thioflavin T (at concentrations up to 10 μM) to enable measurement of Ki values. For phenyl-containing compounds 1–4, no binding was observed for compounds 1 and 4, while the 6-OMe derivative, compound 2, showed a Ki value of 192 nM and the 7-OMe derivative 3 had a Ki value > 500 nM (Table 1, entries 1-4). Among the pyridyl containing compounds 5 – 10, methoxy substitution on the 5- or 6- position of the quinolinyl ring resulted in modest affinity in compounds 7 and 8 with respective Ki values of 56 nM, 52 nM, while substitution on the 3-, 4-, 7-, or 8-position resulted in no measurable binding potency (Table 1, entries 5 – 10). Among the 6-position substituted structures, compounds 11–16 had additional substituents on the pyridyl ring. Compounds 12, 13, and 14 showed high binding affinity with Ki values of 54, 52, and 18 nM respectively; compound 16 displayed moderate binding and compounds 11 and 15 had no binding (Table 1, entries 11–16). For oxadiazole containing compounds 26– 34, one fragment was the 6-position modified quinolinyl moiety while the other included a pyridyl or (pyridyl)imidazole fragment. Compound 28 displayed high binding affinity with a Ki value of 15 nM, but the other compounds had very weak potency (Table 1, entries 17-25). Six of the 25 new compounds displayed modest potency with Ki values < 60 nM; compounds 14 and 28 were slightly more potent with Ki values of 18 and 15 nM respectively.
Among the six compounds with Ki values <60 nM for recombinant α-synuclein fibrils in the Thioflavin T fluorescence assay, compounds 13, 14, and 28 were radiolabeled with carbon-11 or fluorine-18 to further evaluate their performance in a homologous competitive binding assay which measures direct competition between the radioligand and the unlabeled compound. Such assays can provide greater accuracy and sensitivity than the Thioflavin T method. The syntheses of precursors for radiolabeling are illustrated in Scheme 3. Reaction of commercially available 2-methoxy-6-hydroxyquinoline with bromomethyl methyl ether afforded compound 35, which was subjected to condensation with 2-methoxyisonicotinaldehyde to provide compound 36. Acidolysis of the MOM protecting group in compound 36 gave the radiolabeling precursor 37. The oxadiazole containing radiolabeling precursor 38 was obtained from compound 29 using the same procedure used to synthesize compound 37.
Scheme 3.
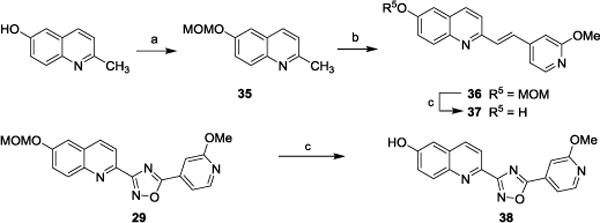
Synthesis of radiolabeling precursors. Reagents and conditions: (a) MOMBr, Et3N, CH2Cl2; (b) 2-methoxyisonicotinaldehyde, TsNH2, toluene, 130 °C; (c) CF3COOH, CH2Cl2.
The radiosyntheses of [11C]13, [18F]14, and [18F]28 are shown in Scheme 4. The one-step procedure for the radiosynthesis of [11C]13 progressed smoothly with [11C]methyl iodide in the presences of aqueous sodium hydroxide solution to afford [11C]13 in good yield and high specific activity (50 ± 10% yield, specific activity >148 GBq/μmol, decay corrected to end of bombardment, EOB). A two-step procedure was used for radiosynthesis of [18F]14 and [18F]28. The radioactive intermediate 2-[18F]fluoroethyl tosylate was synthesized following routine methods,27,28, 29 followed by alkylation of the phenol in precursor compound 37or 38 to respectively afford [18F]14 or [18F]28. We have found this two-step procedure to be much reliable and reproducible than one-step methods; an additional benefit is that phenol precursors are stable during storage. [18F]14 was produced in 21 ± 7% yield, specific activity >27 GBq/μmol (decay corrected to end of synthesis, EOS); [18F]28 was produced in 30 ± 4% yield, specific activity >37 GBq/μmol (decay corrected to EOS).
Scheme 4.
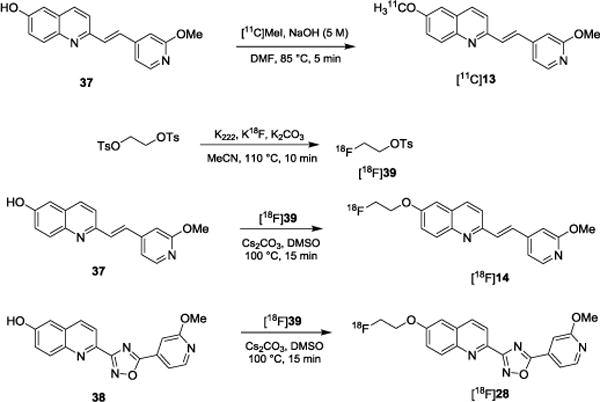
Radiosynthesis of [11C]13, [18F]14, and [18F]28.
Using the direct radioligand competitive binding assay, binding affinities (Kd values) towards recombinant α-synuclein fibrils of compounds [11C]13, [18F]14, and [18F]28 were determined to be 21, 79, and 18 nM respectively. These modest affinities are comparable with the results from the Thioflavin T fluorescence binding assay, which showed similarly modest Ki values of 52, 18, and 15 nM respectively, towards recombinant α-synuclein fibrils. The binding affinities of these three radiotracers towards tissue homogenates from AD cases were also determined in the direct radioligand competitive binding assay; the Kd values of [11C]13, [18F]14, and [18F]28 were determined to be 13, 55, and 25 nM respectively (Figure 3), suggesting that these three radioligands lack selectivity for α-synuclein aggregates over AD.
Significant progress has been made in recent years in the development of PET probes for imaging Aβ plaques in Alzheimer’s disease; [18F]Florbetapir, [18F]Florbetaben, [18F]Flutemetamol, have all been approved by FDA to estimate Aβ neuritic plaque density in adult AD patients with cognitive impairment.12 However, the development of PET radiotracers for selective imaging of α-synuclein aggregation in PD has been more challenging. This may result from the comparatively lower density of aggregated α-synuclein in the brain compared to Aβ or tau proteins.30 Furthermore, α-synuclein aggregates are often accompanied by other pathological Aβ and tau protein aggregates, suggesting that selective detection will be more complicated. As a general rule, a binding potential (BP = Bmax/Kd) >2 is required for clinical translation of a radioligand.31 This implies that identification of highly potent ligands with a Kd value at subnanomolar range will be pivotal for imaging α-synuclein aggregation. In our current work, we designed and synthesized 25 new compounds. Among them, screening with a ThioflavinT assay identified six compounds with modest binding affinities for recombinant α-synuclein fibrils. Three of these leads were subsequently radiolabeled with carbon-11 or fluorine-18. A direct radioligand binding assay also showed all three compounds had modest affinity for recombinant α-synuclein fibrils, which was comparable to the results of the Thioflavin T assay. However, when tissue homogenates from AD cases were used in the direct homologous binding assay, these three radioligands lacked selectivity. Further structural optimization is required to identify new ligands with higher potency and selectivity for α-synuclein aggregation.
Experimental details for the intermediates and target compounds, radiolabeling procedures, and binding assay methods are provided in the Notes.32
In conclusion, we designed and synthesized 25 new quinolinyl analogues for α-synuclein. Thioflavin T fluorescence assay revealed six compounds had binding affinities <60 nM towards recombinant α-synuclein fibrils. Three of these compounds were further radiolabeled with carbon-11 or fluorine-18. A direct radioligand homologous binding assay demonstrated affinities for recombinant α-synuclein fibrils comparable to those determined by Thioflavin T assay. However, the lack of selectivity of these three radioligands toward α-synuclein fibrils over AD tissues in the direct binding assay discouraged further in vivo evaluation. Our structure-activity relationship data may provide useful information for future design and synthesis of new PET tracers for imaging α-synuclein aggregation in patients with PD and other diseases with α-synucleinopathies.
Experimetal details
a) General
All reagents and chemicals were purchased from commercial suppliers and used without further purification unless otherwise stated. 1H NMR and 13C NMR spectra were recorded at 400 MHz on a Varian spectrometer with CDCl3, CD3COCD3, CD3SOCD3 as solvent and tetramethylsilane (TMS) as the internal standard. All chemical shift values are reported in parts per million (ppm) (δ). Under these conditions, the chemical shifts (in ppm) of residual solvents are observed at 7.26 (CDCl3), 2.05 (CD3COCD3), 2.50 (CD3SOCD3) for H1 NMR. The following abbreviations were used to describe peak patterns wherever appropriate: br = broad, d = doublet, t = triplet, m = multiplet. HRMS analyses were conducted in Washington University Resource for Biomedical and Bio-organic Mass Spectrometry. Preparative chromatography was performed on Chemglass chromatography column using 230 – 400 mesh silica gel purchase from Silicycle. Analytical TLC was carried out on Merck 60 F254 silica gel glass plates, and visualization was aided by UV.
General procedure for the synthesis of vinyl quinoline derivatives
A solution of commercially available quinoline derivatives (1 equiv.), p-toluenesulfonamide (1 equiv.) and aldehyde (1 equiv.) in toluene (1 M) was refluxed at 120 °C for 12 h in a reaction tube under N2. After the mixture was cooled to room temperature, the solvent was removed under reduced pressure. Then the residue was purified by silica gel column chromatography to afford the vinyl quinoline derivatives.
General procedure for carboximidamide formation
To a solution of quinoline-2-carbonitrile derivatives (0.4 M, 1 equiv.) in EtOH/H2O (2/1, v/v) was added hydroxylamine hydrochloride (1 equiv.), sodium bicarbonate (2 equiv.) successively. The mixture was stirred at 100 °C overnight. Upon cooling to the room temperature, water was added and the reaction mixture was extracted with EtOAc. The combined organic phase was washed with water, saturated aqueous sodium chloride, and dried over anhydrous Na2SO4. The organic phase was then concentrated to afford the carboximidamide product, which was used without further purification for the next condensation step.
General procedure for oxadiazole formation
To a solution of acid (1 equiv.) in anhydrous DMF (0.18 M) was added EDCI HCl (1 equiv.), and HOBt (1 equiv.) successively in a sealed tube. The mixture was stirred at rt for 30 min. Carboximidamide (1 equiv.) was added in one portion and the reaction was placed in a pre-heated 130 °C oil bath and continued to stir overnight. Water was added to quench the reaction and the mixture was extracted with EtOAc. The combined organic phase was washed with saturated aqueous sodium bicarbonate solution, water, saturated aqueous sodium chloride, and dried over anhydrous Na2SO4. The organic phase was concentrated in vacuum and the residue was subjected to silica gel chromatography to give the target oxadiazole compound.
General procedure for alkylation of 6-hydroxyquinoline
To a solution of 6-hydroxyquinoline-2-carbonitrile (1 equiv.) in anhydrous THF (0.08 M) was added Cs2CO3 (1.8 equiv.), 2-fluoroethyl tosylate (1.8 equiv.). The reaction was stirred at 70 °C overnight in a sealed tube. The mixture was concentrated and the residue was subjected to silica gel chromatography to afford the alkylation product.
(E)-5-Methoxy-2-styrylquinoline (1)
Yellow solid. MP 83 – 85 °C. 1H NMR (400 MHz, CDCl3) δ 8.00 (d, J = 8.4 Hz, 1H), 7.70 – 7.55 (m, 4H), 7.49 (d, J = 8.4 Hz, 1H), 7.43 – 7.33 (m, 4H), 7.30 (t, J = 7.3 Hz, 1H), 7.13 (dd, J = 8.9, 2.3 Hz, 1H), 3.94 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 160.93, 156.08, 149.92, 136.55, 135.96, 134.06, 129.02, 128.75, 128.52, 128.45, 127.20, 122.55, 119.29, 117.26, 107.13, 55.50. HRMS (ESI) calcd. for C18H16NO [M + H]+ 262.1226, found: 262.1152.
(E)-6-Methoxy-2-styrylquinoline (2)
Light yellow solid. MP 137 – 139 °C. 1H NMR (400 MHz, CDCl3) δ 7.93 (dd, J = 17.7, 8.9 Hz, 2H), 7.61 – 7.47 (m, 4H), 7.37 – 7.27 (m, 4H), 7.24 (t, J = 7.3 Hz, 1H), 6.99 (d, J = 2.7 Hz, 1H), 3.86 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 157.61, 153.68, 144.25, 136.67, 135.05, 133.17, 130.62, 129.03, 128.73, 128.35, 128.26, 127.09, 122.28, 119.53, 105.22, 55.52. HRMS (ESI) calcd. for C18H16NO [M + H]+ 262.1226, found: 262.1155.
(E)-7-methoxy-2-styrylquinoline (3)
Light yellow solid. MP 89 – 91 °C. 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 8.4 Hz,1H), 7.66 – 7.49 (m, 4H), 7.43 (d, J = 8.4 Hz,1H), 7.37 – 7.28 (m,4H), 7.24 (dd, J = 13.5, 6.2 Hz, 1H), 7.06 (dd, J = 8.9, 2.3 Hz, 1H), 3.88 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 159.91, 155.07, 148.91, 135.54, 134.95, 133.04, 128.00, 127.73, 127.50, 127.43, 126.18, 121.53, 118.28, 116.24, 106.11, 54.48. HRMS (ESI) calcd. for C18H16NO [M + H]+ 262.1226, found: 262.1154.
(E)-8-Methoxy-2-styrylquinoline (4)
Light yellow solid. MP 96 – 97 °C. 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J = 8.6 Hz, 1H), 7.60 (d, J = 8.6 Hz, 1H), 7.51 (d, J = 7.5 Hz, 2H), 7.45 (d, J = 7.7 Hz, 2H), 7.27 - 7.24 (m, 3H), 7.19 (dd, J = 13.4, 7.3 Hz, 2H), 6.91 (d, J = 7.5 Hz, 1H), 3.98 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 155.14, 155.10, 140.01, 136.56, 136.21, 133.92, 129.69, 128.75, 128.47, 128.33, 127.20, 126.32, 119.41, 119.13, 107.92, 56.07. HRMS (ESI) calcd. for C18H16NO [M + H]+ 262.1226, found: 262.1154.
(E)-3-Methoxy-2-(2-(pyridin-4-yl)vinyl)quinolone (5)
Light yellow solid. MP 152 – 154 °C. 1H NMR (400 MHz, CDCl3) δ 8.54 (d, J = 5.5 Hz, 2H), 7.97 (d, J = 8.4 Hz, 1H), 7.85 (s, 2H), 7.63 (d, J = 8.0 Hz, 1H), 7.49 (t, J = 7.6 Hz, 1H), 7.45 – 7.36 (m, 3H), 7.33 (s, 1H), 3.93 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 151.59, 150.22, 147.28, 144.33, 143.02, 132.17, 129.10, 129.04, 127.10, 126.94, 126.44, 126.28, 121.56, 112.32, 55.50. HRMS (ESI) calcd. for C17H15N2O [M + H]+ 263.1179, found: 263.1109.
(E)-4-Methoxy-2-(2-(pyridin-4-yl)vinyl)quinolone (6)
Light yellow solid. MP 139 – 141 °C. 1H NMR (400 MHz, CDCl3) δ 8.60 (d, J = 5.9 Hz, 2H), 8.13 (d, J = 8.3 Hz, 1H), 8.00 (d, J = 8.5 Hz, 1H), 7.73 – 7.63 (m, 1H), 7.55 (d, J = 16.2 Hz, 1H), 7.45 (dd, J = 15.6, 8.1 Hz, 4H), 6.92 (s, 1H), 4.06 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 162.65, 155.71, 150.31, 149.03, 143.75, 133.73, 131.09, 130.24, 128.81, 125.80, 121.72, 121.29, 120.87, 98.33, 55.67. HRMS (ESI) calcd. for C17H15N2O [M + H]+ 263.1179, found: 263.1127.
(E)-5-Methoxy-2-(2-(pyridin-4-yl)vinyl)quinolone (7)
Light yellow solid. MP 154 – 156 °C. 1H NMR (400 MHz, CDCl3) δ 8.63 – 8.50 (m, 2H), 7.97 (d, J = 8.4 Hz, 1H), 7.59 (d, J = 8.9 Hz, 1H), 7.51 (d, J = 16.3 Hz, 1H), 7.46 – 7.28 (m, 5H), 7.11 (dd, J = 8.9, 2.1 Hz, 1H), 3.89 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 161.06, 154.71, 150.29, 149.89, 143.76, 136.23, 133.18, 131.07, 128.50, 122.89, 121.27, 119.99, 117.64, 107.05, 55.51. C17H15N2O [M + H]+ 263.1179, found: 263.1173.
(E)-6-Methoxy-2-(2-(pyridin-4-yl)vinyl)quinolone (8)
Light yellow solid. MP 132 – 134 °C. 1H NMR (400 MHz, CDCl3) δ 8.60 (d, J = 6.0 Hz, 2H), 8.03 (d, J = 8.6 Hz, 1H), 7.97 (d, J = 9.2 Hz, 1H), 7.60 (d, J = 8.5 Hz, 1H), 7.52 (d, J = 1.2 Hz, 2H), 7.44 (d, J = 6.0 Hz, 2H), 7.37 (dd, J = 9.2, 2.8 Hz, 1H), 7.05 (d, J = 2.7 Hz, 1H), 3.92 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 158.03, 152.32, 150.28, 144.30, 143.98, 135.26, 133.27, 130.82, 130.24, 128.68, 122.73, 121.22, 119.93, 105.10, 55.55. HRMS (ESI) calcd. for C17H15N2O [M + H]+ 263.1179, found: 263.1108.
(E)-7-Methoxy-2-(2-(pyridin-4-yl)vinyl)quinolone (9)
Light yellow solid. MP 104 – 106 °C. 1H NMR (400 MHz, CDCl3) δ 8.55 (d, J = 5.7 Hz, 2H), 8.00 (d, J = 8.4 Hz, 1H), 7.60 (d, J = 8.9 Hz, 1H), 7.54 (d, J = 16.3 Hz, 1H), 7.47 – 7.41 (m, 2H), 7.39 (d, J = 5.8 Hz, 2H), 7.34 (d, J = 2.1 Hz, 1H), 7.11 (dd, J = 8.9, 2.3 Hz, 1H), 3.90 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 161.11, 154.77, 150.31, 149.95, 143.83, 136.25, 133.24, 131.14, 128.50, 122.93, 121.29, 120.02, 117.67, 107.12, 55.53. HRMS (ESI) calcd. for C17H15N2O [M + H]+ 263.1179, found: 263.1108.
(E)-8-Methoxy-2-(2-(pyridin-4-yl)vinyl)quinolone (10)
Brown solid. MP 74 – 76 °C. 1H NMR (400 MHz, CDCl3) δ 8.54 (d, J = 5.8 Hz, 2H), 8.05 (d, J = 8.6 Hz, 1H), 7.66 (d, J = 8.6 Hz, 1H), 7.60 (d, J = 16.4 Hz, 1H), 7.44 (d, J = 16.5 Hz, 1H), 7.41 – 7.34 (m, 3H), 7.30 (d, J = 8.1 Hz, 1H), 6.99 (d, J = 7.6 Hz, 1H), 4.03 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 155.24, 153.80, 150.28, 143.82, 140.03, 136.53, 133.85, 131.02, 128.66, 127.00, 121.26, 119.46, 119.41, 108.12, 56.12. HRMS (ESI) calcd. for C17H15N2O [M + H]+ 263.1179, found: 263.1109.
(E)-2-(2-(2-Fluoropyridin-4-yl)vinyl)-6-methoxyquinoline (11)
Yellow solid. MP 141 – 143 °C. 1H NMR (400 MHz, CDCl3) δ 8.17 (d, J = 5.2 Hz, 1H), 8.02 (d, J = 8.5 Hz, 1H), 7.96 (d, J = 9.2 Hz, 1H), 7.55 (d, J = 8.5 Hz, 1H), 7.49 (d, J = 4.5 Hz, 2H), 7.39 – 7.27 (m, 2H), 7.06 – 6.98 (m, 2H), 3.90 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 164.54 (d, J = 238.4 Hz), 158.17, 151.75, 149.72 (d, J = 8.1 Hz), 147.90 (d, J = 15.2 Hz), 144.27, 135.34, 134.44, 130.83, 128.91 (d, J = 4.0 Hz), 128.83, 122.88, 120.10, 118.94 (d = 4.0 Hz), 106.75 (d, J = 38.4 Hz), 105.05, 55.55. HRMS (ESI) calcd. for C17H14FN2O [M + H]+ 281.1085, found: 281.1008.
(E)-2-(2-(2-Chloropyridin-4-yl)vinyl)-6-methoxyquinoline (12)
Yellow solid. MP 156 – 158 °C. 1H NMR (400 MHz, CDCl3) δ 8.36 (d, J = 5.2 Hz, 1H), 8.05 (d, J = 8.5 Hz, 1H), 7.97 (d, J = 9.2 Hz, 1H), 7.57 (d, J = 8.5 Hz, 1H), 7.51 – 7.46 (m, 3H), 7.39 – 7.35 (m, 2H), 7.05 (d, J = 2.7 Hz, 1H), 3.93 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 158.19, 152.26, 151.74, 149.97, 147.27, 144.34, 135.33, 134.55, 130.91, 128.84, 128.71, 122.90, 121.69, 120.16, 119.85, 105.05, 55.57. HRMS (ESI) calcd. for C17H14ClN2O [M + H]+ 297.0789, found: 297.0709.
(E)-6-Methoxy-2-(2-(2-methoxypyridin-4-yl)vinyl)quinolone (13)
White solid. MP 125 – 127 °C. 1H NMR (400 MHz, CDCl3) δ 8.14 (d, J = 5.4 Hz, 1H), 8.02 (d, J = 8.6 Hz, 1H), 7.96 (d, J = 9.2 Hz, 1H), 7.58 (d, J = 8.5 Hz, 1H), 7.47 (d, J = 2.0 Hz, 2H), 7.36 (dd, J = 9.2, 2.7 Hz, 1H), 7.09 (d, J = 5.3 Hz, 1H), 7.04 (d, J = 2.6 Hz, 1H), 6.86 (s, 1H), 3.95 (s, 3H), 3.91 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 164.97, 157.97, 152.46, 147.10, 146.76, 144.29, 135.21, 133.03, 130.81, 130.38, 128.63, 122.64, 119.92, 114.32, 108.79, 105.10, 55.54, 53.49. HRMS (ESI) calcd. for C18H17N2O2 [M + H]+ 293.1285, found: 293.1207.
(E)-6-(2-Fluoroethoxy)-2-(2-(2-methoxypyridin-4-yl)vinyl)quinolone (14)
White solid. MP 114 – 116 °C. 1H NMR (400 MHz, CDCl3) δ 8.08 (d, J = 5.4 Hz, 1H), 7.92 (dd, J = 8.8, 4.4 Hz, 2H), 7.50 (d, J = 8.6 Hz, 1H), 7.40 (d, J = 4.2 Hz, 2H), 7.34 (dd, J = 9.3, 2.7 Hz, 1H), 7.05 – 6.99 (m, 1H), 6.97 (d, J = 2.7 Hz, 1H), 6.79 (s, 1H), 4.83 – 4.65 (m, 2H), 4.30 – 4.16 (m, 2H), 3.89 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 164.96, 156.71, 152.75, 147.11, 146.68, 144.43, 135.26, 132.91, 131.02, 130.58, 128.45, 122.67, 120.01, 114.31, 108.81, 106.12, 81.70 (d, J = 171.7 Hz), 67.32 (d, J = 20.2 Hz), 53.49. HRMS (ESI) calcd. for C19H18FN2O2 [M + H]+ 325.1347, found: 325.1262.
(E)-2-(2-(3-Chloropyridin-4-yl)vinyl)-6-methoxyquinoline (15)
Light yellow solid. MP 145 – 146 °C. 1H NMR (400 MHz, CDCl3) δ 8.60 (s, 1H), 8.46 (d, J = 5.2 Hz, 1H), 8.05 (d, J = 8.6 Hz, 1H), 7.98 (d, J = 9.2 Hz, 1H), 7.86 (d, J = 16.4 Hz, 1H), 7.69 (d, J = 8.6 Hz, 1H), 7.61 (d, J = 5.2 Hz, 1H), 7.53 (d, J = 16.4 Hz, 1H), 7.37 (dd, J = 9.2, 2.7 Hz, 1H), 7.06 (d, J = 2.6 Hz, 1H), 3.93 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 158.21, 152.16, 150.24, 147.77, 144.30, 141.88, 135.53, 135.29, 130.97, 130.83, 128.81, 126.03, 122.79, 120.11, 119.64, 105.09, 55.57. HRMS (ESI) calcd. for C17H14ClN2O [M + H]+ 297.0789, found: 297.0710.
(E)-2-(2-(3-Bromopyridin-4-yl)vinyl)-6-methoxyquinoline (16)
White solid. MP 104 – 106 °C. 1H NMR (400 MHz, CDCl3) δ 8.74 (s, 1H), 8.49 (d, J = 5.1 Hz, 1H), 8.07 (d, J = 8.6 Hz, 1H), 7.99 (d, J = 9.2 Hz, 1H), 7.82 (d, J = 16.3 Hz, 1H), 7.71 (d, J = 8.6 Hz, 1H), 7.62 (d, J = 5.2 Hz, 1H), 7.51 (d, J = 16.3 Hz, 1H), 7.38 (dd, J = 9.2, 2.7 Hz, 1H), 7.07 (d, J = 2.7 Hz, 1H), 3.94 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 158.22, 152.71, 152.16, 148.31, 144.31, 143.67, 135.66, 135.30, 130.98, 128.81, 128.72, 122.79, 122.02, 120.65, 119.59, 105.10, 55.58. HRMS (ESI) calcd. for C17H14BrN2O [M + H]+ 341.0284, found: 341.0190.
6-Hydroxyquinoline-2-carbonitrile (19)
To a solution of 6-methoxyquinoline-2-carbonitrile (552 mg, 3 mmol) in CH2Cl2 (20 mL) was added AlCl3 (1.0 g, 7.5 mmol). The reaction was stirred at 45 °C overnight in a sealed tube. TLC showed the starting material remained so a second portion of AlCl3 was added (1.0 g, 7.5 mmol). Water was added to quench the reaction and the mixture was extracted with EtOAc. The combined organic phase was washed with saturated bicarbonate solution, saturated sodium chloride, respectively. The concentrated residue was subjected to silica gel chromatography (hexane/EtOAc, 3/1, v/v) to afford 6-hydroxyquinoline-2- carbonitrile (268 mg, 52% yield). 1H NMR (400 MHz, CD3COCD3) δ 9.41 (s, 1H), 8.22 (d, J = 8.5 Hz, 1H), 7.87 (d, J = 9.2 Hz, 1H), 7.68 – 7.62 (m, 1H), 7.44 – 7.38 (m, 1H), 7.20 (d, J = 1.8 Hz, 1H).
(Z)-6-(2-Fluoroethoxy)-N′-hydroxyquinoline-2-carboximidamide (20)
1H NMR (400 MHz, CD3COCD3) δ 8.31 (d, J = 8.5 Hz, 1H), 7.91 (d, J = 9.3 Hz, 1H), 7.72 (d, J = 8.5 Hz, 1H), 7.46 (dd, J = 9.3, 2.8 Hz, 1H), 7.36 (d, J = 2.8 Hz, 1H), 4.81 – 4.67 (m, 2H), 4.41 – 4.32 (m, 2H).
6-(Methoxymethoxy)quinoline-2-carbonitrile (21)
1H NMR (400 MHz, CD3COCD3) δ 8.31 (d, J = 8.4 Hz, 1H), 7.89 (d, J = 9.2 Hz, 1H), 7.69 (d, J = 8.5 Hz, 1H), 7.46 (dd, J = 9.2, 2.7 Hz, 1H), 7.41 (s, 1H), 5.26 (s, 2H), 3.34 (s, 3H).
6-Isopropoxyquinoline-2-carbonitrile (22)
1H NMR (400 MHz, CD3COCD3) δ 8.41 (d, J = 8.5 Hz, 1H), 7.99 (d, J = 9.2 Hz, 1H), 7.81 (dd, J = 8.5, 1.2 Hz, 1H), 7.51 – 7.48 (m, 1H), 7.43 (d, J = 2.3 Hz, 1H), 4.98 – 4.76 (m, 1H), 1.39 (d, J = 6.0 Hz, 6H).
3-(6-Methoxyquinolin-2-yl)-5-(pyridin-3-yl)-1,2,4-oxadiazole (26)
White solid. MP 175 – 177 °C. 1H NMR (400 MHz, CDCl3) δ 9.46 (s, 1H), 8.78 (d, J = 4.7 Hz, 1H), 8.50 (d, J = 8.0 Hz, 1H), 8.19 – 8.12 (m, 3H), 7.45 (dd, J = 7.9, 4.9 Hz, 1H), 7.37 (dd, J = 9.2, 2.6 Hz, 1H), 7.05 (d, J = 2.6 Hz, 1H), 3.88 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 174.47, 169.17, 158.97, 153.44, 149.31, 144.28, 143.36, 135.86, 135.53, 131.90, 130.14, 123.77, 123.31, 120.47, 120.45, 104.86, 55.61. HRMS (ESI) calcd. for C17H13N4O2 [M + H]+ 305.1033, found: 305.0950.
5-(2-Methoxypyridin-4-yl)-3-(6-methoxyquinolin-2-yl)-1,2,4-oxadiazole (27)
White solid. MP 188 – 189 °C. 1H NMR (400 MHz, CDCl3) δ 8.33 (d, J = 5.3 Hz, 1H), 8.24 – 8.04 (m, 3H), 7.64 (d, J = 5.2 Hz, 1H), 7.54 (s, 1H), 7.38 (dd, J = 9.3, 2.7 Hz, 1H), 7.06 (d, J = 2.6 Hz, 1H), 3.95 (s, 3H), 3.90 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 174.68, 169.31, 164.88, 159.01, 148.40, 144.31, 143.32, 135.88, 133.30, 131.93, 130.16, 123.34, 120.45, 114.49, 109.81, 104.86, 55.62, 53.98. HRMS (ESI) calcd. for C18H15N4O3 [M + H]+ 335.1139, found: 335.1051.
3-(6-(2-Fluoroethoxy)quinolin-2-yl)-5-(2-methoxypyridin-4-yl)-1,2,4-oxadiazole (28)
White solid. MP 235 – 237 °C. 1H NMR (400 MHz, CDCl3) δ 8.34 (d, J = 5.3 Hz, 1H), 8.25 – 8.11 (m, 3H), 7.65 (d, J = 5.3 Hz, 1H), 7.55 (s, 1H), 7.44 (dd, J = 9.3, 2.7 Hz, 1H), 7.09 (d, J = 2.7 Hz, 1H), 4.88 – 4.68 (m, 2H), 4.37 – 4.24 (m, 2H), 3.96 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 174.75, 169.28, 164.90, 157.78, 148.42, 144.46, 143.67, 136.00, 133.28, 132.18, 130.00, 123.38, 120.56, 114.49, 109.83, 105.84, 81.63 (d, J = 172.7 Hz), 67.41 (d, J = 21.2 Hz), 54.00. HRMS (ESI) calcd. for C19H16FN4O3 [M + H]+ 367.1201, found: 367.1104.
3-(6-(Methoxymethoxy)quinolin-2-yl)-5-(2-methoxypyridin-4-yl)-1,2,4-oxadiazole (29)
White solid. MP 180 – 181 °C. 1H NMR (400 MHz, CDCl3) δ 8.33 (d, J = 5.2 Hz, 1H), 8.22 – 8.15 (m, 3H), 7.64 (d, J = 5.2 Hz, 1H), 7.54 (s, 1H), 7.44 (d, J = 9.2 Hz, 1H), 7.35 (s, 1H), 5.27 (s, 2H), 3.96 (s, 3H), 3.48 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 174.72, 169.29, 164.88, 156.43, 148.40, 144.56, 143.81, 136.30, 133.28, 132.01, 129.99, 123.55, 120.41, 114.48, 109.81, 108.79, 94.46, 56.30, 53.98. HRMS (ESI) calcd. for C19H17N4O4 [M + H]+ 365.1244, found: 365.1405.
3-(6-Isopropoxyquinolin-2-yl)-5-(2-methoxypyridin-4-yl)-1,2,4-oxadiazole (30)
Light yellow solid. MP 171 – 173 °C. 1H NMR (400 MHz, CDCl3) δ 8.32 (d, J = 5.2 Hz, 1H), 8.20 – 8.03 (m, 3H), 7.63 (dd, J = 5.2, 0.8 Hz, 1H), 7.53 (s, 1H), 7.34 (dd, J = 9.3, 2.6 Hz, 1H), 7.04 (d, J = 2.5 Hz, 1H), 4.66 (dt, J = 12.1, 6.0 Hz, 1H), 3.95 (s, 3H), 1.37 (s, 3H), 1.35 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 174.64, 169.33, 164.87, 157.27, 148.38, 144.02, 143.13, 135.74, 133.31, 131.97, 130.21, 124.21, 120.33, 114.48, 109.79, 106.77, 70.30, 53.96, 21.87. HRMS (ESI) calcd. for C20H19N4O3 [M + H]+ 363.1452, found: 363.1353.
5-(Imidazo[1,2-a]pyridin-7-yl)-3-(6-methoxyquinolin-2-yl)-1,2,4-oxadiazole (31)
Light yellow solid. MP 277 – 279 °C. 1H NMR (400 MHz, CDCl3) δ 8.58 (s, 1H), 8.27 – 8.14 (m, 4H), 7.80 (s, 1H), 7.72 – 7.63 (m, 2H), 7.39 (dd, J = 9.2, 2.8 Hz, 1H), 7.08 (d, J = 2.7 Hz, 1H), 3.91 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 175.02, 169.27, 158.97, 144.31, 144.15, 143.53, 136.47, 135.88, 131.95, 130.15, 126.16, 123.29, 120.50, 119.52, 119.12, 114.18, 110.72, 104.88, 55.64. HRMS (ESI) calcd. for C19H14N5O2 [M + H]+ 344.1142, found: 344.1052.
5-(Imidazo[1,2-a]pyridin-7-yl)-3-(6-isopropoxyquinolin-2-yl)-1,2,4-oxadiazole (32)
Brown yellow solid. MP 203 – 205 °C. 1H NMR (400 MHz, CD3SOCD3) δ 8.84 (d, J = 7.0 Hz, 1H), 8.48 (d, J = 7.4 Hz, 2H), 8.31 – 8.15 (m, 2H), 8.09 (d, J = 8.9 Hz, 1H), 7.90 (s, 1H), 7.65 (d, J = 7.1 Hz, 1H), 7.48 (d, J = 14.6 Hz, 2H), 4.93 – 4.77 (m, 1H), 1.39 (s, 3H), 1.38 (s, 3H). 13C NMR (101 MHz, CD3SOCD3) δ 175.05, 169.14, 157.06, 143.68, 143.38, 143.34, 136.72, 135.96, 131.48, 130.38, 128.64, 124.61, 120.84, 119.40, 117.73, 116.05, 110.34, 107.76, 70.33, 22.10. HRMS (ESI) calcd. for C21H18N5O2 [M + H]+ 372.1455, found: 372.1356.
3-(6-(2-Fluoroethoxy)quinolin-2-yl)-5-(imidazo[1,2-a]pyridin-7-yl)-1,2,4-oxadiazole (33)
Brown yellow solid. MP 188 – 190 °C. 1H NMR (400 MHz, CDCl3) δ 8.66 – 8.44 (m, 1H), 8.24 – 8.11 (m, 4H), 7.75 (d, J = 41.1 Hz, 1H), 7.67 – 7.62 (m, 1H), 7.44 – 7.37 (m, 1H), 7.08 – 7.03 (m, 2H), 4.89 – 4.66 (m, 2H), 4.40 – 4.23 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 175.05, 169.22, 157.38, 144.45, 144.14, 143.86, 136.45, 135.98, 132.18, 129.97, 126.17, 123.30, 121.20, 120.60, 119.11, 114.20, 110.71, 105.86, 81.63 (d, J = 171.7 Hz), 67.41 (d, J = 21.2 Hz). HRMS (ESI) calcd. for C20H15FN5O2 [M + H]+ 376.1204, found: 376.1104.
5-(Imidazo[1,2-a]pyridin-7-yl)-3-(6-(methoxymethoxy)-quinolin-2-yl)-1,2,4-oxadiazole (34)
Light yellow solid. MP 209 – 210 °C. 1H NMR (400 MHz, CDCl3) δ 8.58 (s, 1H), 8.30 – 8.06 (m, 5H), 7.80 – 7.65 (m, 3H), 7.50 – 7.39 (m, 1H), 7.37 (s, 1H), 5.28 (s, 2H), 3.48 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 175.06, 169.25, 156.40, 144.57, 144.16, 144.02, 136.49, 136.29, 132.03, 129.98, 126.16, 123.49, 120.46, 119.49, 119.14, 114.19, 110.71, 108.82, 94.47, 56.31. HRMS (ESI) calcd. for C20H16N5O3 [M + H]+ 374.1248, found: 374.1148.
6-(Methoxymethoxy)-2-methylquinoline (35)
Light yellow liquid. 1H NMR (400 MHz, CDCl3) δ 7.85 (dd, J = 8.7, 4.2 Hz, 2H), 7.31 (dd, J = 9.2, 2.7 Hz, 1H), 7.23 (d, J = 2.7 Hz, 1H), 7.14 (d, J = 8.4 Hz, 1H), 5.19 (s, 2H), 3.43 (s, 3H), 2.62 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 156.83, 154.55, 144.10, 135.36, 129.90, 127.18, 122.23, 109.29, 94.53, 56.09, 24.98. HRMS (ESI) calcd. for C12H14NO2 [M + H]+ 204.1019, found: 204.0962.
(E)-6-(Methoxymethoxy)-2-(2-(2-methoxypyridin-4-yl)-vinyl)quinolone (36)
Light yellow solid. MP 108 – 110 °C. 1H NMR (400 MHz, CDCl3) δ 8.12 (d, J = 5.4 Hz, 1H), 7.97 (dd, J = 8.8, 5.5 Hz, 2H), 7.54 (d, J = 8.6 Hz, 1H), 7.46 – 7.36 (m, 3H), 7.29 – 7.27 (m, 1H), 7.05 (d, J = 5.4 Hz, 1H), 6.83 (s, 1H), 5.26 (s, 2H), 3.92 (s, 3H), 3.49 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 164.94, 155.36, 152.90, 147.08, 146.67, 144.55, 135.53, 132.97, 130.81, 130.61, 128.47, 122.89, 119.84, 114.30, 109.10, 108.80, 94.46, 56.18, 53.47. HRMS (ESI) calcd. for C19H19N2O3 [M + H]+ 323.1390, found: 323.1309.
(E)-2-(2-(2-methoxypyridin-4-yl)vinyl)quinolin-6-ol (37)
To a solution of compound 36 (48 mg, 0.15 mmol) in CH2Cl2 (1 mL) was added CF3COOH (1 mL) at 0 °C. The mixture was stirred at room temperature for 2 h. Saturated sodium bicarbonate was added to neutralize the reaction and the mixture was extracted with EtOAc. The combined organic layer was washed with saturated sodium chloride, dried over anhydrous sodium sulfate, concentrated in vacuo, and the residue was subjected to silica gel chromatography (hexane/EtOAc 1/1) to afford product 37 as a yellow solid (40 mg, 97% yield). MP 244 – 245 °C. 1H NMR (400 MHz, CD3SOCD3) δ 10.17 (s, 1H), 8.27 – 8.13 (m, 2H), 7.91 (d, J = 9.1 Hz, 1H), 7.80 (d, J = 8.6 Hz, 1H), 7.72 – 7.64 (m, 2H), 7.37 (dd, J = 6.8, 4.5 Hz, 2H), 7.19 (d, J = 2.5 Hz, 1H), 7.09 (s, 1H), 3.91 (s, 3H). 13C NMR (101 MHz, CD3SOCD3) δ 164.87, 156.30, 151.92, 147.59, 147.28, 143.25, 135.20, 133.76, 130.85, 130.03, 129.19, 122.87, 120.72, 114.92, 108.80, 108.69, 53.61. HRMS (ESI) calcd. for C17H15N2O2 [M + H]+ 279.1128, found: 279.1052.
2-(5-(2-Methoxypyridin-4-yl)-1,2,4-oxadiazol-3-yl)quinolin-6-ol (38)
To a solution of compound 34 (26 mg, 0.07 mmol) in CH2Cl2 (2 mL) was added CF3COOH (0.5 mL) at 0 °C. The mixture was stirred at room temperature for 3 h. Saturated sodium bicarbonate was added to neutralize the reaction and the mixture was extracted with EtOAc. The combined organic layer was washed with saturated sodium chloride, dried over anhydrous sodium sulfate, concentrated in vacuo, and the residue was subjected to silica gel chromatography (hexane/EtOAc 1/1) to afford product 38 as a yellow solid (18 mg, 79% yield). MP 217 – 219 °C. 1H NMR (400 MHz, CD3COCD3) δ 9.17 (s, 1H), 8.37 (d, J = 5.2 Hz, 1H), 8.23 (d, J = 8.6 Hz, 1H), 8.07 (d, J = 8.6 Hz, 1H), 7.97 (d, J = 9.1 Hz, 1H), 7.63 – 7.58 (m, 1H), 7.43 – 7.35 (m, 2H), 7.21 (d, J = 2.4 Hz, 1H), 3.88 (s, 3H). 13C NMR (101 MHz, CD3SOCD3) δ 172.81, 167.72, 163.16, 155.98, 147.82, 141.58, 140.85, 134.57, 132.06, 130.02, 129.02, 122.26, 119.05, 113.42, 107.64, 107.17, 53.74. HRMS (ESI) calcd. for C17H13N4O3 [M + H]+ 321.0982, found: 321.0982.
b) Radiochemistry
Radiosynthesis of [11C]13
Briefly, [11C]CH3I was produced on-site from [11C]CO2 using a GE PETtrace MeI Microlab. [11C]CH3I was bubbled for a period of 3 – 4 min into a solution of precursor 37 (1 – 2 mg) in anhydrous DMF (0.2 mL) containing 3.0 – 5.0 μL of aqueous sodium hydroxide solution (5.0 M) at room temperature. When the trapping of radioactivity was completed, the sealed reaction vessel was heated at 85 °C for 5 min. After the heating source was removed, 1.7 mL of the HPLC mobile phase (acetonitrile in 0.1 M ammonium formate, 50/50, v/v, pH 4.5) was added into to the reaction vessel. The mixture was loaded onto a reversed phase HPLC system (Agilent Zorbax, SB-C18, 250 × 9.6 mm, 5 μ, UV at 254 nm, flow rate at 4 mL/min) to purify the mixture. The target fraction with retention time of 20 min for [11C]13 was collected into a vial that contained 50 mL sterile water for injection. After finishing the collection, the collected fraction was passed through a C-18 Plus Sep-Pak® cartridge to remove the mobile phase solvent, and the target tracer was retained on the cartridge. Then the Sep-Pak cartridge was rinsed using 20 mL of sterile water. Finally, the [11C]13 trapped on the Sep-Pak was eluted with 0.6 mL of ethanol, following with 5.4 mL 0.9% sodium chloride solution, passing through a 0.22 μ sterile filter into a sterile pyrogen-free glass vial. For quality control (QC), an aliquot was assayed by an analytical HPLC system, UV at 254 nm. [11C]13 was authenticated by co-injecting with the corresponding reference standard, 13. [11C]13 was produced in 50 ± 10% yield with >99% radiochemical purity, specific activity >148 GBq/μmol (decay corrected to EOB). The radiosynthesis typically took 1 hour.
Radiosynthesis of [18F]14 and [18F]28 via [18F]fluoroethyl tosylate
[18F]Fluoride produced in Washington University School of Medicine Cyclotron Facility was first passed through an ion-exchange resin and was then eluted with 0.02 M potassium carbonate (K2CO3) solution. 1.0 – 1.5 mg of the appropriate phenol precursor (37 or 38) was dissolved in Cs2CO3 saturated solution in anhydrous DMSO (200 μL). After vortexing for 1 min, the phenol precursor solution was added into a reaction tube containing [18F]fluoroethyl tosylate, which was readily prepared as described in reference 29. The reaction vessel was capped and briefly swirled, and then kept at 100 °C for 15 min. Subsequently, the residue was diluted with 3 mL of the appropriate HPLC mobile phase (acetonitrile in 0.1 M ammonium formate, 55/45, v/v, pH 4.5 for [18F]14; acetonitrile in 0.1 M ammonium formate, 51/49, v/v, pH 4.5 for [18F]28) then loaded onto a semi-preparative HPLC (Agilent Zorbax, SB-C18, 250 × 9.6 mm, 5 μ) system for purification at 4.0 mL/min flow rate. The HPLC system contains a 5 mL injection loop, a UV detector at 254 nm and a radioactivity detector. After HPLC collection of the desired fraction, (the retention time for [18F]14 was 18 min, for [18F]28 was 22 min), the product fraction was diluted with ~50 mL sterile water. The product was then trapped on a C-18 Sep-Pak Plus cartridge and washed with 20 mL water. The trapped product was eluted with ethanol (0.6 mL) followed by 5.4 mL of 0.9% saline. After passing through a 0.22 μ sterile filter into a sterile pyrogen-free glass vial, the final product was ready for quality control (QC) analysis and direct binding assays. For quality control, an aliquot of sample was assayed by an analytical HPLC system. The sample was authenticated by co-injecting with the reference standard solution. The entire procedure took ~2 h for two-step radiolabeling. [18F]14 was produced in 21 ± 7% yield, specific activity > 27 GBq/μmol (decay corrected to the end of synthesis), [18F]28 was produced in 30 ± 4% yield, specific activity > 37 GBq/μmol (decay corrected to the end of synthesis).
c) In vitro binding assays
Thioflavin T competitive binding fluorescence assay
Indirect affinity determination (Ki) using Thioflavin T fluorescence assay with recombinant α-synuclein fibrils.
The binding affinities of new compounds were determined by a Thioflavin T competitive binding assay. Briefly, the binding assay was conducted in microplates which contained 3 μM Thioflavin T and 0.5 μM α-synuclein fibrils in 30 mM Tris–HCl pH 7.4, 0.1% BSA plus the test compound at concentrations ranging from 3 nM to 10 μM. The mixture was incubated at room temperature for 1.5 h. Fluorescence was determined in a Biotek plate reader using a 440/30 excitation filter and a 485/20 emission filter. All data points were performed in triplicate. Nonspecific fluorescence was measured in parallel reactions containing ThioT plus each concentration of competitor but no fibrils, and these measurements were subtracted from the reactions with fibrils to yield fibril-specific fluorescence. Data were analyzed using Graphpad Prism software (version 4.0) to obtain EC50 values by fitting the data to the equation . Baseline response is (bottom) and maximum response is (top). Ki values were calculated from EC50 values using the equation Ki = EC50/(1 + [radioligand]/Kd). We used an affinity constant value of 1850 nM for Thioflavin-T towards recombinant α-synuclein fibrils.
Direct radioactive competitive binding assay
Affinity determination (Kd) using direct radioactive competitive binding assay with either recombinant α-synuclein fibrils or brain tissue homogenates from AD cases.
These direct binding assays used a fixed concentration of either fibrils or AD tissue and the radioligand ([11C]13, [18F]14, and [18F]28) and varying concentration ranges of corresponding homologous non-radiolabeled standard reference compound. In brief, the standard homologous reference compound was diluted in 30 mM Tris-HCl pH 7.4, 0.1% BSA. Reactions were incubated at 37 °C for 1 h before quantifying bound radioligand. Bound and free radioligand were separated by vacuum filtration through 1.0 μm glass fiber filters in 96-well filter plates (Millipore), followed by three 200 μL washes with ice-cold assay buffer. Filters containing the bound ligand were mixed with 150 μL of Optiphase Supermix scintillation cocktail (PerkinElmer) and counted immediately. All data points were performed in triplicate. The dissociation constant (Kd) and the maximal number of binding sites (Bmax) values were determined by fitting the data to the equation. Bound = (Bmax*[Radioligand])/([Radioligand] + [Unlabeled compound] + Kd) + Bottom by nonlinear regression using Graphpad Prism software (version 4.0), where (Bottom) is nonspecific binding and [Radioligand], [Unlabeled compound], and Kd are expressed in nM.
Acknowledgments
This project was funded by Michael J. Fox Foundation grant supporting a Consortium of Developing α-Synuclein Imaging Agents. United States – the National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (NINDS), National Institute on Aging (NIA) and National Institute of Mental Health (NIMH): NS061025, NS075527, NS103988, NS058714. Mass spectrometry was provided by the Washington University Mass Spectrometry Resource, a National Institute of Health Research Resource (grant P41RR0954). The authors gratefully thank William H. Margenau and Robert Dennett for their excellent assistance in isotope production.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Saeed U, Compagnone J, Aviv RI, et al. Transl Neurodegener. 2017;6:8. doi: 10.1186/s40035-017-0076-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maroteaux L, Campanelli JT, Scheller RH. J Neurosci. 1988;8(8):2804. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng F, Vivacqua G, Yu S. J Chem Neuroanat. 2011;42(4):242. doi: 10.1016/j.jchemneu.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 4.Burre J. J Parkinsons Dis. 2015;5(4):699. doi: 10.3233/JPD-150642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marques O, Outeiro TF. Cell Death Dis. 2012;3:e350. doi: 10.1038/cddis.2012.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.George S, Rey NL, Reichenbach N, Steiner JA, Brundin P. Brain Pathol. 2013;23(3):350. doi: 10.1111/bpa.12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hawkes CH, Del Tredici K, Braak H. Parkinsonism Relat Disord. 2010;16(2):79. doi: 10.1016/j.parkreldis.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 8.Braak H, Del Tredici K. Adv Anat Embryol Cell Biol. 2009;201:1. [PubMed] [Google Scholar]
- 9.Winogrodzka A, Wagenaar RC, Booij J, Wolters EC. Arch Phys Med Rehabil. 2005;86(2):183. doi: 10.1016/j.apmr.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 10.Fahn S, Oakes D, Shoulson I, et al. N Engl J Med. 2004;351(24):2498. doi: 10.1056/NEJMoa033447. [DOI] [PubMed] [Google Scholar]
- 11.Brooks DJ, Frey KA, Marek KL, et al. Exp Neurol. 2003;184:S68. doi: 10.1016/j.expneurol.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 12.Kotzbauer PT, Tu ZD, Mach RH. Clin Transl Imaging. 2017;5(1):3. [Google Scholar]
- 13.Fodero-Tavoletti MT, Mulligan RS, Okamura N, et al. Eur J Pharmacol. 2009;617(1–3):54. doi: 10.1016/j.ejphar.2009.06.042. [DOI] [PubMed] [Google Scholar]
- 14.Levigoureux E, Lancelot S, Bouillot C, et al. Curr Alzheimer Res. 2014;11(10):955. doi: 10.2174/1567205011666141107154201. [DOI] [PubMed] [Google Scholar]
- 15.Maetzler W, Reimold M, Liepelt I, et al. NeuroImage. 2008;39(3):1027. doi: 10.1016/j.neuroimage.2007.09.072. [DOI] [PubMed] [Google Scholar]
- 16.Ono M, Cheng Y, Kimura H, et al. PloS one. 2013;8(9):e74104. doi: 10.1371/journal.pone.0074104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu L, Cui J, Padakanti PK, et al. Bioorg Med Chem. 2012;20(15):4625. doi: 10.1016/j.bmc.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bagchi DP, Yu L, Perlmutter JS, et al. PloS one. 2013;8(2):e55031. doi: 10.1371/journal.pone.0055031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X, Jin H, Padakanti PK, et al. Appl Sci. 2014;4(1):66. doi: 10.3390/app4010066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chu WH, Zhou D, Gaba V, et al. J Med Chem. 2015;58(15):6002. doi: 10.1021/acs.jmedchem.5b00571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koga S, Ono M, Sahara N, Higuchi M, Dickson DW. Mov Disord. 2017;32(6):884. doi: 10.1002/mds.27013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ono M, Haratake M, Saji H, Nakayama M. Bioorg Med Chem. 2008;16(14):6867. doi: 10.1016/j.bmc.2008.05.054. [DOI] [PubMed] [Google Scholar]
- 23.Ettrup A, Mikkelsen JD, Lehel S, et al. J Nucl Med. 2011;52(9):1449. doi: 10.2967/jnumed.111.088815. [DOI] [PubMed] [Google Scholar]
- 24.Rotering S, Scheunemann M, Fischer S, et al. Bioorg Med Chem. 2013;21(9):2635. doi: 10.1016/j.bmc.2013.02.018. [DOI] [PubMed] [Google Scholar]
- 25.Ahamed M, van Veghel D, Ullmer C, Van Laere K, Verbruggen A, Bormans GM. Front Neurosci-Switz. 2016;10 doi: 10.3389/fnins.2016.00431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan YZ, Xu K, Fang Y, Wang ZY. J Org Chem. 2011;76(16):6849. doi: 10.1021/jo2008934. [DOI] [PubMed] [Google Scholar]
- 27.Yue X, Bognar C, Zhang X, et al. Appl Radiat Isot. 2016;107:40. doi: 10.1016/j.apradiso.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yue X, Jin H, Liu H, et al. Org Biomol Chem. 2017;15(24):5197. doi: 10.1039/c7ob00854f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tu Z, Zhang X, Jin H, et al. Bioorg Med Chem. 2015;23(15):4699. doi: 10.1016/j.bmc.2015.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shah M, Seibyl J, Cartier A, Bhatt R, Catafau AM. J Nucl Med. 2014;55(9):1397. doi: 10.2967/jnumed.113.136515. [DOI] [PubMed] [Google Scholar]
- 31.Brust P, van den Hoff J, Steinbach J. Neurosci Bull. 2014;30(5):777–811. doi: 10.1007/s12264-014-1460-6. [DOI] [PMC free article] [PubMed] [Google Scholar]