

SUMMARY
Endoplasmic reticulum (ER) stress is transmitted to mitochondria and is associated with pathologic mitochondrial dysfunction in diverse diseases. The PERK arm of the unfolded protein response (UPR) protects mitochondria during ER stress through the transcriptional and translational remodeling of mitochondrial molecular quality control pathways. Here, we show that ER stress also induces dynamic remodeling of mitochondrial morphology by promoting protective stress-induced mitochondrial hyperfusion (SIMH). ER-stress-associated SIMH is regulated by the PERK arm of the UPR and activated by eIF2α phosphorylation-dependent translation attenuation. We show that PERK-regulated SIMH is a protective mechanism to prevent pathologic mitochondrial fragmentation and promote mitochondrial metabolism in response to ER stress. These results identify PERK-dependent SIMH as a protective stress-responsive mechanism that regulates mitochondrial morphology during ER stress. Furthermore, our results show that PERK integrates transcriptional and translational signaling to coordinate mitochondrial molecular and organellar quality control in response to pathologic ER insults.
Graphical abstract
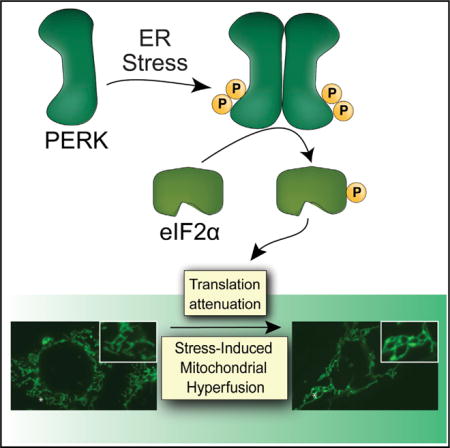
INTRODUCTION
Mitochondrial function is highly sensitive to diverse types of cellular insults, including those that induce the accumulation of misfolded proteins within the endoplasmic reticulum (ER) lumen (i.e., ER stress). ER stress influences many aspects of mitochondrial function and can promote mitochondrial-derived proapoptotic signaling through mechanisms involving mitochondrial depolarization and fragmentation (Malhotra and Kaufman, 2011; Rainbolt et al., 2014; Vannuvel et al., 2013). As such, ER stress and mitochondrial dysfunction are intricately associated in etiologically diverse diseases, including cardiovascular disorders and many neurodegenerative diseases (De Strooper and Scorrano, 2012; Doroudgar and Glembotski, 2013; Mercado et al., 2013; Prell et al., 2013; Verdejo et al., 2012). Considering the pathologic relationship between ER stress and mitochondria, a key question is, “What are the stress-responsive mechanisms that protect mitochondria during ER stress?”
One mechanism by which mitochondria are regulated during stress is through the adaptive remodeling of mitochondrial molecular quality control pathways involved in protein import, folding, and proteolysis (Baker et al., 2011; Quirós et al., 2015). ER stress induces remodeling of these mitochondrial quality control pathways through the PERK arm of the unfolded protein response (UPR) (Rainbolt et al., 2014). Upon activation, PERK selectively phosphorylates the α subunit of eukaryotic initiation factor 2 (eIF2α) (Walter and Ron, 2011). This leads to transient translational attenuation and the activation of stress-responsive transcription factors such as ATF4 (Wek and Cavener, 2007). PERK-dependent ATF4 activation induces expression of mitochondrial chaperones (e.g., the mitochondrial heat shock protein 70 [HSP70] HSPA9) and quality control proteases (e.g., the ATP-dependent AAA protease LON) (Han et al., 2013; Harding et al., 2003; Hori et al., 2002). In addition, PERK-regulated translational attenuation promotes the protective degradation of the TIM17A subunit of the TIM23 import complex to attenuate mitochondrial protein import in response to stress (Rainbolt et al., 2013). This remodeling of mitochondrial molecular quality control pathways afforded by PERK-dependent translational and transcriptional signaling functions to prevent the accumulation of damaged or non-native proteins that can disrupt mitochondrial function in response to ER insults (Rainbolt et al., 2014).
Apart from stress-responsive remodeling of molecular quality control pathways, mitochondria can also be regulated through alterations in mitochondrial morphology (i.e., organellar quality control) (Chan, 2012; Shutt and McBride, 2013; Wai and Langer, 2016). Cellular insults that severely disrupt mitochondrial function can increase mitochondrial fragmentation and subsequent targeting to mitophagy (Youle and Narendra, 2011). Through this mechanism, cells remove damaged mitochondria that can perturb global cellular function and decrease survival. In contrast, other types of stress, including starvation, UV irradiation, and ribosome inhibition, induce protective mitochondrial elongation through mechanisms such as stress-induced mitochondrial hyperfusion (SIMH) (Gomes et al., 2011; Sabouny et al., 2017; Tondera et al., 2009). SIMH is a pro-survival mechanism that suppresses pathologic mitochondrial fragmentation and promotes mitochondrial functions such as ATP production to protect cells in response to acute insults (Chan, 2012; Shutt and McBride, 2013; Wai and Langer, 2016). Consistent with this, genetic inhibition of SIMH increases mitochondrial fragmentation, promotes mitochondrial dysfunction, and/or increases sensitivity to apoptotic stimuli (Gomes et al., 2011; Tondera et al., 2009). However, ER-stress-dependent regulation of mitochondrial morphology is currently poorly defined.
Here, we demonstrate that acute ER stress induces stable remodeling of mitochondrial morphology through SIMH. We show that ER-stress-dependent SIMH is regulated by the PERK arm of the UPR and is promoted downstream of eIF2α phosphorylation-dependent translational attenuation. In addition, we demonstrate that pharmacologic or genetic inhibition of PERK-regulated SIMH is associated with increased mitochondrial fragmentation and impaired mitochondrial respiratory chain activity during ER stress. These results indicate that PERK-dependent SIMH is a protective mechanism to regulate mitochondrial morphology and promote mitochondrial metabolic activity in response to acute ER insults.
RESULTS
Acute ER Stress Promotes SIMH through an SLP2-Dependent Mechanism
We defined the impact of ER stress on mitochondrial morphology in wild-type mouse embryonic fibroblast (MEFWT) cells stably expressing a mitochondrial-targeted GFP (MEFmtGFP) following treatment with the ER stressor thapsigargin (Tg) for 0, 0.5, 6, 12, or 24 hr. We quantified mitochondrial morphology in blinded images by counting cells containing predominantly fragmented, tubular, or elongated mitochondria (see Figure S1A for examples of counting and Experimental Procedures for additional details on the quantification). Short 0.5-hr treatments with Tg increase the population of cells containing fragmented mitochondria (Figures 1A and 1B). This increase in fragmentation has previously been shown to reflect increased Ca2+ influx into mitochondria (Hom et al., 2007). Surprisingly, 6-hr treatments with Tg increase populations of cells containing elongated mitochondria. This increase in elongated mitochondria persisted through the 12-hr treatment, indicating that this elongation reflects a stable remodeling of mitochondrial morphology. However, prolonged 24-hr treatments with Tg induce a second round of fragmentation, which was previously associated with terminal apoptotic signaling (Hom et al., 2007). Mitochondrial elongation was also observed in MEFmtGFP cells treated for 6 hr with the alternative ER stressor tunicamycin (Tm; Figures 1C and S1B) and in Tg-treated HeLa cells transiently expressing a mitochondrial-targeted GFP (mtGFP; Figures S1C and S1D). These results indicate that acute ER stress induces dynamic remodeling of mitochondrial morphology through increased elongation.
Figure 1. ER Stress Induces Stable Remodeling of Mitochondrial Morphology through SLP2-Dependent SIMH.
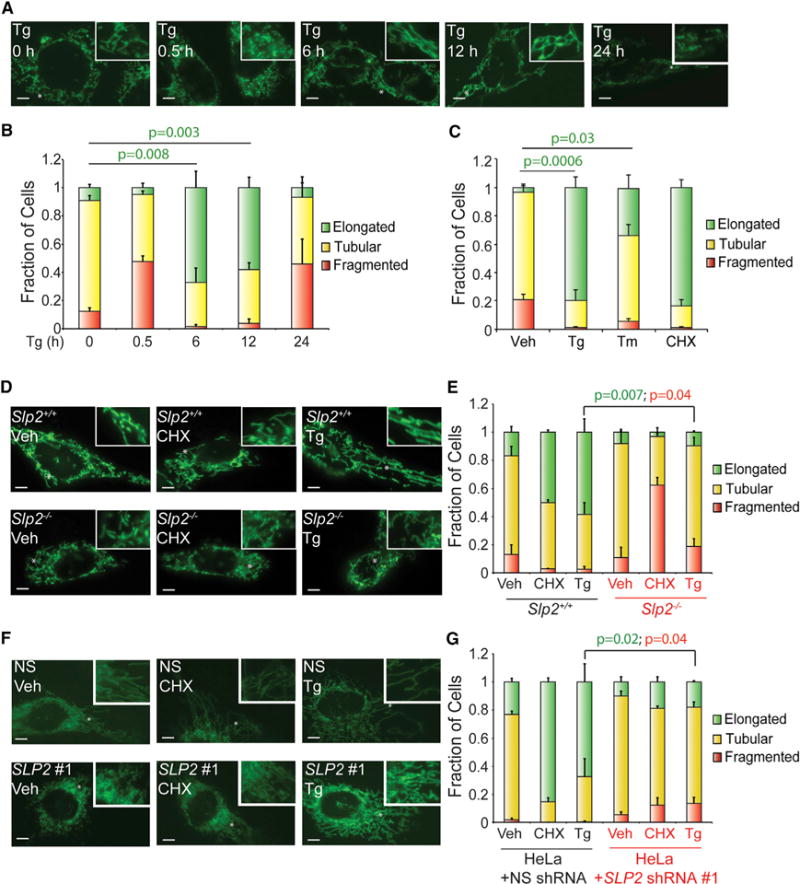
(A) Representative images of MEFmtGFP cells treated for 0, 0.5, 6, 12, or 24 hr with thapsigargin (Tg; 500 nM). The inset shows a 2-fold magnification of the image centered at the asterisk. Scale bars, 5 μm.
(B) Quantification of fragmented (red), tubular (yellow), or elongated (green) mitochondria in MEFmtGFP cells treated for 0, 0.5, 6, 12, or 24 hr with thapsigargin (Tg; 500 nM). Error bars indicate SEM from three independent experiments. p values for elongated (green text) mitochondria from a two-tailed unpaired t test are shown.
(C) Quantification of fragmented (red), tubular (yellow), or elongated (green) mitochondria in MEFmtGFP cells treated for 6 hr with thapsigargin (Tg; 500 nM), tunicamycin (Tm; 1 μM), or cycloheximide (CHX; 50 μg/mL). Error bars indicate SEM from three independent experiments. p values for elongated (green text) mitochondria from a two-tailed unpaired t test are shown.
(D) Representative images of Slp2+/+ and Slp2−/− MEF cells transiently transfected with mitochondrial-targeted GFP (mtGFP) treated for 6 hr with cycloheximide (CHX; 50 μg/mL) or Tg (500 nM). The inset shows a 2-fold magnification of the image centered at the asterisk. Scale bars, 5 μm.
(E) Graph showing the fraction of Slp2+/+ and Slp2−/− MEF cells transiently transfected with mtGFP containing fragmented (red), tubular (yellow), or elongated (green) mitochondria in cells treated for 6 hr with CHX (50 μg/mL) or Tg (500 nM). Error bars indicate SEM for three independent experiments. p values for elongated (green text) or fragmented (red text) mitochondria from a two-tailed unpaired t test are shown.
(F) Representative images of HeLa cells stably expressing non-silencing (NS) shRNA or SLP2 shRNA #1 and transfected with mtGFP. Cells were treated for 6 hr with thapsigargin Tg (500 nM) or CHX (50 μg/mL), as indicated. The inset shows a 2-fold magnification of the image centered at the asterisk. Scale bars, 5 μm.
(G) Graph showing the fraction of HeLa cells stably expressing scrambled shRNA or SLP2 shRNA #1 and transfected with mtGFP containing fragmented (red), tubular (yellow), or elongated (green) mitochondria following treatment for 6 hr with Tg (500 nM) or CHX (50 μg/mL), as indicated. Error bars indicate SEM for three independent experiments. p values for elongated (green text) or fragmented (red text) mitochondria from a two-tailed unpaired t test are shown.
The ribosomal inhibitor cycloheximide (CHX), a compound that potently activates SIMH by promoting mitochondrial fusion (Tondera et al., 2009), increases the population of MEFmtGFP cells containing elongated mitochondria to the same extent observed in Tg-treated cells (Figures 1C and S1B). CHX-dependent SIMH requires the mitochondrial scaffolding protein SLP2 (Tondera et al., 2009). If ER stress induces SIMH through a similar mechanism, we predicted that Tg-induced mitochondrial elongation would also be dependent on SLP2. We transfected Slp2−/− MEFs (Mitsopoulos et al., 2015) with mtGFP and measured mitochondrial morphology following treatment with CHX or Tg. As expected, CHX-dependent SIMH is inhibited in Slp2−/− MEFs (Figures 1D and 1E). Tg-dependent mitochondrial elongation is also inhibited in Slp2−/− MEFs. Similar results were observed in HeLa cells expressing two different SLP2 short hairpin RNAs (shRNAs) (Figures 1F and 1G; Figures S1C–S1E). These results show that ER stress promotes SIMH through a SLP2-dependent mechanism analogous to that observed for CHX.
ER-Stress-Dependent SIMH Is Induced Downstream of PERK-Regulated Translation Attenuation
The predominant stress-responsive signaling pathway activated by ER stress is the UPR (Walter and Ron, 2011). We used a pharmacologic approach to define the contributions of the IRE1, ATF6, and PERK UPR signaling pathways on ER-stress-dependent SIMH. Pharmacologic inhibition of the IRE1 or ATF6 UPR signaling arms using 4μ8c (Cross et al., 2012) or Ceapin-7 (Gallagher et al., 2016; Gallagher and Walter, 2016), respectively, did not prevent Tg-induced SIMH in MEFmtGFP cells (Figure S2A). However, GSK2656157 or ISRIB, two compounds that inhibit the PERK arm of the UPR through distinct mechanisms (Figure 2A) (Axten et al., 2013; Sidrauski et al., 2013), prevented Tg-induced SIMH in both MEFmtGFP cells (Figures 2B–2E) and HeLa cells expressing mtGFP (Figure S2B). We confirmed that these compounds inhibit PERK-regulated transcriptional and translational signaling by qPCR and immunoblotting (Figures S2C–S2F). ISRIB similarly inhibits Tm-induced mitochondrial elongation in MEFmtGFP cells (Figures 2D and 2E). However, CHX-dependent mitochondrial elongation is not affected by co-treatment with ISRIB (Figures 2D and 2E). Thus, pharmacologic inhibition of PERK-dependent eIF2α phosphorylation using two mechanistically distinct compounds selectively disrupts ER-stress-dependent SIMH.
Figure 2. Pharmacologic Inhibition of the PERK UPR Signaling Arm Prevents ER-Stress-Induced SIMH.
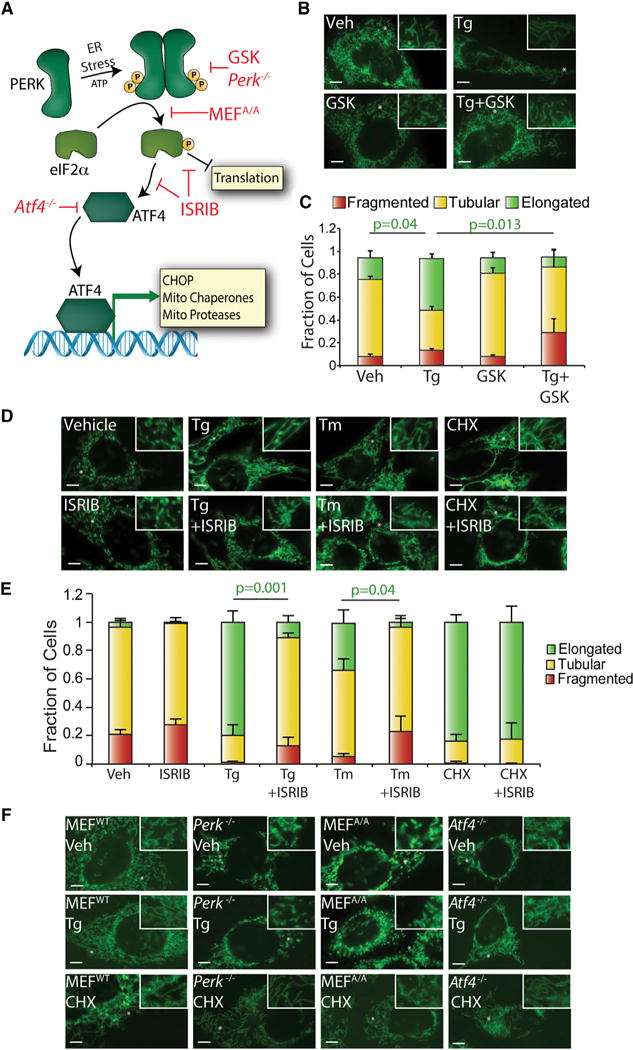
(A) Illustration showing the transcriptional and translational signaling pathways activated by PERK-regulated eIF2α phosphorylation. Red indicates how pharmacologic and genetic disruption of PERK signaling influences PERK-regulated transcriptional and/or translational signaling.
(B) Representative images of MEFmtGFP cells treated for 6 hr with thapsigargin (Tg; 500 nM) and/or the PERK inhibitor GSK2656157 (GSK; 10 μM). The inset shows a 2-fold magnification of the image centered at the asterisk. Scale bars, 5 μm.
(C) Quantification of fragmented (red), tubular (yellow), or elongated (green) mitochondria in MEFmtGFP cells treated for 6 hr with Tg (500 nM) and/or GSK2656157 (GSK; 10 μM). Error bars indicate SEM from three independent experiments. p values for elongated (green text) mitochondria from a two-tailed unpaired t test are shown.
(D) Representative images of MEFmtGFP cells treated for 6 hr with Tg (500 nM), tunicamycin (Tm; 1 μM), cycloheximide (CHX; 50 μg/mL), and/or ISRIB (200 nM), as indicated. The inset shows a 2-fold magnification of the image centered at the asterisk. Scale bars, 5 μm.
(E) Quantification of fragmented (red), tubular (yellow) or elongated (green) mitochondria in MEFmtGFP cells treated for 6 hr with Tg (500 nM), Tm (1 μM), CHX (50 μg/mL), and/or ISRIB (200 nM). Error bars indicate SEM from three independent experiments. p values for elongated (green text) mitochondria from a two-tailed unpaired t test are shown.
(F) Representative images of MEFWT, Perk−/−, MEFA/A, and Atf4−/− cells transiently transfected with mitochondrial-targeted GFP and treated for 6 hr with Tg (500 nM) or CHX (50 μg/mL), as indicated. Scale bars, 5 μm.
We further defined the involvement of PERK-dependent eIF2α phosphorylation in ER-stress-induced mitochondrial elongation using Perk−/− MEFs and knockin MEFs expressing the non-phosphorylatable S51A eIF2α mutant (MEFA/A) (Harding et al., 2000; Scheuner et al., 2001). Transcriptional and translational signaling induced by PERK activation is inhibited in these cells (Figure 2A). Consistent with this, both the Tg-dependent induction of Lon, Hspa9, and Chop and TIM17A degradation are inhibited in Perk−/− and MEFA/A cells (Figures S2G–S2I) (Rainbolt et al., 2013). ER-stress-induced SIMH is also inhibited in Perk−/− and MEFA/A cells, further demonstrating that ER-stress-dependent SIMH requires PERK-regulated eIF2α phosphorylation (Figure 2F).
Next, we determined the importance of PERK-regulated transcriptional signaling for ER-stress-dependent SIMH using Atf4−/− MEFs deficient in the primary upstream PERK-regulated transcription factor ATF4 (Harding et al., 2003). These cells show deficiencies in the transcriptional induction of PERK-regulated genes but not in translation attenuation induced by eIF2α phosphorylation (Figure 2A). As such, Tg-dependent induction of Lon, Hspa9, and Chop is inhibited in Atf4−/− cells (Figure S2J), while TIM17A degradation is not affected (Figure S2K). Interestingly, Tg-induced mitochondrial elongation is not inhibited in Atf4−/− MEFs (Figure 2F). This indicates that PERK-regulated SIMH does not require ATF4 transcriptional activity.
The aforementioned results show that ER stress induces SIMH through a mechanism dependent on PERK-dependent eIF2α phosphorylation but not ATF4. This suggests that PERK promotes SIMH downstream of eIF2α phosphorylation-dependent translation attenuation. Consistent with this, inhibiting translation in Perk−/− and MEFA/A cells using CHX increases mitochondrial elongation in both genotypes (Figure 2F). This indicates that ER-stress-dependent SIMH is regulated by the PERK signaling arm of the UPR and activated by eIF2α phosphorylation-dependent translation attenuation.
Yme1l Depletion Attenuates ER-Stress-Dependent Increases in Elongated Mitochondria
A key question emerging from the aforementioned results is, “How does PERK-dependent translational attenuation regulate SIMH?” Mitochondrial hyperfusion can be induced by diverse mechanisms, including protein kinase A (PKA)-dependent phosphorylation of the pro-fission GTPase DRP1 at position S637 (Gomes et al., 2011), reductions in total DRP1 protein levels (Sabouny et al., 2017), or HDAC6-dependent deacetylation of the outer membrane pro-fusion GTPase MFN1 (Lee et al., 2014). However, Tg treatment does not increase S637 phosphorylation or decrease total DRP1 levels in Slp2+/+ or Slp2−/− MEFs (Figure S3A). Furthermore, co-addition of the PKA inhibitor KT5720 does not inhibit Tg-induced mitochondrial elongation in MEFmtGFP cells (Figure S3B). Similarly, the addition of tubacin—an HDAC6 inhibitor (Haggarty et al., 2003)—does not reduce Tg-dependent mitochondrial elongation (Figure S3C). We also do not observe increased stability of the MFN1 or MFN2 outer membrane GTPases or processing of the inner membrane GTPase OPA1 in Tg-treated cells (Figure S3A)—two stress-responsive posttranslational modifications that can influence mitochondrial morphology (Chan, 2012; Shutt and McBride, 2013; Wai and Langer, 2016). These results suggest that PERK-regulated SIMH is induced through a mechanism different from those discussed above.
Apart from promoting SIMH, PERK-regulated translational attenuation reduces mitochondrial protein import through the degradation of the TIM23 subunit TIM17A (Rainbolt et al., 2013). We defined the relationship between SIMH and TIM17A degradation in MEFmtGFP cells that were shRNA-depleted of Tim17a (Figures 3A and S3D). Depletion of Tim17a did not influence basal mitochondrial morphology or Tg-induced elongation in these cells, demonstrating that TIM17A degradation and SIMH are two distinct events induced by PERK-regulated translational attenuation.
Figure 3. Yme1l Depletion Decreases Elongated Mitochondria during ER Stress.
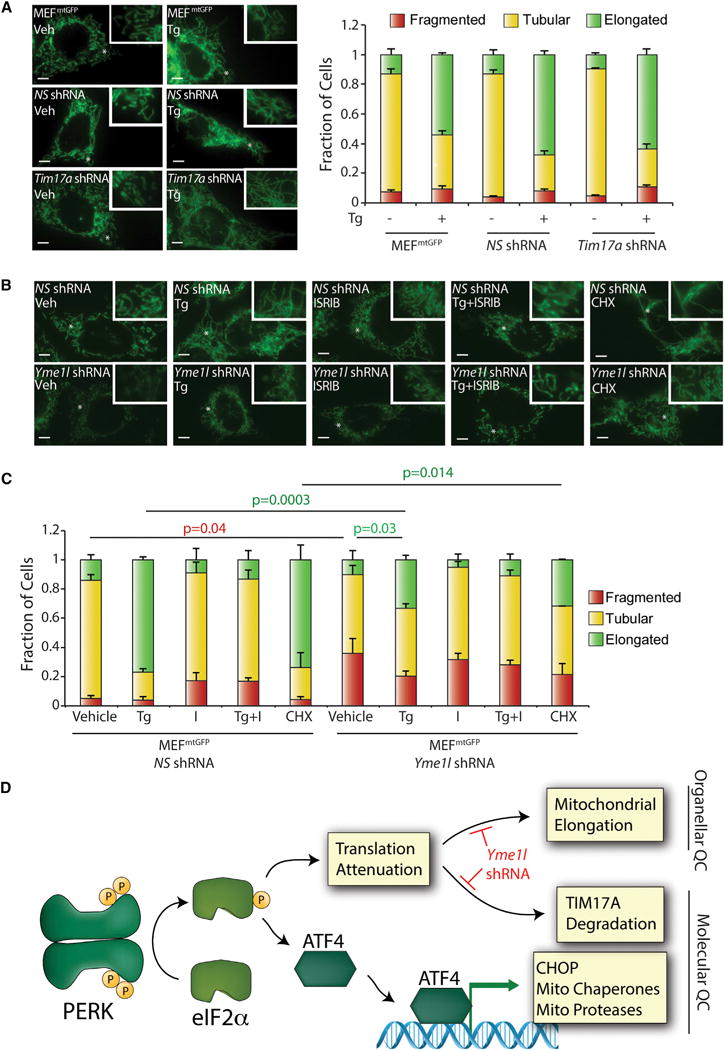
(A) Representative images and quantification of fragmented (red), tubular (yellow), or elongated (green) mitochondria in stable pools of MEFmtGFP cells expressing non-silencing (NS) or Tim17a shRNA treated for 6 hr with thapsigargin (Tg; 500 nM). Uninfected MEFmtGFP cells are also shown as a control. The inset shows a 2-fold magnification of the image centered at the asterisk. Scale bars, 5 μm. Error bars indicate SEM from three independent experiments.
(B) Representative images of stable pools of MEFmtGFP cells expressing non-silencing or Yme1l shRNA treated for 6 hr with Tg. The inset shows a 2-fold magnification of the image centered at the asterisk. Scale bars, 5 μm.
(C) Quantification of fragmented (red), tubular (yellow), or elongated (green) mitochondria in stable pools of MEFmtGFP cells expressing non-silencing or Yme1l shRNA treated for 6 hr with Tg (500 nM). Error bars indicate SEM from three independent experiments. p values for elongated (green text) or fragmented (red text) mitochondria from a two-tailed unpaired t test are shown.
(D) Illustration showing how PERK integrates transcriptional and translational signaling to coordinate mitochondria molecular and organellar quality control during ER stress. Genetic perturbations that disrupt these pathways are indicated in red.
TIM17A is degraded by the ATP-dependent inner membrane protease YME1L downstream of PERK-regulated translation attenuation (Rainbolt et al., 2013). YME1L also regulates inner membrane proteostasis and function through its assembly into a complex with the inner membrane protease PARL and SLP2 (Wai et al., 2016), the latter being a protein whose depletion impairs ER-stress-dependent SIMH (Figures 1D–1G). Thus, we tested whether shRNA depletion of Yme1l impairs mitochondrial elongation in response to ER stress. The depletion of Yme1l in MEFmtGFP inhibited Tg-dependent TIM17A degradation but did not influence PERK-dependent induction of LON, confirming efficient reduction in protease activity (Figures S3E and S3F). In the absence of stress, Yme1l-depleted cells show increased mitochondrial fragmentation (Figures 3B and 3C), consistent with previous results (Anand et al., 2014; Mishra et al., 2014). Interestingly, Yme1l depletion also reduced elongated mitochondria in Tg-treated cells, suggesting that YME1L is involved in ER-stress-dependent SIMH. In contrast, depletion of Parl does not influence Tg-dependent SIMH (Figures S3G and S3H).
Collectively, our results indicate that PERK regulates SIMH downstream of eIF2α phosphorylation-dependent translational attenuation (Figure 3D). This PERK-regulated increase in mitochondrial elongation is independent of PERK-regulated TIM17A degradation but appears to involve the inner membrane protease YME1L, potentially reflecting the importance of degrading a short-lived mitochondrial protein in this process. We are currently focused on further defining the involvement of YME1L and, potentially, other proteases in regulating SIMH during ER stress.
Disruption of PERK-Regulated SIMH Increases Mitochondrial Fragmentation during ER Stress
SIMH suppresses premature fragmentation in response to diverse cellular insults (Chan, 2012; Shutt and McBride, 2013; Wai and Langer, 2016). Consistent with this, ISRIB-dependent inhibition of SIMH in MEFmtGFP cells treated with Tg for 12 or 24 hr increased populations of fragmented mitochondria (Figures 4A and 4B). However, ISRIB inhibits both transcriptional and translational signaling induced by PERK (Figure 2A), preventing us from defining the specific contributions of PERK-regulated SIMH in protecting mitochondria from fragmentation. Therefore, we chose to further define the importance of SIMH in preventing premature ER-stress-induced fragmentation in Slp2−/− MEFs, where PERK-dependent SIMH is inhibited (Figures 1D and 1E). Importantly, PERK-dependent transcriptional induction of Lon, Hspa9, and Chop (Figures S4A and S4B) and TIM17A degradation (induced by PERK-dependent translational attenuation; Figure S4C) are not affected in Slp2−/− MEFs, demonstrating that PERK-dependent SIMH can be selectively inhibited in these cells. Interestingly, Tg treatment for 12 hr induces mitochondrial fragmentation in Slp2−/− MEFs to extents similar to that observed in cells co-treated with Tg and ISRIB (Figures 4C and 4D). This indicates that PERK-regulated SIMH protects mitochondrial morphology and prevents premature fragmentation in response to ER stress.
Figure 4. Pharmacologic or Genetic Inhibition of PERK-Regulated SIMH Disrupts ER-Stress-Dependent Regulation of Mitochondrial Morphology and Metabolism.
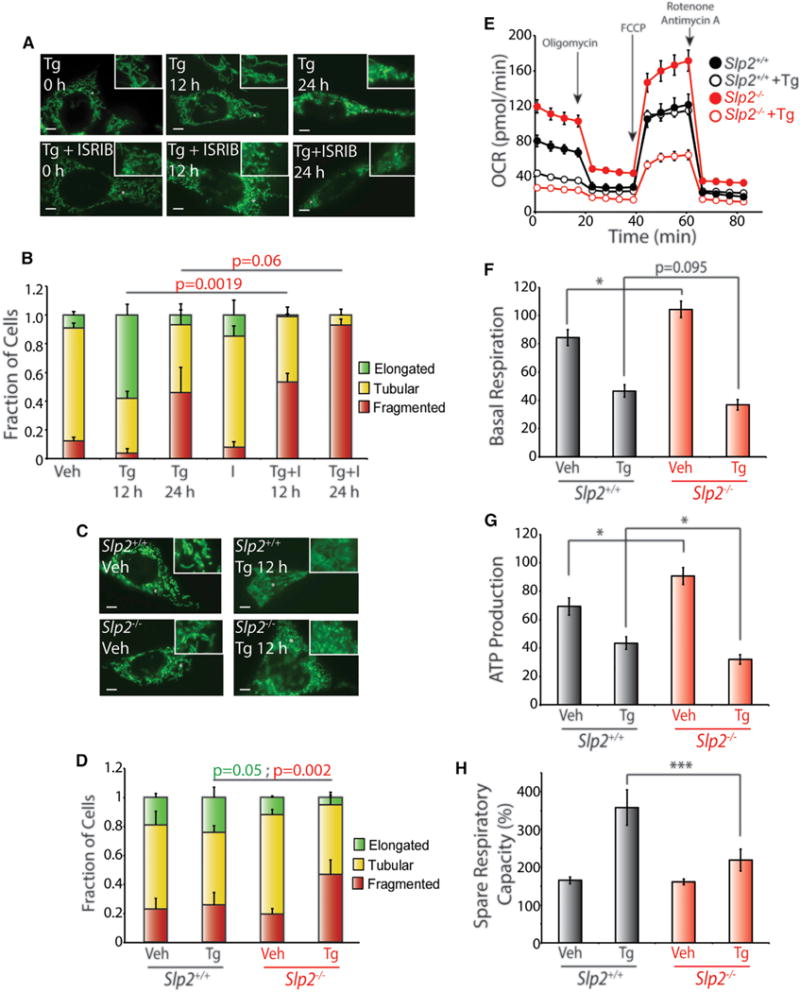
(A) Representative images of MEFmtGFP cells treated for 12 or 24 hr with thapsigargin (Tg; 500 nM) and/or ISRIB (200 nM), as indicated. The inset shows a 2-fold magnification of the image centered at the asterisk. Scale bars, 5 μm.
(B) Quantification of fragmented (red), tubular (yellow), or elongated (green) mitochondria in MEFmtGFP cells treated for 12 or 24 hr with Tg (500 nM) and/or ISRIB (200 nM). Error bars indicate SEM from three independent experiments. p values for fragmented mitochondria (red text) are shown for a one-tailed paired t test.
(C) Representative images of Slp2+/+ and Slp2−/− MEF cells transiently transfected with mitochondrial-targeted GFP treated for 12 hr with Tg (500 nM). The inset shows a 2-fold magnification of the image centered at the asterisk. Scale bars, 5 μm.
(D) Graph showing the fraction of Slp2+/+ and Slp2−/− MEF cells transiently transfected with mtGFP containing fragmented (red), tubular (yellow), or elongated (green) mitochondria in cells treated for 12 hr with Tg (500 nM). Error bars indicate SEM for three independent experiments. p values for fragmented mitochondria (red text) are shown for a two-tailed paired t test.
(E) Representative plot of oxygen consumption rates (OCRs) for Slp2+/+ and Slp2−/− MEFs pre-treated with Tg (500 nM) for 6 hr prior to analysis by Seahorse.
(F) Graph showing basal respiration for Slp2+/+ and Slp2−/− MEFs treated with Tg (500 nM) for 6 hr before OCR measurements. Error bars indicate SEM for n = 30, collected across three independent experiments. *p < 0.05 for a two-tailed unpaired t test.
(G) Graph showing ATP production of Slp2+/+ and Slp2−/− MEFs treated with Tg (500 nM) for 6 hr before OCR measurements. Error bars indicate SEM for n = 30, collected across three independent experiments. *p < 0.05 for a two-tailed unpaired t test.
(H) Graph showing spare respiratory capacity of Slp2+/+ and Slp2−/− MEFs treated with Tg (500 nM) for 6 hr before OCR measurements. Error bars indicate SEM for n = 30, collected across three independent experiments. ***p < 0.005 for a two-tailed unpaired t test.
The increased fragmentation observed in cells deficient in PERK-regulated SIMH could reflect increased sensitivity to ER insults. Consistent with this, pretreatment with Tg and ISRIB for 12 hr reduced the clonal expansion of MEFmtGFP cells, as compared with cells pre-treated with Tg alone (Figure S4D). This is consistent with previous results and highlights that PERK signaling is important for dictating ER stress resistance (Sidrauski et al., 2013). However, we did not observe differences in the clonal expansion of Slp2+/+ and Slp2−/− MEFs in this assay (Figure S4E). This likely reflects the importance of SLP2 in regulating apoptotic signaling through PARL-dependent processing of the pro-apoptotic Smac/Diablo (Saita et al., 2017). Consistent with this, Slp2−/− MEFs show lower levels of active caspase 3 following Tg treatment (Figure S4F). Regardless, our results show that PERK-dependent SIMH protects mitochondria from premature fragmentation and could contribute to decrease cellular sensitivity to ER stress.
Slp2 Deficiency Decreases Mitochondrial Respiratory Chain Capacity during ER Stress
SIMH has been shown to promote mitochondrial metabolic activity in response to other types of stress (Tondera et al., 2009). We used the Seahorse extracellular flux assay to define how SLP2-dependent SIMH impacts mitochondria metabolic activity in response to ER stress. The serum-free conditions required for this experiment induce a mild increase in eIF2α phosphorylation (Figure S4G), which is consistent with previous results showing that reduced serum promotes eIF2α phosphorylation through a mechanism involving the alternative eIF2α kinase GCN2 (Taniuchi et al., 2016). While this mild eIF2α phosphorylation complicates the use of pharmacologic agents to block PERK signaling (e.g., ISRIB), we were able to monitor how ER stress influenced mitochondrial respiration in Slp2−/− MEFs, where PERK-regulated SIMH is selectively inhibited. In the absence of stress, Slp2−/− MEFs showed modest increases in basal respiration and ATP-linked respiration, as compared to Slp2+/+ controls (Figures 4E–4G). Pretreatment with Tg for 6 hr induced a significant reduction in these aspects of mitochondrial respiration in both Slp2+/+ and Slp2−/− cells. However, this reduction was greater in Slp2−/− MEFs, indicating that Tg differentially impacted mitochondrial metabolic activity in these cells. Similar results were observed upon Tm treatment (Figures S4H–S4J). Interestingly, the differential ER-stress-dependent decrease in basal and ATP-linked respiration in Slp2−/− MEFs corresponded with lower levels of spare respiratory capacity in these cells, as compared to Slp2+/+ controls (Figure 4H; Figure S4K). These results demonstrate that ER stress differentially impacts mitochondrial metabolic activity in Slp2−/− cells deficient in ER-stress-induced SIMH and is consistent with previous results showing that SLP2-dependent SIMH promotes mitochondria metabolic activity (Tondera et al., 2009).
DISCUSSION
Here, we show that cells regulate mitochondrial morphology in response to acute ER stress by promoting protective SIMH through a mechanism involving translational attenuation induced by PERK-dependent eIF2α phosphorylation (Figure 3D). PERK integrates transcriptional and translational signaling to remodel quality control pathways involved in protein import, folding, and proteolysis. Our results expand the scope of PERK’s role in adapting mitochondria during ER stress by demonstrating that PERK signaling coordinates the regulation of both mitochondrial molecular quality control and morphology (i.e., organellar quality control). This coordination provides a unique platform to sensitively regulate mitochondria in response to diverse types of ER insults. PERK-dependent remodeling of molecular quality control pathways increases mitochondrial proteostasis capacity to promote proteome integrity and prevent the potentially pathologic accumulation of misfolded or damaged proteins within mitochondria during ER stress (Rainbolt et al., 2014). Our results indicate that PERK-dependent SIMH complements this regulation of mitochondrial molecular quality control by preventing premature mitochondrial fragmentation and promoting mitochondrial metabolic activity. By protecting these aspects of mitochondrial biology, PERK-regulated SIMH increases cellular energetic capacity to facilitate recovery following acute ER insults. Thus, the coordination of mitochondrial molecular and organellar quality control afforded by PERK activity demonstrates that this UPR signaling pathway is a key determinant in regulating mitochondrial function in response to ER stress.
The importance of PERK for regulating both molecular and organellar quality control indicates that alterations in PERK signaling could significantly influence mitochondrial function in disease states. Consistent with this, altered PERK activity is implicated in diverse diseases associated with mitochondrial dysfunction, including cardiovascular disorders, neurodegenerative diseases, and mitochondriopathies such as Leber’s hereditary optic neuropathy (LHON) (Brooks et al., 2014; Cortopassi et al., 2006; Freeman and Mallucci, 2016; Hetz and Mollereau, 2014; Liu and Dudley, 2015; Silva et al., 2009). Mutations in PERK implicated in Wolcott-Rallison syndrome are also suggested to induce pathologic mitochondrial dysfunction (Collardeau-Frachon et al., 2015; Engelmann et al., 2008; Søvik et al., 2008). Aging-dependent alterations in PERK activity (Brown and Naidoo, 2012; Taylor, 2016) could also decrease cellular capacity to regulate mitochondria, potentially contributing to the pathologic mitochondrial dysfunction associated with aging-related diseases. Thus, our identification of PERK as a key stress-responsive signaling pathway involved in the coordinated regulation of mitochondria during ER stress provides a foundation to define the importance of altered PERK signaling in mitochondrial disease pathogenesis. In addition, our results indicate that therapeutic targeting of PERK signaling could provide a unique opportunity to ameliorate pathologic defects in mitochondria associated with diverse diseases.
EXPERIMENTAL PROCEDURES
Cell Culture and Plasmids
MEFmtGFP (a kind gift from Peter Schultz, The Scripps Research Institute; TSRI) (Wang et al., 2012) and HeLa cells (purchased from the ATCC) were cultured in DMEM (Corning-Cellgro) supplemented with 10% fetal bovine serum (FBS; Omega Scientific), 2 mM L-glutamine (GIBCO), 100 U/mL−1 penicillin, and 100 μg/mL−1 streptomycin (GIBCO). MEFWT and the Slp2+/+ and Slp2−/− MEFs (a kind gift from Joaquin Madrenas, McGill University) (Mitsopoulos et al., 2015) were cultured as described earlier and supplemented with non-essential amino acids (GIBCO). Perk+/+, Perk−/−, MEFA/A, Atf4+/+, and Atf4−/− cells (kind gifts from Jonathan Lin) were cultured as described earlier and supplemented with non-essential amino acids and 55 μM 2-mercaptoethanol. Cells were maintained at 37° C and 5% CO2. MEF cells were transfected with MEF Avalanche Transfection Reagent (EZ Biosystems) according to the manufacturer’s protocol. Stable pools of cells expressing non-silencing or gene-specific knockdowns were generated through selection with puromycin (3 μg/mL−1). HeLa cells were transfected by the calcium phosphate co-precipitation method. SLP2, Tim17a, Yme1l, and Parl shRNA in pLKO.1 vector were obtained from Sigma. Stable pools of cells expressing non-silencing or gene-specific knockdowns were generated through selection with puromycin (3 μg/mL−1 for MEF and 1 μg/mL−1 for HeLa).
Fluorescence Microscopy and Mitochondria Scoring
Cells were seeded the day before on glass-bottom dishes coated with poly-D-lysine (Sigma) to reach a confluence of 50% the day of the experiment. Cells were treated in media with reagents for the indicated time and then washed 3 times with HBSS containing 10% FBS, 100 U/mL−1 penicillin, and 100 μg/mL−1 streptomycin and put in the same buffer for imaging. Images were then recorded with an Olympus IX71 microscope with 603 oil objective (Olympus), a Hamamatsu C8484 camera (Hamamatsu Photonics), and HCImage software (Hamamatsu Photonics). For quantification, we collected images for more than 30 cells per condition and then blinded these images. Three researchers then scored the cells for containing primarily fragmented, tubular, or elongated mitochondria (see Figure S1A for examples of scoring). The scores from the three researchers were then averaged and combined with other independent experiments in the data presented in the bar graphs shown throughout the paper.
Statistical Analysis
Data are presented as mean ± SEM and were analyzed by paired one- or two-tailed (as indicated) Student’s t test to determine significance. A one-tailed t test was used when a specific result could be predicted from published reports.
Additional materials and methods are described in the Supplemental Information.
Supplementary Material
Highlights.
Stress-induced mitochondrial hyperfusion (SIMH) is triggered by ER stress
ER-stress-induced SIMH is activated by PERK-dependent translation attenuation
PERK-regulated SIMH promotes electron transport chain activity during ER stress
PERK integrates transcriptional and translational signaling to protect mitochondria
Acknowledgments
We thank T. Kelly Rainbolt for intellectual contributions, experimental support, and critical reading of the manuscript. We thank Danling Wang and Peter Schultz (The Scripps Research Institute [TSRI]) for providing MEFmtGFP cells; Jonathan Lin (UCSD) for providing Perk+/+, Perk−/−, MEFWT, MEFA/A, Atf4+/+, and Atf4−/− MEFs; and Joaquin Madrenas (McGill University) for providing the Slp2+/+ and Slp2−/− MEFs. We thank Peter Walter (UCSF) for providing the ATF6 inhibitor Ceapin. This work was supported by the NIH (NS095892 to R.L.W.). J.L. was supported by a George E. Hewitt Foundation postdoctoral fellowship.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures and four figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.02.055.
AUTHOR CONTRIBUTIONS
J.L., J.M.S., V.W.R.M., A.M., and R.L.W. conceived, designed, performed, and interpreted the experiments. N.M. and M.C.A. performed experiments. R.L.W. supervised the project. All authors were involved in the drafting and revision of the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol. 2014;204:919–929. doi: 10.1083/jcb.201308006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axten JM, Romeril SP, Shu A, Ralph J, Medina JR, Feng Y, Li WH, Grant SW, Heerding DA, Minthorn E, et al. Discovery of GSK2656157: an optimized PERK inhibitor selected for preclinical development. ACS Med Chem Lett. 2013;4:964–968. doi: 10.1021/ml400228e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker MJ, Tatsuta T, Langer T. Quality control of mitochondrial proteostasis. Cold Spring Harb Perspect Biol. 2011;3:a007559. doi: 10.1101/cshperspect.a007559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks AC, Guo Y, Singh M, McCracken J, Xuan YT, Srivastava S, Bolli R, Bhatnagar A. Endoplasmic reticulum stress-dependent activation of ATF3 mediates the late phase of ischemic preconditioning. J Mol Cell Cardiol. 2014;76:138–147. doi: 10.1016/j.yjmcc.2014.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MK, Naidoo N. The endoplasmic reticulum stress response in aging and age-related diseases. Front Physiol. 2012;3:263. doi: 10.3389/fphys.2012.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–287. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- Collardeau-Frachon S, Vasiljevic A, Jouvet A, Bouvier R, Senée V, Nicolino M. Microscopic and ultrastructural features in Wolcott-Rallison syndrome, a permanent neonatal diabetes mellitus: about two autopsy cases. Pediatr Diabetes. 2015;16:510–520. doi: 10.1111/pedi.12201. [DOI] [PubMed] [Google Scholar]
- Cortopassi G, Danielson S, Alemi M, Zhan SS, Tong W, Carelli V, Martinuzzi A, Marzuki S, Majamaa K, Wong A. Mitochondrial disease activates transcripts of the unfolded protein response and cell cycle and inhibits vesicular secretion and oligodendrocyte-specific transcripts. Mitochondrion. 2006;6:161–175. doi: 10.1016/j.mito.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Cross BC, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM, Silverman RH, Neubert TA, Baxendale IR, Ron D, Harding HP. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci USA. 2012;109:E869–E878. doi: 10.1073/pnas.1115623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Scorrano L. Close encounter: mitochondria, endoplasmic reticulum and Alzheimer’s disease. EMBO J. 2012;31:4095–4097. doi: 10.1038/emboj.2012.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doroudgar S, Glembotski CC. New concepts of endoplasmic reticulum function in the heart: programmed to conserve. J Mol Cell Cardiol. 2013;55:85–91. doi: 10.1016/j.yjmcc.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelmann G, Meyburg J, Shahbek N, Al-Ali M, Hairetis MH, Baker AJ, Rodenburg RJ, Wenning D, Flechtenmacher C, Ellard S, et al. Recurrent acute liver failure and mitochondriopathy in a case of Wolcott-Rallison syndrome. J Inherit Metab Dis. 2008;31:540–546. doi: 10.1007/s10545-008-0867-0. [DOI] [PubMed] [Google Scholar]
- Freeman OJ, Mallucci GR. The UPR and synaptic dysfunction in neurodegeneration. Brain Res. 2016;1648(Pt B):530–537. doi: 10.1016/j.brainres.2016.03.029. [DOI] [PubMed] [Google Scholar]
- Gallagher CM, Walter P. Ceapins inhibit ATF6α signaling by selectively preventing transport of ATF6α to the Golgi apparatus during ER stress. eLife. 2016;5:e11880. doi: 10.7554/eLife.11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher CM, Garri C, Cain EL, Ang KK, Wilson CG, Chen S, Hearn BR, Jaishankar P, Aranda-Diaz A, Arkin MR, et al. Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6α branch. eLife. 2016;5:e11878. doi: 10.7554/eLife.11878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13:589–598. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci USA. 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15:481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci. 2014;15:233–249. doi: 10.1038/nrn3689. [DOI] [PubMed] [Google Scholar]
- Hom JR, Gewandter JS, Michael L, Sheu SS, Yoon Y. Thapsigargin induces biphasic fragmentation of mitochondria through calcium-mediated mitochondrial fission and apoptosis. J Cell Physiol. 2007;212:498–508. doi: 10.1002/jcp.21051. [DOI] [PubMed] [Google Scholar]
- Hori O, Ichinoda F, Tamatani T, Yamaguchi A, Sato N, Ozawa K, Kitao Y, Miyazaki M, Harding HP, Ron D, et al. Transmission of cell stress from endoplasmic reticulum to mitochondria: enhanced expression of Lon protease. J Cell Biol. 2002;157:1151–1160. doi: 10.1083/jcb.200108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Kapur M, Li M, Choi MC, Choi S, Kim HJ, Kim I, Lee E, Taylor JP, Yao TP. MFN1 deacetylation activates adaptive mitochondrial fusion and protects metabolically challenged mitochondria. J Cell Sci. 2014;127:4954–4963. doi: 10.1242/jcs.157321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Dudley SC., Jr Role for the unfolded protein response in heart disease and cardiac arrhythmias. Int J Mol Sci. 2015;17:52. doi: 10.3390/ijms17010052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra JD, Kaufman RJ. ER stress and its functional link to mitochondria: role in cell survival and death. Cold Spring Harb Perspect Biol. 2011;3:a004424. doi: 10.1101/cshperspect.a004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado G, Valdés P, Hetz C. An ERcentric view of Parkinson’s disease. Trends Mol Med. 2013;19:165–175. doi: 10.1016/j.molmed.2012.12.005. [DOI] [PubMed] [Google Scholar]
- Mishra P, Carelli V, Manfredi G, Chan DC. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014;19:630–641. doi: 10.1016/j.cmet.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsopoulos P, Chang YH, Wai T, König T, Dunn SD, Langer T, Madrenas J. Stomatin-like protein 2 is required for in vivo mitochondrial respiratory chain supercomplex formation and optimal cell function. Mol Cell Biol. 2015;35:1838–1847. doi: 10.1128/MCB.00047-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prell T, Lautenschläger J, Grosskreutz J. Calcium-dependent protein folding in amyotrophic lateral sclerosis. Cell Calcium. 2013;54:132–143. doi: 10.1016/j.ceca.2013.05.007. [DOI] [PubMed] [Google Scholar]
- Quirós PM, Langer T, López-Otín C. New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol. 2015;16:345–359. doi: 10.1038/nrm3984. [DOI] [PubMed] [Google Scholar]
- Rainbolt TK, Atanassova N, Genereux JC, Wiseman RL. Stress-regulated translational attenuation adapts mitochondrial protein import through Tim17A degradation. Cell Metab. 2013;18:908–919. doi: 10.1016/j.cmet.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainbolt TK, Saunders JM, Wiseman RL. Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends Endocrinol Metab. 2014;25:528–537. doi: 10.1016/j.tem.2014.06.007. [DOI] [PubMed] [Google Scholar]
- Sabouny R, Fraunberger E, Geoffrion M, Ng AC, Baird SD, Screaton RA, Milne R, McBride HM, Shutt TE. The Keap1-Nrf2 stress response pathway promotes mitochondrial hyperfusion through degradation of the mitochondrial fission protein Drp1. Antioxid Redox Signal. 2017;27:1447–1459. doi: 10.1089/ars.2016.6855. [DOI] [PubMed] [Google Scholar]
- Saita S, Nolte H, Fiedler KU, Kashkar H, Venne AS, Zahedi RP, Krüger M, Langer T. PARL mediates Smac proteolytic maturation in mitochondria to promote apoptosis. Nat Cell Biol. 2017;19:318–328. doi: 10.1038/ncb3488. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- Shutt TE, McBride HM. Staying cool in difficult times: mitochondrial dynamics, quality control and the stress response. Biochim Biophys Acta. 2013;1833:417–424. doi: 10.1016/j.bbamcr.2012.05.024. [DOI] [PubMed] [Google Scholar]
- Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, Gamache K, Gallagher CM, Ang KK, Wilson C, et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. eLife. 2013;2:e00498. doi: 10.7554/eLife.00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JM, Wong A, Carelli V, Cortopassi GA. Inhibition of mitochondrial function induces an integrated stress response in oligodendroglia. Neurobiol Dis. 2009;34:357–365. doi: 10.1016/j.nbd.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Søvik O, Njølstad PR, Jellum E, Molven A. Wolcott-Rallison syndrome with 3-hydroxydicarboxylic aciduria and lethal outcome. J Inherit Metab Dis. 2008;31(Suppl 2):S293–S297. doi: 10.1007/s10545-008-0866-1. [DOI] [PubMed] [Google Scholar]
- Taniuchi S, Miyake M, Tsugawa K, Oyadomari M, Oyadomari S. Integrated stress response of vertebrates is regulated by four eIF2α kinases. Sci Rep. 2016;6:32886. doi: 10.1038/srep32886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RC. Aging and the UPR(ER) Brain Res. 2016;1648(Pt B):588–593. doi: 10.1016/j.brainres.2016.04.017. [DOI] [PubMed] [Google Scholar]
- Tondera D, Grandemange S, Jourdain A, Karbowski M, Mattenberger Y, Herzig S, Da Cruz S, Clerc P, Raschke I, Merkwirth C, et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009;28:1589–1600. doi: 10.1038/emboj.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannuvel K, Renard P, Raes M, Arnould T. Functional and morphological impact of ER stress on mitochondria. J Cell Physiol. 2013;228:1802–1818. doi: 10.1002/jcp.24360. [DOI] [PubMed] [Google Scholar]
- Verdejo HE, del Campo A, Troncoso R, Gutierrez T, Toro B, Quiroga C, Pedrozo Z, Munoz JP, Garcia L, Castro PF, Lavandero S. Mitochondria, myocardial remodeling, and cardiovascular disease. Curr Hypertens Rep. 2012;14:532–539. doi: 10.1007/s11906-012-0305-4. [DOI] [PubMed] [Google Scholar]
- Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. 2016;27:105–117. doi: 10.1016/j.tem.2015.12.001. [DOI] [PubMed] [Google Scholar]
- Wai T, Saita S, Nolte H, Müller S, König T, Richter-Dennerlein R, Sprenger HG, Madrenas J, Mühlmeister M, Brandt U, et al. The membrane scaffold SLP2 anchors a proteolytic hub in mitochondria containing PARL and the i-AAA protease YME1L. EMBO Rep. 2016;17:1844–1856. doi: 10.15252/embr.201642698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang D, Wang J, Bonamy GM, Meeusen S, Brusch RG, Turk C, Yang P, Schultz PG. A small molecule promotes mitochondrial fusion in mammalian cells. Angew Chem Int Ed Engl. 2012;51:9302–9305. doi: 10.1002/anie.201204589. [DOI] [PubMed] [Google Scholar]
- Wek RC, Cavener DR. Translational control and the unfolded protein response. Antioxid Redox Signal. 2007;9:2357–2371. doi: 10.1089/ars.2007.1764. [DOI] [PubMed] [Google Scholar]
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.