

ABSTRACT
In comparison to sporadic Creutzfeldt–Jakob disease (sCJD) with MM1-type and MM2- cortical (MM2C)-type, genetic CJD with a prion protein gene V180I mutation (V180I gCJD) is clinically characterized by onset at an older age, slower progress, and the absence of visual disturbances or cerebellar symptoms. In terms of pathological characteristics, gliosis and neuronal loss are generally milder in degree, and characteristic spongiform change can be observed at both the early and advanced stages. However, little is known on the progress of spongiform change over time or its mechanisms. In this study, to elucidate the pathological course of V180I gCJD, statistical analysis of the size and dispersion of the major diameters of vacuoles in six V180I gCJD cases was performed, with five MM1-type sCJD and MM2C-type sCJD cases as controls. As a result, V180I gCJD showed no significant difference in vacuolar diameter regardless of disease duration. In addition, the dispersion of the major diameters of vacuoles in V180I gCJD was larger than that in the MM1-type, which was smaller than that in the MM2C-type. We speculated that the absence of difference in the size of the vacuoles regardless of disease duration suggests that tissue rarefaction does not result from the expansion of vacuole size and increase in number of vacuoles in V180Ig CJD. These features were considered to be significant pathological findings of V180I gCJD.
KEYWORDS: Creutzfeldt–Jakob disease, genetic Creutzfeldt–Jakob disease, V180I mutation, neuropathology, spongiform change, prion protein
Introduction
Genetic Creutzfeldt–Jakob disease (gCJD) with a prion protein (PrP) gene V180I mutation (V180I gCJD) is the most common type of genetic CJD in Japan [1]. Although there have been several past reports on the pathological findings observed in V180I gCJD, much of its pathological progression remains unknown.
V180I gCJD is distinguished by the following clinical characteristics in comparison to sporadic CJD (sCJD): the age of onset is older (mean age: 72.8 years, range: 58–81 years), the progress is slower, higher cortical dysfunctions such as aphasias are observed, and visual disturbances or cerebellar symptoms are absent, which are important for diagnosing sCJD [2–4]. V180I gCJD is also characterized by the absence of a family history or notable myoclonus, and the lack of periodic sharp-wave complexes (PSWC) on the electroencephalogram (EEG) [2–4].
Fine small round vacuoles with clear boundaries have been observed in MM1-type sCJD. At cerebral pathologic stage II of MM1-type sCJD (disease duration 2–5 months) proposed by our previous report, spongiform change is the most typical and prominent [5] In contrast, large confluent vacuoles in small coalescing groups, resembling grape-like clusters, have been observed in MM2-cortical (MM2C)-type sCJD. In MM2C-type, this pathological change proceeds slowly with clinical symptoms, as compared to the MM1-type. In contrast, the following pathological characteristics of V180I gCJD have been reported in autopsied cases: [3,6–12] spongiform change can be observed clearly in patients with V180I gCJD of not only short disease duration, but also a long disease duration, as compared to the MM1- and MM2C-types of sCJD. In V180I gCJD, characteristic spongiform changes are observed in the morphology of vacuoles, which are called various-sized and non-confluent (VaSNoC) vacuoles [13]. In comparison to sCJD, gliosis and neuronal loss are generally milder in V180I gCJD. The condition is also characterized by weaker, synaptic-type PrP immunoreactivity.
However, no studies have investigated the correlation between vacuole morphology and duration of disease in V180I gCJD, and the pathological progression of V180I gCJD, especially in terms of how spongiform change occurs remains unclear. In this study, we aimed to clarify the pathological progression by examining the size and number of vacuoles and dispersion of vacuole size (variation in vacuolar size) of V180I gCJD for each disease duration in comparison with that of sCJD (MM1-type and MM2C-type).
Results
Clinical findings
The clinical features of the six patients with V180I gCJD included in the study are shown in Table 1. Mean age at onset was 79.1 ± 5.2 years (mean ± standard deviation (SD), range: 73–87 years), and mean duration of disease was 47.8 ± 38.5 months (mean ± SD, range: 10–102 months) and symptoms progressed slowly. PSWC was not observed by EEG. As regard to the cranial magnetic resonance imaging examination, T2-weighted imaging revealed swelling of the cerebral cortex, and diffusion-weighted imaging (DWI)-examined cases showed high-signal areas mainly in the cerebral cortex and basal ganglia.
Table 1.
Clinicopathological findings of the six patients with V180I genetic Creutzfeldt–Jakob disease.
| |
Patient 1 |
Patient 2 |
Patient 3 |
Patient 4 |
Patient 5 |
Patient 6 |
|---|---|---|---|---|---|---|
| Clinical features | ||||||
| Age at onset | 87 | 84 | 80 | 78 | 73 | 73 |
| Age at death | 87 | 85 | 82 | 81 | 81 | 81 |
| Sex | Female | Female | Male | Female | Female | Female |
| Family history | − | − | − | − | − | − |
| Total disease duration | 10 months | 20 months | 21 months | 33 months | 101 months | 102 months |
| Initial symptoms | Slow reaction | Numbness, tremor | Right hemiparesis, motor aphasia, | Disorientation | Disorientation | Aphasia |
| Major symptoms and signs | Dementia, gait disturbance | Dementia | Dementia, tremor | Dementia, abnormal behavior | Dementia | Dementia |
| Cerebral cortical dysfunction at early disease stage | + | + | + | + | + | + |
| Visual symptoms at early disease stage | − | − | − | − | − | − |
| Parkinsonism at early disease stage | − | + | + | + | − | + |
| Cerebellar symptoms at early disease stage | − | − | − | − | − | − |
| Myoclonus | − | + | + | + | + | + |
| Akinetic mutism state (Time to reach) | − | + (5 months) | + (9 months) | + (16 months) | + (20 months) | + (22 months) |
| Cause of death | Respiratory failure | Respiratory failure | Pneumonia | Respiratory failure | Respiratory failure | Respiratory failure |
| MRI study | ||||||
| MRI study | + (1 months after the onset) | + (4 months after the onset) | + (10 months after the onset) | + (8 months after the onset) | N.E. | + (42 months after the onset) |
| Swelling of the cerebral cortex (T2-weighted image) | + | + | + | + | N.E. | + |
| DWI hyperintensity (Observed region) | + (cerebral cortex, basal ganglia) | + (cerebral cortex, basal ganglia) | N.E. | + (cerebral cortex, basal ganglia) | N.E. | + (cerebral cortex) |
| Cerebral white matter lesion | − | − | − | + | N.E. | − |
| CSF study | ||||||
| NSE (ng/mL) | N.E. | N.E. | 29.9 | 25 | N.E. | 17.8 |
| 14-3-3 protein (μg/mL) | N.E. | 697 | N.E. | 1435 | N.E. | 3423 |
| Total tau (pg/mL) | N.E. | > 1200 | N.E. | 1370 | N.E. | 5474 |
| EEG study | ||||||
| PSWC | − | − | − | − | − | − |
| Slowing | + | + | + | + | + | + |
| PrP gene analysis | ||||||
| Codon 129 polymorphism | Met/Met | Met/Met | Met/Val | Met/Met | Met/Met | Met/Met |
| Codon 219 polymorphism | Glu/Glu | Glu/Glu | Glu/Glu | Glu/Glu | Glu/Glu | Glu/Glu |
| Pathological findings | ||||||
| Brain weight (g) | 1050 | 1150 | 1060 | 750 | 600 | 720 |
| Spongiform degeneration | Widely | Widely | Widely | Widely | Widely | Widely |
| in the cerebral cortex | observed | observed | observed | observed | observed | observed |
| PrP immunostaining | Very weak and synaptic | Very weak and synaptic | Very weak and synaptic | Very weak and synaptic | Very weak and synaptic | Very weak and synaptic |
| Neurofibrillary tangles | Braak stage II | Braak stage I | Braak stage II | Braak Stage II | Braak stage II | Braak stage IV |
| Senile plaques | CERAD stage C | CERAD stage B | CERAD stage 0 | CERAD stage 0 | CERAD stage 0 | CERAD stage A |
| Western blot analysis of PrP | Characteristic pattern | Characteristic pattern | Characteristic pattern | Characteristic pattern | Characteristic pattern | Characteristic pattern |
DWI, diffusion-weighted image; NE, not examined; NSE, neuron-specific enolase; EEG, electroencephalogram; PSWC, periodic sharp-wave complexes; Met, methionine; Val, valine; Glu, glutamic acid; CSF, cerebrospinal fluid; MRI, magnetic resonance imaging; PrP, prion protein; CERAD, Consortium to Establish a Registry for Alzheimer's disease
Five patients with sCJD (three with the MM1-type and two with the MM2C-type) were also investigated as representative cases of CJD. The clinical features of the patients with sCJD are shown in Table 2.
Table 2.
Clinicopathological findings of the three patients with MM1 type and two patients with MM2C type sporadic Creutzfeldt–Jakob disease.
| |
Patient 7 |
Patient 8 |
Patient 9 |
Patient 10 |
Patient 11 |
|---|---|---|---|---|---|
| Type of CJD | MM1 | MM1 | MM1 | MM2C | MM2C |
| Clinical features | |||||
| Age at onset | 88 | 82 | 80 | 68 | 67 |
| Age at death | 88 | 82 | 81 | 68 | 67 |
| Sex | Female | Male | Female | Male | Male |
| Family history | − | − | − | − | − |
| Total disease duration | 4 months | 5 months | 5 months | 5 months | 5 months |
| Initial symptoms | Left hemiparesis | Disorientation | Aphasia, right hemiparesis | Dementia | Dementia |
| Major symptoms and signs | Dementia, dysbasia | Dementia | Right hemiparesis, dementia | Dementia | Dementia |
| Cerebral cortical dysfunction at early disease stage | + | + | + | + | + |
| Visual symptoms at early disease stage | − | − | − | − | − |
| Parkinsonism at early disease stage | − | − | − | − | − |
| Cerebellar symptoms at early disease stage | − | − | − | − | − |
| Myoclonus | + | + | + | + | + |
| Akinetic mutism state (Time to reach) | + (1 month) | + (3 month) | + (2 month) | + (5 months) | + (5 months) |
| Cause of death | Pneumonia | Pneumonia | Respiratory failure | Respiratory failure | Respiratory failure |
| MRI study | |||||
| MRI study | + (2 months after the onset) | + (1 months after the onset) | + (3 months after the onset) | + (5 months after the onset) | + (2 months after the onset) |
| DWI hyperintensity (Observed region) | + (cerebral cortex) | + (cerebral cortex) | + (cerebral cortex) | + (cerebral cortex) | + (cerebral cortex) |
| Cerebral white matter lesion | − | − | − | − | − |
| CSF study | |||||
| NSE (ng/mL) | 29.9 | 145.4 | 126.3 | N.E. | 22.4 |
| 14-3-3 protein (μg/mL) | 4044 | 4014 | 12250 | N.E. | N.E. |
| Tau (pg/mL) | 2400 | > 1200 | 27870 | N.E. | N.E. |
| EEG study | |||||
| PSWC | + | + | + | + | + |
| Slowing | + | + | + | + | + |
| PrP gene analysis | |||||
| Codon 129 polymorphism | Met/Met | Met/Met | Met/Met | Met/Met | Met/Met |
| Codon 219 polymorphism | Glu/Glu | Glu/Glu | Glu/Glu | Glu/Glu | Glu/Glu |
| Pathological findings | |||||
| Brain weight (g) | 910 | 960 | 940 | 1300 | 1385 |
| Spongiform degeneration | Widely | Widely | Widely | Widely | Widely |
| in the cerebral cortex | observed | observed | observed | observed | observed |
| PrP immunostaining | synaptic | synaptic | synaptic | perivacuolar and coarse plaques | perivacuolar and coarse plaques |
| Neurofibrillary tangles | Braak stage II/III | Braak stage II/III | Braak stage II | Braak stage I | Braak stage I |
| Senile plaques | CERAD C | CERAD 0 | CERAD 0 | CERAD 0 | CERAD 0 |
| Western blot analysis of PrP | Type 1 | Type 1 | Type 1 | Type 2 | Type 2 |
DWI, diffusion-weighted image; NE, not examined; NSE, neuron-specific enolase; EEG, electroencephalogram; PSWC, periodic sharp-wave complexes; Met, methionine; Val, valine; Glu, glutamic acid; CJD, Creutzfeldt–Jakob disease; CSF, cerebrospinal fluid; MRI, magnetic resonance imaging; PrP, prion protein; CERAD, Consortium to Establish a Registry for Alzheimer's disease
Macroscopic findings in V180I gCJD cases
Mean brain weight before formalin fixation was 888.3 ± 206.0 g (mean ± SD, range: 600–1150 g). Representative macroscopic findings for patient 5 with disease duration of 101 months are shown in Fig. 1. In this case, although markedly high diffuse atrophy was observed in the cerebrum, the precentral gyrus was relatively preserved (Fig. 1A). Coronal cut surface findings also revealed highly diffuse atrophy, and cortical thinning and severe white matter atrophy were noted (Fig. 1B). The hippocampus (Fig. 1B) and the brainstem (Fig. 1C) were well preserved. The color tone of the substantia nigra and the locus coeruleus were well preserved (Fig. 1C). The cerebellum was preserved (Fig. 1D).
Figure 1.
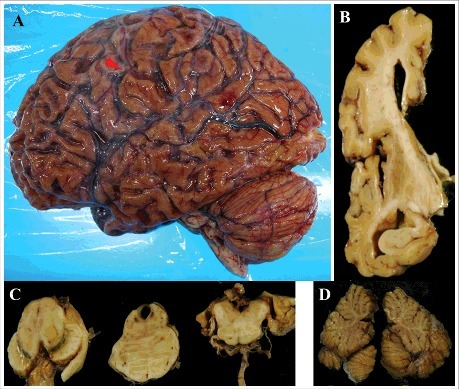
Macroscopic findings in patient 5. (A) the lateral aspect of the left cerebral hemisphere. The arrowhead indicates the central sulcus. (B) the left coronal section of the frontotemporal lobe through the anterior commissure from the posterior side. (C) the horizontal cut surface of the brainstem, from the left to the midbrain, pons, and medulla oblongata. (D) the sagittal division plane of the cerebellar vermis.
Although the degree of the cerebral atrophy of each case depended on disease duration, there were essentially similar findings in all patients regarding the cerebellum and brainstem.
Microscopic findings in V180I gCJD cases
Microscopic findings of the frontal neocortex of patients 1 through 6 are shown in Figure 2. A high degree of spongiform change was extensively observed. The morphology of spongiform changes showed VaSNoC vacuoles. Mild-to-severe tissue rarefaction was observed dependent on disease duration, but neurons relatively remained. There were similar findings at the temporal, parietal, and occipital neocortices. In all patients, the degrees of neuronal loss and gliosis were mild compared to the findings in the sCJD cases. Neurons remained despite a disease duration of 101 and 102 months (Fig. 2E, F). The approximate degree of neocortical pathological findings in the six patients with V180I gCJD as observed with HE staining and immunostaining, is shown in Table 3. In all patients, the neuropil was well preserved and spongiform change was mild in the hippocampus, subiculum, and parahippocampal gyrus in comparison to that in the cerebral cortex. Microscopic findings of the hippocampus (CA1) and cerebellum of patient 5 are shown in Figure 3. In the hippocampus, neurons were well preserved. Gliosis was mild and there was limited vacuolation. In patients 1 through 4, mild gliosis was found in the basal ganglia, thalamus, and amygdala, but there was no neuronal loss. The cerebral white matter was well preserved. In contrast, in patients 5 and 6, moderate gliosis and mild neuronal loss were found in the basal ganglia, thalamus, and amygdala. Severe tissue rarefaction and gliosis were found in the cerebral white matter. In the cerebellum, limited to mild spongiform change was noted in the molecular layer, without thinning. The granule-cell layer was also well preserved, as were Purkinje cells (Fig. 3B). In all patients, the brainstem was generally well preserved against neuronal loss and gliosis.
Figure 2.
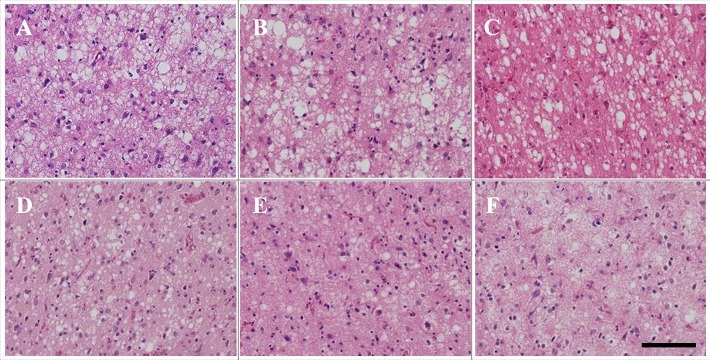
Microscopic findings of frontal lobe HE staining in patients with V180I genetic Creutzfeldt–Jakob disease (CJD) (A: patient 1, B: patient 2, C: patient 3, D: patient 4, E: patient 5, F: patient 6). Spongiform change is noted. However, neuronal loss and gliosis are mild compared to in sporadic CJD (MM1-type, MM2C-type). Even in patients 5 and 6, neurons remain. Scale bars: 100 μm HE, hematoxylin and eosin.
Table 3.
The approximate degree of neocortical pathological findings in the six patients with V180I gCJD.
| Spongiform change | Gliosis | Neuropil rarefaction | Neuronal loss | |
|---|---|---|---|---|
| Patient 1 | Severe | Moderate | Mild | Mild |
| Patient 2 | Severe | Moderate | Mild | Mild |
| Patient 3 | Severe | Moderate | Mild | Moderate |
| Patient 4 | Severe | Moderate | Mild | Moderate |
| Patient 5 | Moderate | Severe | Moderate | Severe |
| Patient 6 | Moderate | Severe | Severe | Severe |
gCJD, genetic Creutzfeldt–Jakob disease.
Figure 3.
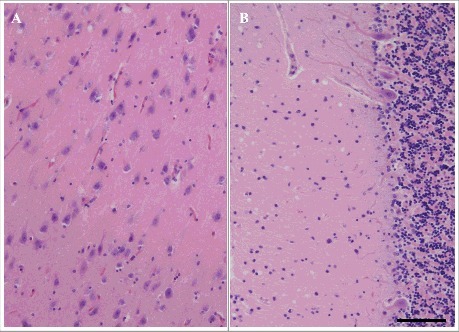
Microscopic findings of HE staining of the hippocampus (CA1) (A) and cerebellum (B) in patient 5. Mild neuronal loss, gliosis, and spongiform change are shown. Scale bars: 100 μm HE, hematoxylin and eosin.
Microscopic findings in sCJD cases
Microscopic findings of the frontal neocortex of patients 7 through 11 are shown in Fig. 4. Spongiform change is noted. Fine small round vacuoles with clear boundaries were observed in MM1-type sCJD (Fig. 4A–C). Large confluent vacuoles in grape-like clusters were observed in MM2C-type sCJD (Fig. 4D, E).
Figure 4.
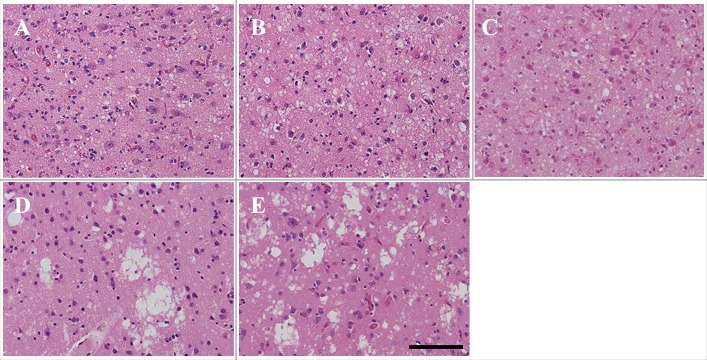
Microscopic findings of frontal lobe HE staining in patients 7–11 (A: patient 7, B: patient 8, C: patient 9, D: patient 10, E: patient 11). Fine vacuoles are observed in MM1-type sporadic Creutzfeldt–Jakob disease (sCJD) (A–C). Large confluent vacuoles are observed in MM2C-type sCJD (D, E). Scale bars: 100 μm HE, hematoxylin and eosin.
Quantitative analysis of cerebral cortical pathology
In the present study, observations and measurement of the major diameters of vacuoles in patients with V180I gCJD and patients with sCJD (three MM1-type patients and two MM2C-type patients) at the frontal, temporal, parietal, and occipital neocortices were quantitatively analyzed. There was a significant difference among V180I gCJD and the MM1-type sCJD and MM2C-type sCJD (p < 0.001; Kruskal-Wallis test). Then, pairwise comparisons were performed with the Steel-Dwass test for each patient. Table 4 shows the total number, mean major diameters, and the coefficient of variation of measurable and clear vacuoles in all patients. Fig. 5 shows the box-whisker plot of the vacuole major diameter for each patient.
Table 4.
The total number of vacuoles, mean vacuolar major diameter, and the coefficient of variation in all patients.
| Type of CJD | disease duration (months) | Total number of vacuoles | Vacuolar major diameter, Mean ± SD (μm) (range) | Coefficient of variation | |
|---|---|---|---|---|---|
| Patient 1 | V180I gCJD | 10 | 1202 | 9.92 ± 4.11 (3.12–34.37) | 0.41 |
| Patient 2 | V180I gCJD | 20 | 1347 | 9.94 ± 5.26 (3.12–37.50) | 0.53 |
| Patient 3 | V180I gCJD | 21 | 1250 | 9.97 ± 4.46 (3.12–34.37) | 0.45 |
| Patient 4 | V180I gCJD | 33 | 1088 | 9.74 ± 4.46 (3.12–29.69) | 0.46 |
| Patient 5 | V180I gCJD | 101 | 726 | 9.77 ± 3.842 (3.12–26.56) | 0.39 |
| Patient 6 | V180I gCJD | 102 | 1166 | 9.75 ± 4.22 (3.12–29.68) | 0.43 |
| Patient 7 | sCJD (MM1) | 4 | 947 | 5.99 ± 1.93 (1.56–15.62) | 0.32 |
| Patient 8 | sCJD (MM1) | 5 | 965 | 6.16 ± 1.93 (3.12–17.19) | 0.31 |
| Patient 9 | sCJD (MM1) | 5 | 1142 | 5.63 ± 2.00 (1.56–15.62) | 0.36 |
| Patient 10 | sCJD (MM2C) | 5 | 452 | 13.06 ± 12.76 (3.12–85.94) | 0.97 |
| Patient 11 | sCJD (MM2C) | 5 | 505 | 12.68 ± 9.57 (3.12–93.75) | 0.76 |
CJD, Creutzfeldt–Jakob disease; g, genetic; s, sporadic.
Coefficient of variation signifies the evaluation of dispersion of vacuole size.
Figure 5.
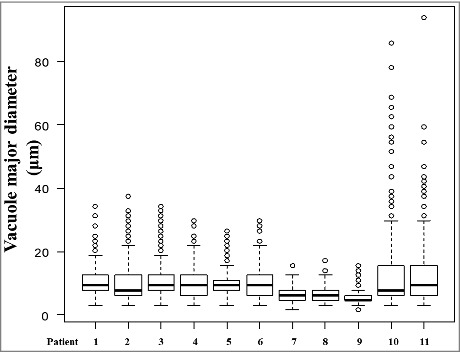
Comparison of vacuolar diameters in all patients with V180I genetic Creutzfeldt–Jakob disease (gCJD), and MM1-type and MM2C-type sporadic CJD by box-and-whisker plots of vacuole major diameter. The horizontal line inside each box indicates the median value, and the length of the box is the interquartile range (25th–75th percentile). The extremes of the whiskers contain 95% values. Open circles indicate outliers.
Comparison of vacuoles observed in V180I gCJD
No significant differences were observed in the vacuolar major diameters of the cerebral neocortex among patient 1 through 6 in multiple comparisons (Fig. 5) (each was P > 0.05; Steel-Dwass test). In V180I gCJD, the vacuole size showed similar major diameter regardless of disease duration (Fig. 5). No significant difference was observed in the number of vacuoles depending on the duration of the disease; however, the number of vacuoles tended to decrease as the disease duration became longer (Table 4). The major diameter of the vacuoles observed with V180I gCJD was 9.86 ± 4.42 µm (mean ± SD). In V180I gCJD, there was no apparent difference in the coefficient of variation of vacuolar major diameter in each patient. Therefore, in V180I gCJD, no apparent dispersion in vacuole size was observed regardless of disease duration (Table 4).
Comparison of vacuoles in V180I gCJD and sCJD
The vacuolar major diameters in patients with V180I gCJD (patients 1–6) were larger than those in patients with the MM1-type (patients 7–9) and the difference was statistically significant (Fig. 5) (each was P < 0.05; Steel-Dwass test). The major diameter of the vacuoles observed in patients with the MM1-type was 5.94 ± 1.99 µm (mean ± SD). By contrast, the vacuolar major diameter in patients with V180I gCJD was not significantly different from that of patients with the MM2C-type (Fig. 5) (each was P > 0.05; Steel-Dwass test). The major diameter of the vacuoles observed in patients with the MM2C-type was 12.86 ± 11.19 µm (mean ± SD). The coefficient of variation of vacuolar diameter was generally highest in the MM2C-type, followed by V180I gCJD, and the MM1-type (Table 4). Therefore, the dispersion in vacuole major diameter was relatively larger in the MM2C-type, followed by V180I gCJD, and the MM1-type. The number of vacuoles observed was the largest in V180I gCJD, followed by the MM1-type, and was the lowest in the MM2C-type (Table 4).
Discussion
In the present study, we compared vacuolar features according to disease duration of V180I gCJD, as well as between V180I gCJD and sCJD cases (MM1-type, MM2C-type). The results revealed that V180I gCJD showed no statistically significant change in vacuolar major diameter despite disease duration. Furthermore, the vacuole size observed in V180I gCJD was larger than that observed in MM1-type sCJD, and the difference was statistically significant; in addition, unlike the vacuoles found in MM2C-type sCJD, no confluent vacuole was remarkable. The major diameter of the vacuole seen in the MM1-type was relatively uniform; however, the major diameter in V180I gCJD was relatively more varied than that in the MM1-type. In addition, the major diameter of the vacuoles seen in the MM2C-type was generally more varied than that in the MM1-type and V180I gCJD.
The clinical features of the six patients examined in the present study were consistent with those previously reported [2–4]. The pathological examination of 17 patients with CJD revealed that neuronal loss and gliosis are directly proportional to disease duration, whereas spongiform change is inversely proportional to disease duration [14]. However, in that report, the CJD disease type was not described. The results of our present study showed that in V180I gCJD, the size of vacuoles and dispersion of vacuole size did not change, regardless of disease duration. This is considered to be a characteristic finding in the pathological progression of V180I gCJD. In this study, for the first time, various-sized vacuoles were observed in V180I gCJD, and the dispersion of vacuole size was larger than that in the MM1-type and smaller than that in the MM2C-type, with the differences being statistically significant.
To date, no conclusion on the localization of vacuoles in CJD has been reached. However, electron microscopy revealed that vacuoles exist in neurons in human CJD [15], and electron microscopy of V180I gCJD revealed membrane-bound intracellular extranuclear vacuolar inclusions in residual neurons [10]. It is thought that vacuoles are likely to be present in neuronal cell bodies and some of their neurites, such as dendrites, and anterior and postsynaptic terminals.
The findings of the size were similar for any duration of disease in V180I gCJD, suggesting the possibility that expansion of vacuole diameter did not result in tissue rarefaction. Based on our results, the pathological progression of V180I gCJD may be explained by the following four mechanisms: 1. At some point of the disease course, vacuoles formed in neurons. 2. The vacuoles did not become larger and their number did not become increased. However, neurons with vacuoles slowly diminished. As neurons diminished, rarefaction of the neuropil slowly occurred. 3. Then, similar size vacuoles were formed in the neurons that did not previously have them. 4. Because neuronal loss progresses very slowly, at this time, the total number of vacuoles did not significantly differ depending on the duration of the disease. However, when the disease duration becomes long, neuronal loss progresses, the number of neurons that have vacuoles decreases, and so the total number of vacuoles also gradually decreases. Neuronal loss and rarefaction of the neuropil lead to pathological progression of V180I gCJD, such as tissue rarefaction and decrease in cerebral weight. The difference in morphology of vacuoles seen in V180I gCJD and MM1-type and MM2C-type sCJD may be due to the biochemical difference of PrP, as shown, for instance, with Western blot analysis. We previously hypothesized that unknown factors may be acting protectively against the rapid progression of the clinical course and the development of pathological involvement associated with V180I gCJD [3,6,18]. Moreover, in V180I gCJD, we speculate the possibility that the amount of PrP accumulated is small because immunoreactivity of PrP is weak and the neurotoxicity of PrP may be lower than that of sCJD. The mechanism involved in the vacuolation of CJD remains unknown. However, based on the present observations, the possibility was speculated that the size, number, and dispersion of vacuoles may influence the progression of neuronal loss, at least in part.
We should note, as a limitation to this study that artifacts in the specimens associated with fixation, preparation, and staining may have affected the actual features. In addition, spongiform change and oligodendrocyte perivacuoles may be difficult to distinguish. Moreover, blindness could not be maintained because the form of the vacuole varies depending on the type of CJD disease.
In conclusion, VaSNoC vacuoles were observed in patients with V180I gCJD, and the statistical analysis showed that the number and size of vacuoles did not change with disease duration. In our speculation, these pathologic observations seem to influence the clinical progression of V180I gCJD.
Materials and methods
This study was approved by the Ethics Committee of Aichi Medical University.
Subjects
In the present study, we included six consecutive autopsied patients with V180I gCJD, whose pathological examination was performed at the Institute for Medical Science of Aging, Aichi Medical University. All patients were Japanese, and none had a family history of prion disease. We followed protocols for prion gene analysis and Western blot analysis of PrP as previously reported [16]. Typing of PrP was performed based on the sCJD classification system proposed by Parchi et al. [17]. In all six patients with V180I gCJD, genetic analysis was performed, and a valine to isoleucine mutation was confirmed in PrP gene codon 180. In patient 3, codon 129 showed methionine/valine heterozygosity, and the valine polymorphism existed on a different allele of the V180I mutation. In patients 1, 2, 4, 5, and 6, codon 129 showed methionine/ methionine homozygosity. Codon 219 showed glutamic acid/ glutamic acid homozygosity in all cases. Western blot analysis of the PrPs revealed a characteristic pattern in all patients with V180I gCJD. A diglycoform band could not be detected, and the molecular weight of the nonglycoform band was equivalent to type 2 PrP, but was slightly greater than the band characteristic of sCJD. The cases of patient 33, 4 [13], and 66 have been previously reported. We included five patients with MM1-type and MM2C-type as representative cases of CJD; the CJD classification of these cases was confirmed by PrP gene analysis and Western blot analysis. Because in the patients with MM1-type sCJD with disease duration more than 11 months, clear vacuoles could not be observed due to tissue rarefaction [5], we could not compare them to patients with MM1-type sCJD with disease duration that corresponded to that of the patients with V180I gCJD that we studied. For this reason, we compared the vacuolar findings of the cases that were at Stage II of cerebral cortical pathology with the most marked and apparent vacuolization reported [5], and were at similar ages with the V180I gCJD cases that we examined. We included all three patients with MM1-type sCJD that met these investigation requirements in the pathological examination performed at our institute. No mutation was noted in the PrP gene. Codon 129 showed methionine/ methionine homozygosity and codon 219 showed glutamic acid/ glutamic acid homozygosity in all cases. Western blot analysis of PrP revealed a pattern of type-1 PrP. MM2C-type was examined in patient with the same disease duration as MM1-type. We included all two patients with MM2C-type sCJD whose disease duration was similar to that of the MM1-type sCJD cases. No mutation was noted in the PrP gene. Codon 129 showed methionine/ methionine homozygosity and codon 219 showed glutamic acid/ glutamic acid homozygosity. Western blot analysis of PrP revealed a type-2 PrP pattern.
Histopathological examination
Autopsy was performed on the whole brain and tissues were fixed with 20% neutral buffered formalin for 1 month. The brain tissue block was fixed with 95% formic acid for 1 h to inactivate PrP. Sections of 8-μm thickness were mounted, deparaffinized, dehydrated, and stained. For routine neuropathological examination, the sections were stained with hematoxylin and eosin (HE), Klüver–Barrera (KB), and modified Gallyas-Braak silver staining. Immunohistochemical analysis of selected sections was performed with antibody 3F4 against PrP (Dako, Glostrup, Denmark, mouse monoclonal, diluted 1:100) after hydrolytic autoclaving to retrieve the antigen. The PrP immunostaining protocol using the En Vision amplified method (EnVision plus kit, Dako) was conducted as previously described [19,20]. Anti-β-protein (Dako, mouse monoclonal, diluted 1:1000), anti-human GFAP antibody (Dako, mouse monoclonal, diluted 1:1000), anti-Iba-1 antibody (WAKO, rabbit monoclonal, diluted 1:400), anti-CD68 antibody (Dako, mouse monoclonal, diluted 1:200), and phosphorylated-tau (Innogenetics, Ghent, Belgium, AT-8, mouse monoclonal, diluted 1:3000) immunostaining were also used as appropriate. Immunostained sections were lightly counterstained with Mayer's hematoxylin.
Pathologic investigation of the vacuole findings was performed in the following cerebral cortical regions, as referred to in the investigation by Parchi et al. [17]: the frontal (middle frontal gyri), temporal (middle temporal gyri), parietal (inferior parietal lobule), and occipital (lateral occipital gyrus) neocortices. At these four sites, HE-stained specimens were observed at 200 × magnification in all patients. For each site, one field of representative view was printed on paper and the major diameter of each vacuole was measured manually. The number of vacuoles measured in the frontal, parietal, temporal, and occipital neocortices were summated as the total number of vacuoles as shown in Table 4. In the irregularly shaped or confluent vacuoles, we measured the maximum diameter. The indistinct vacuoles without clear boundary were excluded from the measurement.
Statistical analysis
The Bartlett test was used, and we confirmed that the vacuolar size did not follow a normal distribution in any of the patients. To compare the vacuole major diameters of all patient, we used the Kruskal-Wallis test with post-hoc multiple comparisons using the Steel-Dwass test. Evaluation of dispersion of vacuole size in the data was performed using the coefficient of variation. Significance was defined as a P value of < 0.05. All statistical analyses were performed with EZR (Saitama Medical Center, Jichi Medical University, Saitama, Japan; http://www.jichi.ac.jp/saitama-sct/SaitamaHP.files/statmedEN.html; Kanda, 2012), which is a graphical user interface for R (The R Foundation for Statistical Computing, Vienna, Austria, version 2.13.0). More precisely, it is a modified version of R commander (version 1.6-3) designed to add statistical functions frequently used in biostatistics [21].
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by the Research Committee of Prion Disease and Slow Virus Infection, Research on Policy Planning and Evaluation for Rare and Intractable Diseases, Health and Labour Sciences Research Grants from the Ministry of Health, Labour, and Welfare of Japan, and JSPS KAKENHI Grant Number 15K08369. This research was partially supported by the Strategic Research Program for Brain Sciences from the Japan Agency for Medical Research and development.
References
- [1].Nozaki I, Hamaguchi T, Sanjo N, et al. et al.. Prospective 10-year surveillance of human prion diseases in Japan. Brain. 2010;133(10):3043–3057. doi: 10.1093/brain/awq216 [DOI] [PubMed] [Google Scholar]
- [2].Jin K, Shiga Y, Shibuya S, et al.. Clinical features of Creutzfeldt-Jakob disease with V180I mutation. Neurology. 2004;62(3):502–505. [DOI] [PubMed] [Google Scholar]
- [3].Iwasaki Y, Sone M, Kato T, et al.. Clinicopathological characteristics of Creutzfeldt-Jakob disease with a PrP V180I mutation and M129V polymorphism on different alleles. Rinsho Shinkeigaku. 1999;39(8):800–806. [Article in Japanese with English abstract] [PubMed] [Google Scholar]
- [4].Qina T, Sanjo N, Hizume M, et al. et al.. Clinical features of genetic Creutzfeldt-Jakob disease with V180I mutation in the prion protein gene. BMJ Open. 2014;4(5):e004968. doi: 10.1136/bmjopen-2014-004968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Iwasaki Y, Tatsumi S, Mimuro M, et al.. Relation between clinical findings and progression of cerebral cortical pathology in MM1-type sporadic Creutzfeldt-Jakob disease: proposed staging of cerebral cortical pathology. J Neurol Sci. 2014;341(1-2):97–104. doi: 10.1016/j.jns.2014.04011 [DOI] [PubMed] [Google Scholar]
- [6].Iwasaki Y, Mori K, Ito M, et al.. An autopsied case of V180I Creutzfeldt-Jakob disease presenting with panencephalopathic-type pathology and a characteristic prion protein type. Neuropathology. 2011;31(5):540–548. doi: 10.1111/j.1440-1789.2010.01192.x [DOI] [PubMed] [Google Scholar]
- [7].Yoshida H, Terada S, Ishizu H, et al.. An autopsy case of Creutzfeldt-Jakob disease with a V180I mutation of the PrP gene and Alzheimer-type pathology. Neuropathology. 2010;30(2):159–164. doi: 10.1111/j.1440-1789.2009.01048.x [DOI] [PubMed] [Google Scholar]
- [8].Suzuki K, Matsumura N, Suzuki T, et al.. Creutzfeldt-Jakob disease with V180I mutation and senile plaque. Geriatr Gerontol Int. 2009;9(2):210–212. doi: 10.1111/j.1447-0594.2009.00517.x [DOI] [PubMed] [Google Scholar]
- [9].Honda H, Ishii R, Hamano A, et al.. Microsphere formation in a subtype of Creutzfeldt-Jakob disease with a V180I mutation and codon 129 MM polymorphism. Neuropathol Appl Neurobiol. 2013;39(7):844–848. doi: 10.1111/nan.12047 [DOI] [PubMed] [Google Scholar]
- [10].Kobayashi S, Saito Y, Maki T, et al.. Cortical propagation of Creutzfeldt-Jakob disease with codon 180 mutation. Clin Neurol Neurosurg. 2010;112(6):520–523. doi: 10.1016/j.clineuro.2010.03.015 [DOI] [PubMed] [Google Scholar]
- [11].Matsumura T, Kojima S, Kuroiwa Y, et al.. An autopsy-verified case of Creutzfeldt-Jakob disease with codon 129 polymorphism and codon 180 point mutation. Rinsho Shinkeigaku. 1995;35(3):282–285. [Article in Japanese] [PubMed] [Google Scholar]
- [12].Suzuki K, Matsumura N, Suzuki T, et al.. A case of Creutzfeldt-Jakob disease with codon 129 polymorphism and codon 180 point mutation. Nihon Ronen Igakkai Zasshi. 2008;45(1):107–111. [Article in Japanese] [DOI] [PubMed] [Google Scholar]
- [13].Iwasaki Y, Mori K, Ito M, et al.. An autopsy case of Creutzfeldt-Jakob disease with a prion protein gene codon 180 mutation presenting with pathological laughing and an exaggerated startle reaction. Neuropathology. 2017;37:575–581. doi: 10.1111/neup.12399 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- [14].Masters CL, Richardson EP Jr.. Subacute spongiform encephalopathy (Creutzfeldt-Jakob disease). The nature and progression of spongiform change Brain. 1978;101(2):333–344. [DOI] [PubMed] [Google Scholar]
- [15].Liberski PP, Streichenberger N, Giraud P, et al.. Ultrastructural pathology of prion diseases revisited: brain biopsy studies. Neuropathol Appl Neurobiol. 2005;31(1):88–96. [DOI] [PubMed] [Google Scholar]
- [16].Iwasaki Y, Yoshida M, Hashizume Y, et al.. Clinicopathologic characteristics of sporadic Japanese Creutzfeldt-Jakob disease classified according to prion protein gene polymorphism and prion protein type. Acta Neuropathol. 2006;112(5):561–571. [DOI] [PubMed] [Google Scholar]
- [17].Parchi P, Giese A, Capellari S, et al.. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46(2):224–233. [PubMed] [Google Scholar]
- [18].Iwasaki Y. Three cases of Creutzfeldt-Jakob disease with prion protein gene codon180 mutation presenting with pathological laughing and crying. J Neurol Sci. 2012;319(1-2):47–50. doi: 10.1016/j.jns.2012.05.023 [DOI] [PubMed] [Google Scholar]
- [19].Iwasaki Y, Hashizume Y, Yoshida M, et al.. Neuropathologic characteristics of brainstem lesions in sporadic Creutzfeldt-Jakob disease. Acta Neuropathol. 2005;109(6):557–566. doi: 10.1007/s00401-005-0981-0 [DOI] [PubMed] [Google Scholar]
- [20].Kitamoto T, Shin RW, Doh-ura K, et al.. Abnormal isoform of prion proteins accumulates in the synaptic structures of the central nervous system in patients with Creutzfeldt-Jakob disease. Am J Pathol. 1992;140(6):1285–1294. [PMC free article] [PubMed] [Google Scholar]
- [21].Kanda Y. Investigation of the freely-available easy-to-use software “EZR” (Easy R) for medical statistics. Bone Marrow Transplant. 2013;48(3):452–458. doi: 10.1038/bmt.2012.244 [DOI] [PMC free article] [PubMed] [Google Scholar]