

ABSTRACT
TDP-43 and FUS are DNA/RNA binding proteins associated with neuronal inclusions in amyotrophic lateral sclerosis (ALS) patients. Other neurodegenerative diseases are also characterized by neuronal protein aggregates, e.g. Huntington's disease, associated with polyglutamine (polyQ) expansions in the protein huntingtin. Here we discuss our recent paper establishing similarities between aggregates of TDP-43 that have short glutamine and asparagine (Q/N)-rich modules and are soluble in detergents, with those of polyQ and PIN4C that have large Q/N-rich domains and are detergent-insoluble. We also present new, similar data for FUS. Together, we show that like overexpression of polyQ or PIN4C, overexpression of FUS or TDP-43 causes inhibition of the ubiquitin proteasome system (UPS) and toxicity, both of which are mitigated by overexpression of the Hsp40 chaperone Sis1. Also, in all cases toxicity is enhanced by the [PIN+] prion. In addition, we show that the Sis1 mammalian homolog DNAJBI reduces toxicity arising from overexpressed FUS and TDP-43 respectively in human embryonic kidney cells and primary rodent neurons. The common properties of these proteins suggest that heterologous aggregates may enhance the toxicity of a variety of disease-related aggregating proteins, and further that chaperones and the UPS may be key therapeutic targets for diseases characterized by protein inclusions.
KEYWORDS: TDP-43, FUS, yeast, amyotrophic lateral sclerosis, chaperone, Sis1, DNAJB1, [PIN+], Ubiquitin Proteasome System (UPS)
Many neurodegenerative diseases are linked to a conformational change of a protein from a soluble form to an insoluble, amyloid-like aggregate that accumulates in neurons. For each disease, different aggregating protein(s) are involved. For example Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis (ALS), Huntington's disease and Creutzfeldt-Jakob disease respectively are associated with aggregated forms of: Aβ; α-synuclein; TDP-43, FUS and others; huntingtin; and PrP. Similar to prions, aggregates assembled from several of these proteins seed conformational change and aggregation of soluble forms of their protein [1−3].
Although homologs of these proteins are not found in yeast, when they are expressed in yeast they form aggregates and are toxic, modeling disease [4−8]. In some cases, e.g. huntingtin, aggregation and toxicity in yeast depends upon the presence of the prion form, [PIN+], of the endogenous yeast protein RNQ1 that contains a region rich in glutamines and asparagines (Q/N) [9,10]. We previously showed that the [PIN+] prion facilitates the de novo appearance of other yeast prions and aggregates of proteins with Q/N-rich domains, presumably by a cross-seeding mechanism [11−14].
Using the power of yeast genetics, modifiers of Aβ, FUS, TDP-43, α-synuclein and huntingtin toxicity have been identified in yeast. Importantly, by virtue of the modifiers’ homologies to human proteins, these modifiers have successfully identified human risk-factors for disease. These observations confirm the relevance and value of yeast in the study of neurodegenerative disease. Interestingly, there has been very little overlap between the modifiers isolated for the different proteins, suggesting some unique mechanisms for the various disease proteins [15−24].
TDP-43 is an RNA/DNA binding protein normally found in cell nuclei, but also found in neuronal cytoplasmic aggregates characteristic of a number of disorders including Alzheimer's disease, ALS, frontotemporal dementia, and chronic traumatic encephalopathy. A region of TDP-43 has been identified as “prion-like” on the basis of its amino acid sequence [25]. However TDP-43 aggregates are not typical amyloids and have limited detergent resistance [8,26].
Although Sis1, a conserved Hsp40 chaperone, had not been uncovered in large scale screens for modifiers of TDP-43 or FUS toxicity in yeast [19,27,28], overexpression of Sis1 rescues yeast from toxicity associated with polyQ-containing proteins whose toxicity and aggregation was dependent upon the presence of the [PIN+] prion. Furthermore, ubiquitin-mediated protein degradation is inhibited when two of these proteins, polyQ and PIN4C, are aggregated [9,29,30].
In our recent paper [31] we showed that the presence of [PIN+] increases, while overexpression of Sis1 reduces, TDP-43 toxicity (Fig. 1a). Additionally, overexpression of the mammalian homologue for Sis1, DNAJB1 reduced the toxicity of overexpressed TDP-43 in primary rodent neurons [31] (Fig. 1b). We also showed that TDP-43 inhibits ubiquitin-mediated degradation of a reporter protein especially in the presence of [PIN+], and that overexpression of Sis1 restores reporter degradation (Fig. 1c).
Figure 1.
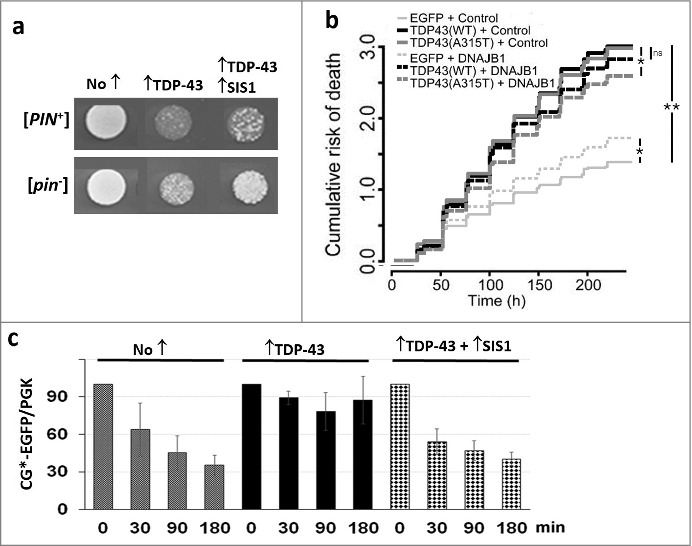
Overexpression of Sis1 mitigates the effects of TDP-43 in yeast and primary rodent cortical neurons. (Adapted from [31) a. Overexpression of TDP-43 is more toxic in the presence of the [PIN+] prion and is less toxic in the presence of overexpressed Sis1. The growth of normalized suspensions of [PIN+] and [pin−] versions of the same yeast strain transformed with pGAL1-TDP-43-YFP (↑TDP-43) and pGAL1-SIS1 (↑SIS1), or empty control plasmids (No ↑), on plasmid selective galactose medium is shown. b. Expression of DNAJB1 reduces toxicity of TDP-43 overexpression in rodent primary cortical neurons. A cumulative risk of death plot is shown for neurons transfected with vectors shown. While DNAJB1 displays some toxicity in control neurons when overexpressed, it also reduces the risk of death in neurons expressing TDP-43. *p < 0.005; **p < 2 × 10−16, by Cox proportional hazards analysis. N = 725-979 neurons per condition, assembled from three separate experiments. c. TDP-43 inhibits proteolysis of cytosolic misfolded proteins and this is reversed by overexpression of Sis1. We used degradation of CG*-GFP, the mutant version of the secretory protein carboxypeptidase Y lacking its signal sequence (ΔssCPY*) tagged with EGFP as a reporter to test for ubiquitin proteasome system (UPS) activity. New protein synthesis was inhibited with cycloheximide, after which levels of CG*-GFP were determined in cell lysates harvested at the times indicated. Data for a [PIN+] yeast strain is shown. All cells expressed CG*-EGFP from a GAL1 plasmid and expressed TDP-43 and Sis1 as indicated. Normalized cell lysates run on SDS-PAGE were immunoblotted with anti-GFP for CG*-EGFP level and anti-Pgk1 as an internal loading control. Quantification of three blots showing normalized ratio of CG*-EGFP and Pgk1 is shown with standard error.
In the extra-view we now present similar data for the FUS protein (Figs. 2–4). FUS, like TDP-43, is a nuclear RNA/DNA binding protein that forms neuronal cytoplasmic aggregates in patients with ALS and frontotemporal dementia. Also like TDP-43, FUS has short modular Q/N-rich domains and forms detergent-sensitive aggregates [8,32]. This work highlights common properties of these disease proteins.
Figure 2.
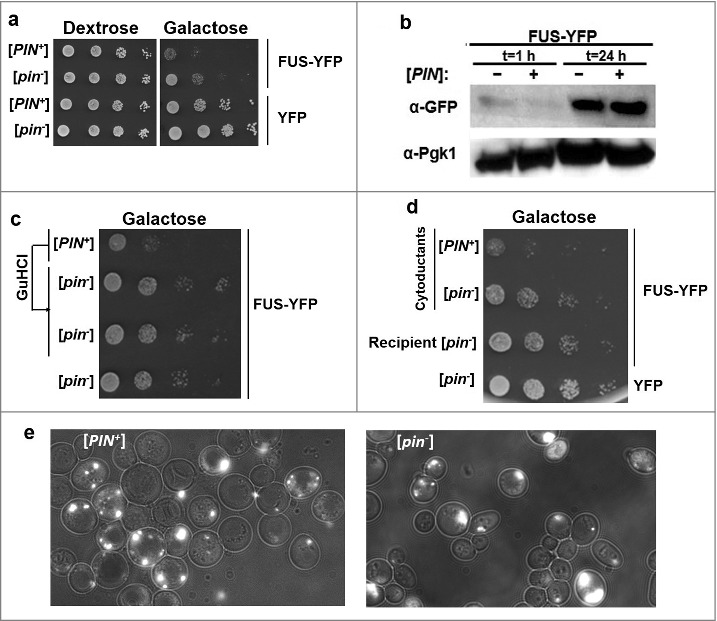
[PIN+] exacerbates FUS toxicity. a. [PIN+] exacerbates FUS toxicity. High [PIN+] (L1749) and isogenic [pin−] (L2910) cells harboring GAL1-controlled URA3 FUS-YFP (p2043), or YFP (p1752) plasmids were grown in liquid plasmid selective raffinose (2%) medium overnight. Cells were then transferred to plasmid selective Dextrose (2%) and Galactose (2%) media by serial dilution and photographed after 4–5 days. b. The presence of [PIN+] does not significantly enhance expression levels of FUS-YFP. Lysates of [pin−] or [PIN+] cells overexpressing FUS-YFP in 2% Galactose for 1 hour, or 24 hours were analyzed by western blot. Expression levels of FUS-YFP were detected by α-GFP (Roche), while Pgk1 was used as an internal loading control. c. Curing of [PIN+] which converts [PIN+] cells into [pin−] cells, relieves FUS toxicity. [PIN+] cells harboring FUS-YFP were grown on YPD+5 mM guanidine hydrochloride (GuHCl) plates for three passages, which causes loss of prions [36]. Two subclones of GuHCl-treated cells (middle two rows), untreated [PIN+] (top row) and [pin−] cells with FUS-YFP were transferred to plasmid selective galactose plates by serial dilution where they were allowed to grow. d. Cytoduction of [PIN+] into [pin−] cells exacerbates FUS toxicity. [PIN+] or [pin−] donor cells (respectively, L1749 and L2910) were mated on YPD with recipient [pin−] cells (L2599) harboring FUS-YFP. Representative cytoductants (recipients containing some cytoplasm from the donor) that became [PIN+] due to the [PIN+] donor (row 1) or remained [pin−] due to the [pin−] donor (row 2) as well as uncytoduced recipients with FUS-YFP (row 3) or YFP (row 4) are shown for comparison as a serial dilution on plasmid selective galactose plates. e. FUS-YFP forms similar aggregates in [PIN+] and [pin−] cells. Isogenic [PIN+] and [pin−] cells transformed with GAL-FUS-YFP plasmid were grown in 2% galactose medium before being imaged.
Figure 3.
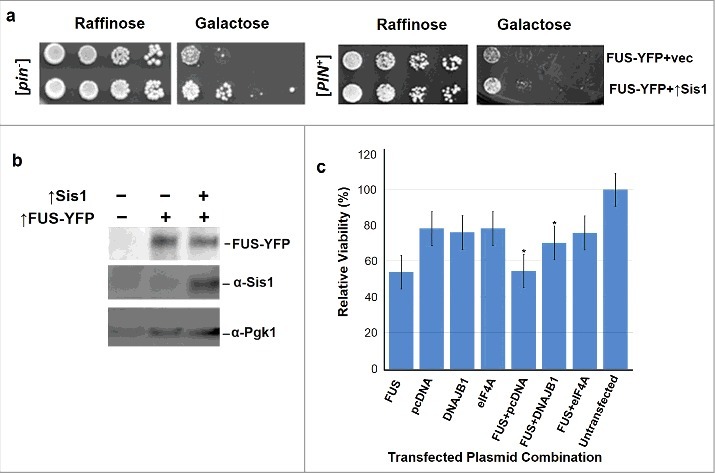
Excess Sis1 or DNAJB1 respectively relieve FUS toxicity in yeast and HEK cells. a. Overexpressed Sis1 rescues FUS associated toxicity. Isogenic [PIN+] and [pin−] versions of W303 yeast cells containing GAL1-controlled FUS-YFP (p2043) in addition to GAL1-controlled Sis1 (p1767) or vector (p1768), were grown overnight in plasmid selective raffinose (2%) media. Serial dilutions of cells were spotted and grown on plasmid selective non-inducing raffinose (2%) or inducing galactose (2%) plates. In this experiment [PIN+] and [pin−] cells were spotted on different plates and are not directly comparable. b. Sis1 overexpression does not enhance FUS protein levels. W303 [pin−] cells transformed with different plasmids were grown in plasmid selective galactose for 48 hours. Cells were then lysed and equal amounts of proteins were loaded on SDS-PAGE. Control vectors in lane 1 did not express either FUS or Sis1; FUS-YFP was overexpressed in lane 2; and FUS-YFP and Sis1 were co-overexpressed in lane 3. The blot was probed with anti-GFP (1:10,000, Roche) to detect FUS-YFP, anti-Sis1 (1:1000, Gift from E. Craig) and anti-Pgk1 (1:10,000) as an internal loading control. c. Mammalian DNAJB1 relieves FUS toxicity in human embryonic kidney (HEK) cells. HEK293T cells were transfected with plasmid(s) shown in the horizontal axis of the graph according to the manufacturer's instructions (Life technologies). Cell viability was measured with the MTT assay at 48 h post-transfection. Values represent means ± S.D. (n = 3). Empty vector (pcDNA) was used as a negative control, and eIF4A was used as the positive control since previous studies [28] demonstrated its ability to rescue FUS associated toxicity in HEK293T cells (*p < 0.05 using one way ANOVA with Dunnett's).
Figure 4.
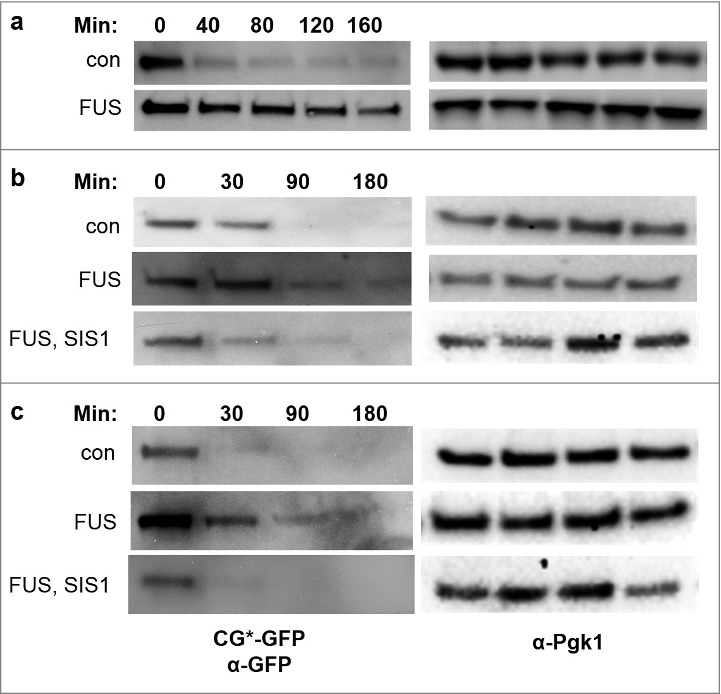
Degradation of UPS reporter protein CG*-GFP, is inhibited in yeast in the presence of FUS and overexpression of Sis1 mitigates this effect. Cells were grown in liquid plasmid selective 2% galactose medium for 48 hours to overexpress control (p2039) or FUS (p2049). In b and c GAL1-controlled Sis1 (p1767) or vector (p1768) were also expressed. Cycloheximide (0.5 or 0.7 mg/mL) was added, and cells were collected at the minutes (Min) indicated. Protein was extracted, and run on SDS-PAGE. The blots were probed with anti-GFP (1:10,000, Roche) to detect CG*-GFP levels (left) and anti-Pgk1 (1:10,000) as an internal loading control (right). Strains used were L1749 [PIN+] (a and b) and L2910 [pin-] (c).
[PIN+] Enhances Toxicity of Overexpressed FUS
A comparison of the growth of [pin−] vs. [PIN+] cells with a GAL1-controlled plasmid expressing FUS-YFP showed that [PIN+] increased toxicity (Fig. 2a). This difference was not due to an increase in the cellular level of FUS in [PIN+] vs. [pin−] cells (Fig. 2b). Also no dramatic differences in the appearance of FUS-YFP aggregates was noted in [PIN+] vs. [pin−] cells (Fig. 2e). After cells were cured of [PIN+] by growth on plates containing guanidine hydrochloride (GuHCl), FUS-associated toxicity was reduced to the levels seen in [pin−] cells (Fig. 2c). Likewise, cytoduction of [PIN+] (transfer of [PIN+] along with donor cytoplasm) into recipient [pin−] cells harboring FUS-YFP, exacerbated FUS toxicity (Fig. 2d). Thus [PIN+] aggravates FUS toxicity.
Sis1 and DNAJB1 Overexpression Respectively Relieves FUS Toxicity in Yeast and Mammalian Cells
We co-overexpressed Sis1 and FUS in [pin−] and [PIN+] yeast cells to compare growth. Excess Sis1 relieved FUS toxicity (Fig. 3a) without changing FUS levels (Fig. 3b). We frequently observed a slightly increased rescue of FUS toxicity by overexpressed Sis1 in [pin−] vs. [PIN+] cells. We observed the same phenomenon for TDP-43 rescue [31]. In both cases we believe the difference is caused by enhanced toxicity in [PIN+] cells, making rescue by Sis1 less efficient. FUS is toxic in mammalian HEK293T cells (Sun et al., 2011) and the mammalian homolog of Sis1, DNAJB1 relieves this toxicity in a MTT cell viability assay (Fig. 3c).
FUS Overexpression Inhibits the Degradation of Cytosolic Misfolded Proteins and Overexpression of Sis1 Moderates this Effect
To ask if FUS aggregates affect clearance of misfolded cytosolic proteins, we monitored the degradation of a reporter protein— CG*-GFP, a GFP-tagged mutant version of the secretory protein carboxypeptidase Y lacking its signal sequence (∆ssCPY*)— in the presence and absence of FUS. Normally, CG*-GFP is rapidly degraded via the ubiquitin proteasome system (UPS), dependent on Hsp40 and Hsp70 chaperones [29]. We measured the stability of CG*-GFP in cells overexpressing control vector or FUS by detecting the CG*-GFP level after adding cycloheximide to inhibit new protein synthesis (Fig. 4). CG*-GFP levels decreased over time after cycloheximide treatment in control cells, while the degradation of CG*-GFP was almost entirely blocked by FUS overexpression. This suggests that aggregates of FUS, like aggregates of TDP-43, interfere with the UPS. In addition, when Sis1 was concurrently overexpressed with FUS, degradation of CG*-GFP returned to about the level seen in the absence of FUS. (Fig. 4).
Discussion
The formation of neuronal protein aggregates is a common characteristic of neurodegenerative diseases, suggesting a similar underlying pathogenic mechanism [33]. However, important differences exist. While many of these aggregates exhibit properties characteristic of amyloids, including detergent-insolubilities, others such as TDP-43 and FUS aggregates remain soluble in detergents [8,26]. Also, some disease proteins contain large, easily-identifiable Q/N-rich regions (e.g. huntingtin), while other disease proteins (e.g. Aβ, PrP and α-synuclein) are capable of aggregating and forming amyloid without a Q/N-rich segment.
Although Q/N-rich regions were not initially identified in FUS and TDP-43, it now appears that small modular Q/N-rich domains in these proteins are involved in their ability to aggregate [32]. In our recent paper [31] and this extra-view, we respectively establish additional similarities between aggregates of TDP-43/FUS and those of huntingtin polyQ/PIN4C. We show that overexpression of TDP-43 or FUS, like that of polyQ or PIN4C, result in several shared downstream events, including: a) inhibition of the UPS that is rescued by simultaneous overexpression of the Hsp40 chaperone, Sis1; b) toxicity that is rescued by Sis1; and c) enhanced toxicity in the presence of the [PIN+] prion.
Proteasomal impairment has been associated with the appearance of protein aggregates in neurodegenerative diseases, and our results support and extend this correlation [2,3]. [PIN+], the aggregated prion form of RNQ1, a protein with a Q/N-rich region, dramatically enhances the aggregation of several Q/N-rich proteins, including polyQ and PIN4C [9,29,30]. Since these proteins have large Q/N-rich aggregating regions, it is likely that [PIN+] cross-seeds their aggregation. There is less similarity between [PIN+] and the small Q/N modules in TDP-43 or FUS. Also, we could not detect [PIN+]-induced enhanced aggregation of TDP-43 or FUS, which already aggregate very efficiently in [pin−] cells. Nonetheless, there could be a subtle cross-seeding effect of [PIN+] leading to the appearance of more toxic TDP-43 and FUS aggregates. Indeed, [PIN+] enhances aggregation of the prion protein HET-S [34], which does not contain a Q/N-rich region, establishing that [PIN+] can act on proteins lacking Q/N-rich domains.
Alternatively, the toxicity of TDP-43 and FUS could be enhanced by [PIN+] because [PIN+] aggregates sequester Sis1 and other essential chaperones [35]. In support of this hypothesis, Sis1 overexpression reduces TDP-43 and FUS toxicity. Possibly the reduced amount of free Sis1 chaperone in [PIN+] cells makes the cells more susceptible to proteasome inhibition mediated by TDP-43 or FUS. Indeed we showed that Sis1 overexpression relieves UPS inhibition caused by TDP-43 or FUS. However, it is important to remember that Sis1 overexpression could enhance proteasome function independently of TDP-43 or FUS.
In summary, our results suggest that the presence of heterologous aggregates in mammalian cells may influence the toxicity of a variety of disease-related proteins. Our work also highlights chaperones and the UPS as key therapeutic targets with broad neuroprotective potential not only in ALS, but also in other neurodegenerative diseases characterized by protein inclusions.
Funding Statement
NIH, Grant to Susan Liebman, R01GM056350 NIH, Grant to Sami Barmada, R01NS097542.
Acknowledgments
We thank E. Craig, U of Wisconsin, for anti-Sis1 antibody. This work was supported by National Institutes of Health grants 5R01GM056350 (to S.W.L.) and R01NS097542 (to S.J.B.).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Cushman M, Johnson BS, King OD, et al.. Prion-like disorders: blurring the divide between transmissibility and infectivity. J Cell Sci. 2010;123:1191–201. doi: 10.1242/jcs.051672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nonaka T, Masuda-Suzukake M, Arai T, et al.. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013;4:124–34. doi: 10.1016/j.celrep.2013.06.007 [DOI] [PubMed] [Google Scholar]
- [3].Nonaka T, Watanabe ST, Iwatsubo T, et al.. Seeded aggregation and toxicity of {alpha}-synuclein and tau: cellular models of neurodegenerative diseases. J Biol Chem. 2010;285:34885–98. doi: 10.1074/jbc.M110.148460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Johnson BS, McCaffery JM, Lindquist S, et al.. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:6439–44. doi: 10.1073/pnas.0802082105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ju S, Tardiff DF, Han H, et al.. A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biol. 2011;9:e1001052. doi: 10.1371/journal.pbio.1001052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Khurana V, Lindquist S. Modelling neurodegeneration in Saccharomyces cerevisiae: why cook with baker's yeast? Nat Rev Neurosci. 2010;11:436–49. doi: 10.1038/nrn2809 [DOI] [PubMed] [Google Scholar]
- [7].Kryndushkin D, Ihrke G, Piermartiri TC, et al.. A yeast model of optineurin proteinopathy reveals a unique aggregation pattern associated with cellular toxicity. Molecular microbiology. 2012;86:1531–47. doi: 10.1111/mmi.12075 [DOI] [PubMed] [Google Scholar]
- [8].Kryndushkin D, Shewmaker F. Modeling ALS and FTLD proteinopathies in yeast: an efficient approach for studying protein aggregation and toxicity. Prion. 2011;5:250–7. doi: 10.4161/pri.17229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Meriin AB, Zhang X, He X, et al.. Huntington toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1. J Cell Biol. 2002;157:997–1004. doi: 10.1083/jcb.200112104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Osherovich LZ, Weissman JS. Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI(+)] prion. Cell. 2001;106:183–94. doi: 10.1016/S0092-8674(01)00440-8 [DOI] [PubMed] [Google Scholar]
- [11].Vitrenko YA, Gracheva EO, Richmond JE, et al.. Visualization of aggregation of the Rnq1 prion domain and cross-seeding interactions with Sup35NM. J Biol Chem. 2007;282:1779–87. doi: 10.1074/jbc.M609269200 [DOI] [PubMed] [Google Scholar]
- [12].Derkatch IL, Uptain SM, Outeiro TF, et al.. Effects of Q/N-rich, polyQ, and non-polyQ amyloids on the de novo formation of the [PSI+] prion in yeast and aggregation of Sup35 in vitro. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:12934–9. doi: 10.1073/pnas.0404968101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Derkatch IL, Bradley ME, Hong JY, et al.. Prions affect the appearance of other prions: the story of [PIN(+)]. Cell. 2001;106:171–82. doi: 10.1016/S0092-8674(01)00427-5 [DOI] [PubMed] [Google Scholar]
- [14].Du Z, Li L. Investigating the interactions of yeast prions: [SWI+], [PSI+], and [PIN+]. Genetics. 2014;197:685–700. doi: 10.1534/genetics.114.163402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bonini NM, Gitler AD. Model organisms reveal insight into human neurodegenerative disease: ataxin-2 intermediate-length polyglutamine expansions are a risk factor for ALS. Journal of molecular neuroscience: MN. 2011;45:676–83. doi: 10.1007/s12031-011-9548-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Treusch S, Hamamichi S, Goodman JL, et al.. Functional links between Abeta toxicity, endocytic trafficking, and Alzheimer's disease risk factors in yeast. Science. 2011;334:1241–5. doi: 10.1126/science.1213210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Couthouis J, Hart MP, Erion R, et al.. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21:2899–911. doi: 10.1093/hmg/dds116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Couthouis J, Hart MP, Shorter J, et al.. A yeast functional screen predicts new candidate ALS disease genes. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:20881–90. doi: 10.1073/pnas.1109434108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Elden AC, Kim HJ, Hart MP, et al.. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–75. doi: 10.1038/nature09320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tardiff DF, Jui NT, Khurana V, et al.. Yeast reveal a “druggable” Rsp5/Nedd4 network that ameliorates alpha-synuclein toxicity in neurons. Science. 2013;342:979–83. doi: 10.1126/science.1245321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Auburger G, Sen NE, Meierhofer D, et al.. Efficient Prevention of Neurodegenerative Diseases by Depletion of Starvation Response Factor Ataxin-2. Trends Neurosci. 2017;40:507–16. doi: 10.1016/j.tins.2017.06.004 [DOI] [PubMed] [Google Scholar]
- [22].Becker LA, Huang B, Bieri G, et al.. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature. 2017;544:367–71. doi: 10.1038/nature22038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gitler AD, Dhillon P, Shorter J. Neurodegenerative disease: models, mechanisms, and a new hope. Dis Models Mech. 2017;10:499–502. doi: 10.1242/dmm.030205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sproviero W, Shatunov A, Stahl D, et al.. ATXN2 trinucleotide repeat length correlates with risk of ALS. Neurobiol Aging. 2017;51:178. e1- e9. doi: 10.1016/j.neurobiolaging.2016.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012;1462:61–80. doi: 10.1016/j.brainres.2012.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Neumann M, Kwong LK, Sampathu DM, et al.. TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: protein misfolding diseases without amyloidosis. Archives of neurology. 2007;64:1388–94. doi: 10.1001/archneur.64.10.1388 [DOI] [PubMed] [Google Scholar]
- [27].Gitler ADaEAC Modulators of TDP-43 Mediated Toxicity and Methods of Use Thereof for Identifying Agents Having Efficacy for the Treatment and Prevention of Proteinopathies. USA, 2012. [Google Scholar]
- [28].Sun Z, Diaz Z, Fang X, et al.. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 2011;9:e1000614. doi: 10.1371/journal.pbio.1000614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Park SH, Kukushkin Y, Gupta R, et al.. PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell. 2013;154:134–45. doi: 10.1016/j.cell.2013.06.003 [DOI] [PubMed] [Google Scholar]
- [30].Yang Z, Stone DE, Liebman SW. Prion promoted phosphorylation of heterologous amyloid is coupled with ubiquitin-proteasome system inhibition and toxicity. Mol Microbiol. 2014; doi: 10.1111/mmi.12716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Park SK, Hong JY, Arslan F, et al.. Overexpression of the essential Sis1 chaperone reduces TDP-43 effects on toxicity and proteolysis. PLoS Genet. 2017;13:e1006805. doi: 10.1371/journal.pgen.1006805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Udan M, Baloh RH. Implications of the prion-related Q/N domains in TDP-43 and FUS. Prion. 2011;5:1–5. doi: 10.4161/pri.5.1.14265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lee SJ, Lim HS, Masliah E, et al.. Protein aggregate spreading in neurodegenerative diseases: problems and perspectives. Neurosci Res. 2011;70:339–48. doi: 10.1016/j.neures.2011.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Taneja V, Maddelein ML, Talarek N, et al.. A non-Q/N-rich prion domain of a foreign prion, [Het-s], can propagate as a prion in yeast. Mol Cell. 2007;27:67–77. doi: 10.1016/j.molcel.2007.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sondheimer N, Lopez N, Craig EA, et al.. The role of Sis1 in the maintenance of the [RNQ+] prion. EMBO J. 2001;20:2435–42. doi: 10.1093/emboj/20.10.2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liebman SW, Chernoff YO. Prions in yeast. Genetics. 2012;191:1041–72. doi: 10.1534/genetics.111.137760 [DOI] [PMC free article] [PubMed] [Google Scholar]