

Abstract
Progress in colloidal synthesis in the last two decades has enabled high-quality semiconductor, plasmonic, and magnetic nanocrystals (NCs). As synthesized, these NCs are usually capped with long-chain apolar ligands. Postsynthetic surface functionalization is required for rendering such NCs colloidally stable in polar media such as water. However, unlike small anionic molecules and polymeric coatings, producing positively charged stable NCs, especially at high ionic strengths, has remained challenging. Here, we present a general approach to achieve aqueously stable cationic NCs using a set of small (<2.5 nm long) positively charged ligands. The applicability of this method is demonstrated for a variety of materials including semiconductor CdSe/CdS core/shell NCs, magnetic Fe@Fe3O4, Fe3O4, and FePt NCs, and three different classes of plasmonic Au NCs including large nanorods. The obtained cationic NCs typically have zeta potential values ranging from +30 to +60 mV and retain colloidal stability for days to months, depending on NC/ligand pair, in several biological buffers at elevated pH and in concentrated salt solutions. This allowed us to demonstrate site-specific staining of cellular structures using fluorescent cationic NCs with several different surface chemistries. Furthermore, colloidal stability of the obtained NCs in the presence of other charged species allowed the assembly of cationic and anionic counterparts driven primarily by electrostatic attraction. With this approach, we prepare highly uniform 3D and 2D binary mixtures of NCs through induced homogeneous aggregation and alternating-charge layer-by-layer deposition, respectively. Such binary mixtures may provide a new route in the engineering of nanocrystalline solids for electronics, thermoelectrics, and photovoltaics.
Introduction
Inorganic nanocrystals (NCs) are important in many fields, including biosensing, drug delivery, magnetic resonance imaging, electronics, and optoelectronics.1−6 Their successful utilization often requires a high degree of monodispersity owing to their unique size-dependent properties. There are many well-developed approaches to produce high quality monodisperse semiconductor, magnetic, plasmonic, and dielectric NCs.7 Most of these employ high temperature reactions in organic solvents in the presence of long-chain apolar molecules that passivate the NC surface (surface capping ligands).8,9 Such ligands impart colloidal stability to NCs in apolar solvents such as toluene or hexane. However, many applications, especially those that are bio-oriented, require aqueous solutions of NCs. Alternative approaches have been developed to generate high-quality, water-soluble NCs for certain systems,7,10−13 i.e., Au and Ag, but they are not suitable for many others, especially covalent compounds such as CdSe semiconductor NCs (also known as quantum dots, QDs) and Fe3O4 NCs, since low reaction temperatures lead to poor crystallinity of such NCs. Furthermore, aqueous synthetic techniques often result in NCs that are very sensitive to the pH and ionic strength of the solution.7 Therefore, many state-of-the-art approaches to high quality aqueous NCs involve two steps: synthesis of hydrophobic NCs followed by functionalization of their surfaces with hydrophilic molecules.
There exist four common postsynthetic strategies for rendering hydrophobic NCs water-soluble. The first technique is to interlock amphiphilic ligands with the long alkyl chains from the original capping layer, forming a hydrophilic bilayer.14−16 This method preserves the native coating, which is often advantageous; for example, it maintains the pristine electronic quality of the QD surface and hence high quantum yields.17 However, this micelle-like structure significantly increases the hydrodynamic diameter of NCs and exhibits several shortcomings due to the dynamic nature of the interlocking bilayer: reduced colloidal stability upon dilution and difficulties with subsequent chemical modification.18 The second approach utilizes ligand cleavage and is only feasible for a limited range of NCs with rather chemically inert surfaces.19−21 The third approach is based on the growth of an inert silica shell around the NCs, through a sol–gel process.22,23 Such SiO2-coated NCs are very stable in aqueous solutions; however, the silica shell is often inhomogeneous and porous, limiting its chemical passivation ability.24 The fourth, perhaps most frequently used, approach is ligand exchange, wherein the original surfactant is displaced by a new incoming molecule.
There are many ligands to choose from for the ligand exchange reaction: dendrimers,25,26 polymers,16,24,27−36 peptides,37,38 inorganic ligands,39−45 and small charged organic molecules.46−49 Among them, bulky polymers, especially polyethylene glycol (PEG) derivatives, are often preferred because they provide NCs better colloidal stability in high ionic strength solutions. High salt concentrations shrink the electric double layer around NCs, destabilizing the colloid. Under these conditions, colloids relying only on electrostatic stabilization tend to flocculate, whereas bulky hydrophilic polymer ligands, which additionally contribute by steric repulsion, prevent aggregation more efficiently.24 However, bulky polymeric substituents substantially increase the hydrodynamic diameter of the NCs.50 Often this is undesirable, for example, in NC-mediated catalysis,51 for further encapsulation into nanocages,52 or when the exact distance between inorganic NC core and outer conjugates is decisive for function (e.g., for Förster energy transfer).53−55 Shorter, ∼1–2.5 nm long, organic molecules do not increase the hydrodynamic diameter of NCs substantially but they are able to provide sufficient steric repulsion for colloidal stabilization.56−59 In this work, we propose several short organic ligands that provide colloidal stability for various classes of NCs in high ionic strength buffer solutions. We focus only on ligands with cationic functionality because of their limited characterization in the literature.46,60−62
Fundamentally, it is easier to achieve stable anionic surface functionalization due to more facile solvation of small cations compared to large anionic counterions.63 In nature, negatively charged surfaces are somewhat more abundant, with clays and cell membranes as typical examples. On the atomic scale, this can be traced to the very essence of coordination chemistry, where metal cations are typically surrounded by shells of anions.
There is a great variety of potential applications for cationic NCs. For instance, the combination of cationic and anionic NCs could enable novel self-assembled hybrid materials with unique optical and chemical properties.64−66 In addition, cationic NCs have been demonstrated to be important in biology, particularly for in vivo applications. For example, cationic NCs have a very different suborgan biodistribution in the kidneys, spleen, and liver, compared to their neutral and anionic counterparts.67 Surface charge also plays an important role in protein adsorption and cellular uptake. It is known that both anionic and cationic NCs delivered in vivo are coated by a corona of adsorbed blood proteins and that the corona composition is directly influenced by the surface chemistry and charge of the NCs.68,69 Cationic NCs also exhibit high uptake by macrophages and other cells; it is thought that the slight negative charge of the cell membrane encourages internalization.70 The affinity of cationic Au NCs for cell membranes has been used recently in X-ray radiation therapy.71 Other biological functions affected by surface charge include blood retention times, cytotoxicity, and immune system activation.70,72−74
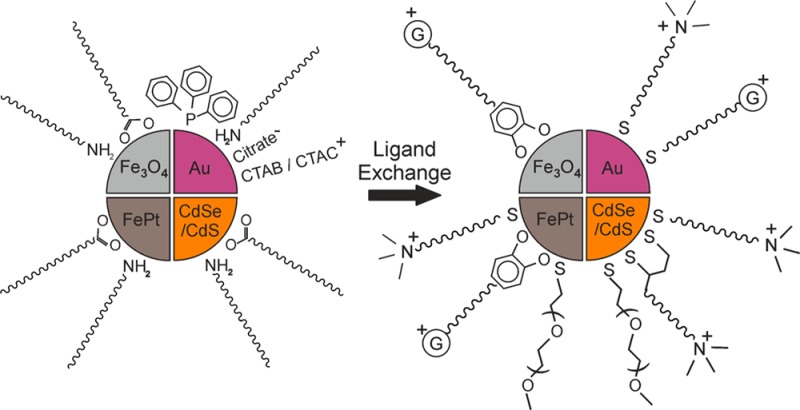
Many of these aforementioned applications necessitate NC stability in polar solvents, biological media, and highly concentrated salt solutions without using bulky polymeric ligands. At the same time, the commercial supply of short organic cationic ligands suitable for NC capping is rather limited and protocols for surface functionalization are less common than for anionic coatings. This motivated us to develop a collection of cationic ligands (Figure 1), three of which are newly designed molecules, which can be attached to the NC surface via ligand exchange reactions. We showcase a strategy for the selection of short cationic ligands suited for specific kinds of NCs (semiconductor CdSe/CdS, magnetic oxide Fe3O4 and alloy FePt, and plasmonic Au of various shapes) or specific initial capping ligands (carboxylates, amines, phosphines, CTAB, and citrate). We present ligand exchange protocols that fully retain the integrity and properties of NCs and ensure long-term colloidal stability. Initial trials using these cationic NCs for three different applications, site-specific cellular staining, layer-by-layer self-assembly, and induced homogeneous aggregation of oppositely charged NCs, demonstrate the high versatility and the broad utility of these cationic surface chemistries.
Figure 1.

Structure of the ligands employed in this study. At neutral pH, the top six ligands are cationic. DA-Arg, H-CALNR-NH2, and DHLA-Bet are newly designed ligands described in the main text. HS-PEG550 (n ≈ 11) was used as a neutral reference ligand or to create binary ligand layer compositions with varied surface charge.
Experimental Section
Materials, Ligands, Nanocrystals, and Buffer Solutions
All chemicals were used as received without further purification. The following chemicals were purchased from Sigma-Aldrich: 1-dodecanethiol (DDT), 2-dimethylaminoethanethiol hydrochloride (DMAETCl), 3-mercaptopropionic acid (MPA), (11-mercaptoundecyl)-N,N,N-trimethylammonium bromide (TMABr), acetone, ethyl acetate, dopamine hydrochloride (DA), formamide, methanol (MeOH), 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), fuming hydrochloric acid (HCl), poly(sodium 4-styrenesulfonate) (PSS, Mw = 70 000) sodium citrate tribasic dihydrate, sulfuric acid (H2SO4), and tetrahydrofuran (THF). Chloroform was purchased from Fisher Chemicals, and hydrogen peroxide (H2O2) and sodium chloride (NaCl) were from Merck. The HS-PEG550 ligand was provided by Creative PEGWorks. Phosphate buffered saline (PBS) was purchased from Life Technologies. Tris(hydroxymethyl)aminomethane hydrochloride (Tris-HCl) was supplied by Fluka Chemicals.
The ligands used in this study are listed in Figure 1. Details regarding their synthesis and characterization are provided in the Supporting Information.
All NCs intended for ligand exchange were synthesized according to previously reported procedures. Details and references can be found in the Supporting Information. All NCs, including cationic NCs with exchanged ligands, were stored at 4 °C.
The following buffer solutions were tested in this work: 1× PBS at pH 7.4, 10 mM HEPES at pH 8, 50 mM Tris-HCl at pH 8, and 50 mM trisodium phosphate (Na3PO4) containing 200 mM NaCl at pH 8. Salt solutions of 50 and 500 mM NaCl were used at neutral pH. The pH measurements were made using a VWR pHenomenal 1000 H pH meter. The pH was adjusted by stock solutions of 1 M HCl and NaOH if necessary.
Ligand Exchange Reactions
Fe@Fe3O4 and Fe3O4 Nanocrystals: Oleic acid (Oleylamine and Oleic Acid) to DA or DA-Arg
. Exchange of the original ligands for DA and DA-Arg was achieved following the procedure reported previously for DA.60 An additional washing step using an excess of a 2:1 chloroform/acetone solution was utilized. The final DA-Arg-capped NCs were easily dissolved in H2O in contrast to DA-capped ones which required a small amount of acid to be solubilized in H2O.
FePt Nanocrystals: Oleylamine and Oleic Acid to DA-Arg and TMABr or HS-PEG550
Exchange of the original hydrophobic ligands for a mixed layer containing a catechol (DA-Arg) and a thiol ligand (HS-PEG550 or TMABr) was achieved using an adapted procedure reported elsewhere.75 1 mg of FePt NCs (∼15 nmol) was precipitated using an equal amount of ethanol, followed by redispersion in 0.4 mL of a ligand solution. The latter was composed of 7:1 THF/H2O and contained 1.5 mg of DA-Arg and either 2 mg of TMABr or 2 μL of HS-PEG550. The reaction was stirred for 1 day at 35 °C, followed by precipitation with 10:3 acteone/ethyl acetate. The resulting pellet was easily solubilized in H2O.
CdSe/CdS Core/Shell Quantum Dots: Oleylamine and Oleic Acid to TMABr or DMAETCl
1 mg (∼0.8 nmol) of hydrophobic QDs was dissolved in 100 μL of chloroform containing 1 mg of TMABr. The mixture was shaken for 1 h at room temperature, after which the turbid solution was centrifuged. The pellet was suspended in 100 μL of methanol. The resulting solution was washed by addition of 300 μL of ethyl acetate to induce precipitation of TMABr-capped QDs. After centrifugation, the pellet was easily redispersed in H2O. The same procedure was used for DMAETCl except a mixture of 1:6 methanol/chloroform was used as a reaction solvent.
CdSe/CdS Core/Shell Quantum Dots: Oleylamine and Oleic Acid to HS-PEG550 (HS-PEG550 and TMABr)
Cap exchange of oleic acid and oleylamine for HS-PEG550 was achieved by following a published procedure for heavier analogues.27,35 A mixed layer containing HS-PEG550 and TMABr was obtained using a modified version of the same protocol. Briefly, 1 mg (∼0.8 nmol) of the original hydrophobic QDs was precipitated with an equal amount of ethanol and resuspended in 100 μL of methanol containing 0.75 μL of HS-PEG550 and 0.5 mg of TMABr. The reaction was carried out at 60 °C for 2.5 h resulting in a clear orange solution. The QDs were precipitated using 10:1 hexane/chloroform and redispersed in H2O.
CdSe/CdS Core/Shell Quantum Dots: Oleylamine and Oleic Acid to DHLA-Bet
A variety of ligand exchange conditions were investigated to produce QDs capped by DHLA-Bet, varying the temperature, reaction time, solvent, and pH. The most stable samples were obtained by the two following methods. 1 mg (∼0.8 nmol) of the original hydrophobic QDs was dissolved in 100 μL of hexane. 100 μL of H2O containing 1.5 mg of DHLA-Bet was added to this solution, followed by slow addition of 300 μL of THF. The reaction was shaken for 2 h resulting in QD transfer into the polar solvent phase. This solution was washed 3 times with hexane. The DHLA-Bet-capped QDs were precipitated from solution using a mixture of 1:1 THF/hexane and redispersed in H2O. Alternatively, ligand exchange could be achieved by redispersing the 1 mg pellet of QDs in a methanol solution containing 1 mg of DHLA-Bet and stirring for 2 h at room temperature, followed by the same washing procedure.
Au Nanocrystals: Oleylamine (Oleylamine and TOPO) to TMABr
The desired amount of hydrophobic Au NCs was redispersed in 100 μL of methanol containing TMABr and shaken for 1 h to form a turbid solution. The amount of ligand was carefully adjusted for the total surface area of the Au NCs. For example, with 0.5 mg of Au NCs of diameter 3.5 nm (∼1.7 nmol), 1.68 mg of TMABr was added (corresponds to ∼30 molecules per 1 nm2 of NC surface). The obtained turbid solution was mixed with 300 μL of ethyl acetate and centrifuged. The pellet was redispersed in H2O.
Au Nanocrystals: Oleylamine to H-CALNR-NH2
1 mg (∼3.4 nmol) of Au NCs was dissolved in 100 μL of hexane. A ligand solution of 100 μL of H2O, 1 mg of H-CALNR-NH2, and 2 μL of 100 mM HCl was added, forming a biphasic mixture. 500 μL of THF was slowly added to the solution inducing phase transfer after 20 min of stirring at room temperature. The aqueous layer was washed 3 times with hexane, followed by precipitating the Au NCs with chloroform and acetone. The resulting pellet was redispersed in H2O.
Au Nanocrystals: Triphenylphosphine to TMABr
Small 1.5 nm Au NCs capped by triphenylphosphine were functionalized with TMABr directly after synthesis according to a published procedure but with an additional washing step.61 Briefly, 3 mg (∼130 nmol) of Au NCs in 450 μL of DCM was combined with 3 mg of TMABr in 450 μL of H2O. The biphasic mixture was stirred overnight, resulting in the purple color transferring into the aqueous layer. The Au NCs were washed 3 times with DCM, precipitated by 5:1 THF/H2O, and redispersed in H2O.
Spherical Au Nanocrystals and Nanorods: CTAB/CTAC to TMABr
Round-trip ligand exchange (from hydrophilic to hydrophobic and then back to hydrophilic) was used to functionalize water-soluble Au NCs capped by a double layer of CTAB/CTAC with TMABr.76 In a typical reaction, 1 mg (∼0.02 nmol) of Au nanorods (NRs) in 300 μL of H2O was combined with 600 μL of 10 mM DDT in chloroform. 600 μL of acetone was slowly added causing the Au NRs to transfer to the organic layer. Small additional amounts of acetone or ethanol were added to help induce phase transfer for reactions where the particles were stuck at the phase boundary. The aqueous layer was discarded, and the DDT-capped Au NRs were removed from solution through centrifugation. For smaller Au NCs prepared by this method, acetone was added to aid precipitation. The resulting pellet was washed twice with 5:1 ethanol/chloroform. In a plastic centrifuge tube, the Au NRs were redispersed in 1 mL of chloroform containing 5 mg of TMABr. The solution was stirred at 50 °C for 20 min resulting in precipitation of the Au NRs. The precipitate was extracted into H2O and washed three times with chloroform, followed by precipitation with a mixture of ethanol and ethyl acetate and redispersion in H2O. This reaction should be carried out in plastic vessel because the resulting particles are highly charged and stick irreversibly to glass.77
Au Nanocrystals: CTAB/CTAC to Citrate
The CTAB/CTAC double layer could be converted to citrate ions by following a published procedure.78 Briefly, approximately 10 mg (∼0.2 nmol) of Au NRs coated with CTAB was diluted in 40 mL of 0.15% wt. PSS solution and incubated for 1 h. After removal of residual CTAB and excess PSS by centrifugation, this procedure was repeated twice more. The PSS-capped Au NRs were centrifuged and redispersed in 20 mL of 5 mM sodium citrate tribasic dihydrate and left to incubate for 12 h. This was repeated once more, resulting in citrate-capped Au NRs. This procedure was also extended to CTAC-capped Au nanospheres.
Au Nanocrystals: Citrate to TMABr
The citrate to TMABr ligand exchange was achieved using DDT as an intermediary ligand. In a typical reaction, 1 mg (∼0.02 nmol) of citrate-capped Au NRs in 1 mL of H2O was added to 1 mL of 10 mM DDT in chloroform. Acetone was then added until the solution became a single phase (approximately 5 mL). The solution was shaken for 30 s and centrifuged to remove the Au NRs capped by DDT. The resulting pellet was washed twice with 5:1 ethanol/chloroform before dispersion in pure chloroform. Functionalization with TMABr was carried out in the same manner as above. The same procedure was used for smaller Au NCs synthesized directly by reduction with sodium citrate using larger initial volumes of H2O and acetone.
Layer-by-Layer Deposition
Glass substrates were rendered hydrophilic and negatively charged by treatment in boiling piranha solution (3:1 of 98% H2SO4 and 30% H2O2) for 1 h. Caution: Piranha solution is highly corrosive and reactive toward organic compounds and must be handled with care. After washing the glass substrates with H2O, they were placed in a methanol solution of positively charged core/shell QD-TMABr for 1 min. The substrates were then washed twice by methanol and dried under a flow of air. The deposition procedure was then repeated using a formamide solution of negatively charged core/shell QD–S2– which was prepared by a method published elsewhere.39 Up to 5 positively and negatively charged alternating layers were deposited onto the substrates in this manner, measuring photoluminesence after each step. Core/shell QD concentrations were approximately 5 mg/mL.
Cell Imaging
HeLa cells were seeded the day before the imaging experiment in 8-well chambers and grown in DMEM Glutamax media containing 10% fetal bovine serum. Cells were fixed prior to QD incubation with 3.7% paraformaldehyde and permeabilized with 0.25% Triton X-100. The CdSe/CdS QDs functionalized with TMABr, HS-PEG550, or a mixed layer of the same two ligands were diluted in PBS, pH 7.4, to a final concentration of 10 nM. Cells were incubated for 10 or 30 min, followed by two washings with PBS. The chambers were removed, and the cells were mounted with Mowiol. Imaging was performed with a Leica DMI6000B epifluorescence microscope with 40× magnification using a DIC and a Cy5 channel operated with excitation and emission wavelengths ranging from 590 to 650 nm and from 662 to 738 nm, respectively.
Characterization
Absorbance
The UV–vis absorbance spectra were measured on a Jasco V-670 UV–vis–NIR spectrophotometer.
Dynamic Light Scattering (DLS)
The hydrodynamic diameter of the NCs was determined from a number distribution using Zetasizer Nano-ZS from Malvern Instruments. Solvents and buffers were filtered through a 0.45 μm filter before use. Values presented are averaged from either three or five measurements (as stated).
Zeta Potential Measurements
Zeta potentials were derived from electrophoretic mobility measurements performed on a Zetasizer Nano-ZS (Malvern Insturments). Three series of 100 measurement cycles each were performed for each sample in order to check sample stability. Results of the electrophoretic mobility measurements were converted to zeta potentials using the Hückel approximation. Stock solutions of 1 M HCl and 1 M NaOH were used to adjust the pH to the desired value.
Transmission Electron Microscopy (TEM)
The NC images were recorded using a JEOL JEM 2200 fs microscope operated at 200 kV. TEM grids were first immersed in toluene for 20 s to remove formvar protection. NCs dissolved in apolar solvents were directly drop-casted on prewashed grids. TEM grids were hydrophilized by short plasma treatment prior to deposition of NCs dissolved in polar solvents.
Scanning Electron Microscopy (SEM)
Silicon substrates were sonicated in isopropanol for 15 min followed by the same deposition treatment as described earlier. Scanning electron microscopy (SEM) measurements were carried out using a Nova Nano SEM 230 microscope.
Atomic Force Microscopy (AFM)
The AFM measurements were conducted with a Bruker Dimension Icon 3 in Peak Force Tapping Mode. Horizontal scars correction was applied to the images. All images were leveled using a plane with 3 pixels.
Results and Discussion
Exchange of ligands on the NC surface requires high chemical affinity of the anchoring group of the incoming ligand. Thiol anchoring groups were employed for NCs with soft surface cations, e.g., Au, Cd, and Pt, whereas catechols were utilized for Fe-based NCs with harder cations (Figure 2). Aqueous colloidal solutions of all tested NC samples were generally stable on the time scale of weeks to months. Solubility in several other polar solvents was tested as well (Table S1). Table 1 and Figure 3 summarize the various NC and ligand combinations with typical zeta potentials and hydrodynamic diameters in aqueous solutions. Figure 4 shows illustrative TEM images of a variety of the NC systems before and after ligand exchange. Figure S2 shows FTIR spectra of representative NC systems that confirm ligand exchange.
Figure 2.

Cartoon outlining the ligand exchange reactions and buffer stability conferred by the new ligands. The original surfactants are shown on the left and the incoming cationic ligands in the center. The positively charged “G” in the center image represents the distal guanidinium group. The colloidal stability of Fe@Fe3O4–DA-Arg NCs (top right) and CdSe/CdS–TMABr QDs (bottom right) in different buffer and salt solutions is provided as an example. The QDs are shown under UV irradiation. The solutions are (1) PBS, (2) 10 mM HEPES, (3) 50 mM Tris-HCl, (4) 500 mM NaCl, (5) 50 mM NaCl, and (6) 50 mM Na3PO4 containing 200 mM NaCl.
Table 1. Summary of the Water-Soluble Cationic NCs and Corresponding Ligands Used in This Study.
| material | original surfactanta | diameter by TEMb (nm) | incoming ligand | zeta potentialc (mV) | hydrodynamic diameterd (nm) |
|---|---|---|---|---|---|
| Fe@Fe3O4 | oleic acid + oleylamine | 5.0 | DA | +33.4 | 5.8 |
| Fe@Fe3O4 | oleic acid + oleylamine | 5.0 | DA-Arg | +37.7 | 6.5 |
| Fe3O4 | oleic acid | 18.0 | DA-Arg | +37.0 | 19.7 |
| FePt | oleic acid + oleylamine | 3.6 | DA-Arg + HS-PEG550 | +18.9 | 9.0 |
| FePt | oleic acid + oleylamine | 3.6 | DA-Arg + TMABr | +25.8 | 8.3 |
| CdSe/CdS | oleic acid + oleylamine | 8.8 | DMAETCl | +35.7 | 9.8 |
| CdSe/CdS | oleic acid + oleylamine | 8.8 | TMABr | +44.6 | 10.2 |
| CdSe/CdS | oleic acid + oleylamine | 8.8 | DHLA-Bet | +45.4 | 14.7 |
| CdSe/CdS | oleic acid + oleylamine | 8.8 | HS-PEG550 | +3.0 | 17.3 |
| CdSe/CdS | oleic acid + oleylamine | 8.8 | HS-PEG550 + TMABr | +33.5 | 15.0 |
| Au | triphenylphosphine | 1.5 | TMABr | +35.8 | |
| Au | oleylamine | 3.5 | TMABr | +31.4 | 7.1 |
| Au | oleylamine | 3.5 | H-CALNR-NH2 | +30.8 | 5.4 |
| Au | oleylamine + TOPO | 11.9 | TMABr | +52.9 | 12.1 |
| Au | citrate → DDT | 4.1 | TMABr | +16.1 | 4.4 |
| Au | CTAC → DDT | 7.3 | TMABr | +41.8 | 7.6 |
| Au | CTAC → DDT | 19.5 | TMABr | +34.8 | 20.4 |
| Au | CTAC → PSS → citrate → DDT | 19.5 | TMABr | +17.2 | 21.0 |
| Au | CTAB → DDT | 13.0 by 49.0 | TMABr | +40.9 | |
| Au | CTAB → PSS → citrate → DDT | 13.0 by 49.0 | TMABr | +44.7 |
Ligands shown in the second column after the “→” symbol indicate preliminary ligand exchange steps.
The diameter was determined by TEM following synthesis with the original surfactant.
The zeta potential values are averaged from three trials of 100 runs.
The hydrodynamic diameters of NCs after ligand exchange are reported as averages from five trials using a number distribution.
Figure 3.
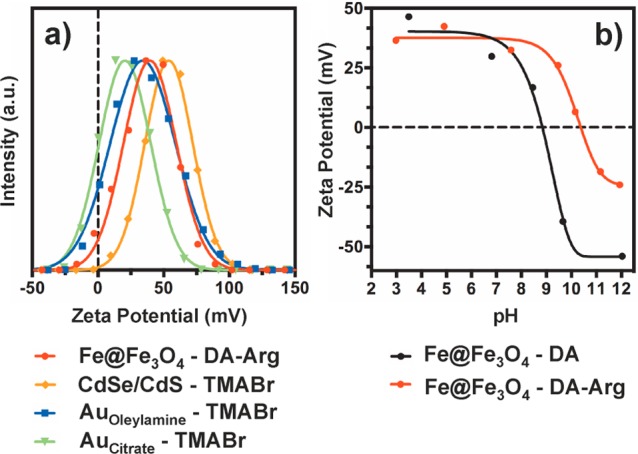
(a) Zeta potential measurements for various cationic NCs in water at neutral pH and (b) zeta potential values plotted against pH for the Fe@Fe3O4 NCs functionalized with the two catechol ligands. The original surfactant for the Au samples is indicated by the subscript.
Figure 4.

TEM images of the NCs used in this study before and after functionalization with the cationic ligands depicted in Figure 1. The first and third columns show the starting NCs capped with hydrophobic ligands in apolar solvents, whereas the second and fourth columns present cationic NCs in aqueous solution. NCs shown in (k) were originally capped with CTAC which was exchanged to DDT before functionalization with the cationic ligand. All scale bars are 20 nm.
Fe@Fe3O4 and Fe3O4 Nanocrystals
Hard Fe3+ sites require hard Lewis bases as incoming ligands, typically containing an oxygen-based anchoring group.40 Catechols represent ideal incoming ligands for Fe3O4 NCs because the two hydroxyl groups allow for strong bidentate attachment to the surface.79,80 Dopamine (DA) has been extensively studied as a water solubilizing ligand for Fe@Fe3O4 NCs.81−83 The resulting particles are stable for months in aqueous solution but precipitate at elevated pH because the NCs fail to retain their positive charge due to deprotonation of the primary amine of DA (pKa1 = 9.05), HO– adsorption on the surface of the NC, and deprotonation of the pre-existing surface Fe–OH groups (Figure 3b).84−86 However, many potential applications of aqueous Fe@Fe3O4 NCs require high colloidal stability of NCs in high ionic strength buffer solutions. This motivated us to design a new ligand, DA-Arg, which would remain positively charged at high pH values. The new ligand linked the amino acid arginine to DA, which was retained as an anchor, via an amide bond (Figure 1). This structure shifted the isoelectric point of the resulting NCs to pH ∼ 10.5 due to the higher pKa of the guanidinium side chain (12.10)87 (Figure 3b) and improved their colloidal stability in high ionic strength solutions due to the greater steric demands of DA-Arg compared to DA. The ligand exchange procedure remained almost the same as for DA except that acid was no longer required to resolubilize the particles in H2O. TEM images show that the resulting NCs retain their size and shape and do not aggregate after ligand exchange (Figure 4a,b).
FePt Nanocrystals
The DA-Arg ligand was also exploited to stabilize FePt alloy NCs. This bimetallic alloy has the ability to simultaneously bind two types of ligands: catechols bind at the hard acidic Fe sites and thiols, at the soft Pt sites.75 Therefore, DA-Arg was tested in conjunction with either TMABr or HS-PEG550. As expected, DA-Arg/TMABr-capped NCs have slightly higher zeta potentials than samples capped with DA-Arg/HS-PEG550 (Table 1). Despite their relatively low zeta potentials, neither sample showed signs of aggregation in DLS experiments (Table 1), and both were stable in aqueous solution for months. Figure 4e,f shows the starting FePt NCs and the sample functionalized with a DA-Arg/HS-PEG550 mixed ligand layer. Comparison of these TEM images confirms that the NCs preserve their shape and size throughout the ligand exchange procedure. Attempts to functionalize FePt NCs with HS-PEG550 alone did not result in aqueously stable NCs, as previously reported.75 In contrast, and somewhat surprisingly, a monolayer composed solely of DA-Arg solubilized the particles in water with a zeta potential of +21.7 mV; however, this sample was less stable in salt solutions than NCs possessing binary ligand layers.
CdSe/CdS Core/Shell Quantum Dots
Hydrophobic core/shell QDs were rendered cationic using one of three thiol molecules: DMAETCl, TMABr, or DHLA-Bet. Each ligand readily produced water-soluble samples with zeta potentials of about +40 mV (Table 1). Notably, ligand exchange with the strongly amphiphilic TMABr molecule has to be performed in apolar media, e.g., chloroform, where TMABr forms inverse micelles with the anchor thiol groups directed outward. Though ligand exchange in polar solvents is possible as well, it results in NCs with lower surface charge and only short-term colloidal stability. In contrast, QDs capped with TMABr that were prepared in chloroform were stable for approximately one month or longer and had zeta potentials higher than +40 mV. The resulting QDs preserved their bright photoluminescence with a quantum yield of 46% in water compared to 52% prior to ligand exchange.
Motivated to improve colloidal stability further, we synthesized a bidentate TMABr analogue, hereafter called DHLA-Bet (Figure 1). Most of the conditions tested for ligand exchange gave water-soluble QDs that had positive zeta potentials and did not aggregate according to DLS (Table 1). However, the aqueous solutions of DHLA-Bet-capped QDs were colloidally stable for a few days only when kept at room temperature. In polar organic solvents, these same cationic CdSe/CdS–DHLA-Bet QDs were stable for months (Table S1). Analogous behavior was observed for Au–DHLA-Bet NCs. We assume that the weak colloidal stability of NCs capped by DHLA-Bet is a result of noncompact capping by the ligand, facilitating diffusion of various ionic species to the surface of the NCs. This effect may originate from the larger footprint of DHLA-Bet relative to a single thiol and from the less hydrophobic nature of the DHLA-Bet side chain compared to TMABr. DHLA-Bet-capped QDs were not stable in any buffer or salt solution tested, so further trials were abandoned.
Au Nanocrystals
The wide range of reported synthetic methods for producing Au NCs allowed different surface chemistries and particle sizes to be investigated. The most commonly used surface chemistries for Au NCs can be divided into three classes: (i) hydrophobic NCs capped with bulky amines, phosphines, or TOPO (typically spherical NCs of 1.5–12 nm in size); (ii) water-soluble Au NCs capped by a double layer of CTAB or CTAC (typically larger NCs, 5–30 nm, of various shapes, e.g., spheres and nanorods); (iii) water-soluble Au NCs capped with citrate anions (synthesis results in rather small, 4–12 nm, spherical NCs). We found that all three classes of Au NCs can be rendered cationic and stable in aqueous solutions, though the different surface chemistries of the starting NCs necessitate different ligand exchange strategies for functionalization.
The first class of Au NCs, hydrophobic particles, could be rendered cationic through direct ligand exchange with either TMABr or H-CALNR-NH2. The former ligand was attached simply by NC redispersion in a methanol solution of TMABr followed by NC washing. Zeta potentials obtained for this class of particles typically ranged from +30 to +60 mV (Table 1). A similar biphasic ligand exchange strategy was extended to small 1.5 nm Au NCs capped by triphenylphosphine and resulted in highly stable aqueous Au NCs (Table 1, Figure 4c,d). The peptide ligand H-CALNR-NH2 was chosen on the basis of a previous investigation into optimal peptide sequences for enhancing the aqueous stability of Au NCs.88 Although earlier attempts to utilize the H-CALNR-OH sequence as a ligand were unsuccessful for citrate-capped Au NCs and led to aggregation,88 we further optimized the ligand structure by replacing the terminal carboxylic acid with an amide in order to avoid electrostatically driven aggregation of oppositely charged NCs. The new ligand allowed us to produce Au NCs that were stable in aqueous solution and showed no signs of aggregation (Table 1, Figure 4h). We found that successful ligand exchange reaction with H-CALNR-NH2 required a biphasic approach under acidic conditions. The same conditions also enabled production of H-CALNR-NH2-capped QDs.
Larger Au NCs and anisotropic Au NRs are usually produced in water and capped with a double layer of CTAB/CTAC (the second class of Au NCs discussed above). Complete exchange of this double layer with chemically anchored ligands is not trivial because CTAB/CTAC molecules are soluble in both polar and apolar solvents and their removal from the NC surface leads to immediate NC aggregation. A round-trip ligand exchange protocol has emerged to produce water-soluble, CTAB/CTAC-free Au NCs.76 In this approach, NCs are first transferred to an organic phase by CTAB/CTAC exchange with a hydrophobic molecule, such as 1-dodecanethiol (DDT). The NCs can be subsequently washed to remove most of the remaining CTAB/CTAC. Then, a second ligand exchange step transfers the NCs back to the aqueous phase. We adapted this approach for three different sizes of Au NCs: 7.3 and 19.5 nm spheres and larger NRs. All samples retain their morphology throughout ligand exchange (Figure 4k,l). We note that steric colloidal stabilization provided by DDT only prevents NC flocculation but is not capable of dispersing heavy Au NCs in apolar solvents because of weak ligand-to-solvent interactions. As a result, the 19.5 nm Au NCs and Au NRs stabilized by DDT gradually settle in solution. However, any colloidal instability was completely alleviated after the second ligand exchange step when NCs were rendered cationic and dissolved in water, as evidenced by TEM, zeta potential, and the characteristic pink color of Au NC colloidal solutions (Table 1, Figures 4k,l and 6b). TMABr-capped Au NCs exhibited zeta potential values of +40 mV and upward and remained stable in water for months.
Figure 6.
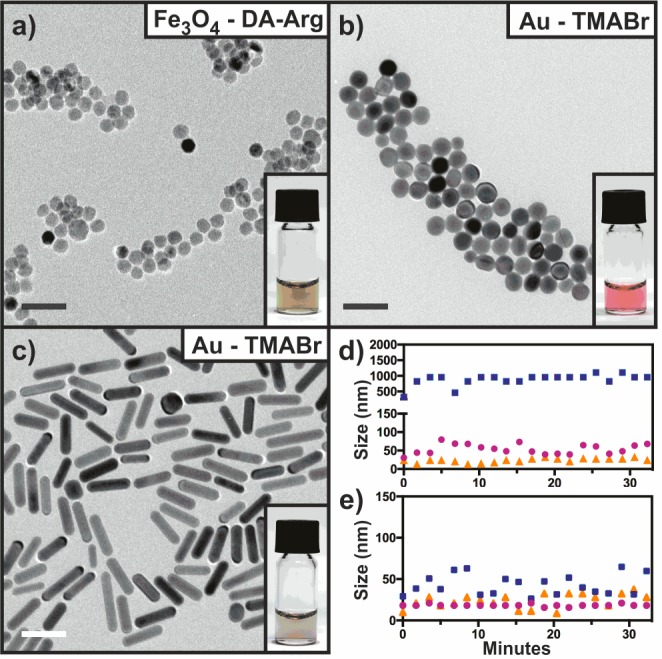
TEM images of large cationic NCs dispersed in H2O. (a) 18.0 nm Fe3O4 NCs with DA-Arg. (b) 19.5 nm Au NCs functionalized with TMABr via the citrate pathway. (c) 49.0 by 13.0 nm Au nanorods functionalized with TMABr via the citrate pathway. All scale bars are 50 nm. Graphs (d) and (e) present aggregation profiles of NCs from (a) and (b), respectively. The blue, purple, and orange data points correspond to 50 mM Tris-HCl, 10 mM HEPES, and 50 mM NaCl, respectively.
The two-step ligand exchange strategy employed here transferred the CTAB/CTAC-coated Au NCs first from water to chloroform and then back to water. The first step of ligand exchange, CTAB/CTAC-to-DDT, and subsequent purification significantly reduce the CTAB/CTAC concentration. However, thorough purification of poorly stable, large DDT-capped Au NCs is complicated, and residual amounts of CTAB/CTAC have been reported to persist after ligand exchange.78,89 Even submicromolar amounts of CTAB/CTAC are cytotoxic and can be undesirable for further processing.90 Therefore, we decided to introduce intermediary ligand exchange steps using poly(sodium 4-styrenesulfonate) (PSS) and citrate anions to eliminate any residual CTAB/CTAC. It has been shown previously that such exchange can reduce CTAB/CTAC concentrations to levels undetectable by X-ray photoelectron spectroscopy (XPS),78 but conversion of Au NCs bearing an anionic citrate capping layer to a cationic ligand shell is not trivial due to electrostatically induced aggregation. We found that the modified ligand exchange protocol overcame this problem and enabled efficient replacement of the citrate groups with suitable cations. Comparisons of samples directly functionalized in the two-step CTAB/CTAC → DDT → TMABr process and the same NCs produced by the CTAB/CTAC → PSS → citrate → DDT → TMABr process showed that both methods yielded stable aqueous solutions of cationic Au NCs, though the second method typically resulted in NCs with slightly lower zeta potentials (Table 1). The latter protocol was also successfully applied to smaller 4.1 nm Au NCs produced directly by reduction with sodium citrate. Thus, the ligand exchange strategies developed here can be used to produce cationic versions of all three classes of Au NCs.
Colloidal Stability in Biological Buffers and High Ionic Strength Solutions
The colloidal stability of the cationic NCs presented above was investigated in six biologically relevant solutions: PBS at pH 7.4, 10 mM HEPES at pH 8, 50 mM Tris-HCl at pH 8, 50 mM NaCl, 500 mM NaCl, and Na3PO4 containing 200 mM NaCl at pH 8.
Stability of Cationic Fe@Fe3O4 and FePt Nanocrystals
Cationic Fe@Fe3O4–DA-Arg and FePt–DA-Arg/thiol NCs were stable in Tris-HCl and HEPES buffers and the 50 mM NaCl solution for up to several weeks. In contrast, Fe@Fe3O4 NCs capped with DA instead of DA-Arg were not stable in any of the buffered solutions, likely due to deprotonation of the ammonium groups at high pH. DA-Arg-capped Fe@Fe3O4 and FePt NCs were not stable in solutions containing phosphate ions, probably because phosphate anions can compete for hard Fe3+ sites.40 No noticeable difference in buffer stability was found for the FePt NCs capped with DA-Arg and either TMABr or HS-PEG550.
Stability of Cationic CdSe/CdS Core/Shell Quantum Dots
The same buffer and salt solutions were tested with core/shell QDs functionalized with DMAETCl, TMABr, HS-PEG550, and a mixed layer of TMABr and HS-PEG550, All samples, with the notable exception of DMAETCl-capped QDs, exhibited significant colloidal stability in the buffer and salt solutions (Figure 5c,d). Typically, they were stable for approximately 2 weeks. The lower stability of DMAETCl-capped QDs can be attributed to the relatively short chain length of the ligand and the comparatively low pKa of tertiary amines. We note that TMABr-capped QDs have notably smaller hydrodynamic diameters than QDs coated with either HS-PEG550 or a mixed layer of TMABr and HS-PEG550 (Table 1, Figure 5d) but nevertheless displayed the same high stability in all buffer and salt solutions tested.
Figure 5.

Colloidal stability of cationic NC dispersions in PBS, Tris-HCl, HEPES, and salt solutions: (a–h) the evolution of the hydrodynamic diameter measured by DLS for the corresponding NC–ligand pairs; (i) the evolution of the characteristic plasmonic absorbance at λabs= 400 nm for the small 1.5 nm Au NCs. Changes in the absorbance were used to investigate NC stability because of the insufficient accuracy of DLS for such small sizes. The original surfactant of the Au NCs is indicated by the subscript (TPP = triphenylphosphine). Fe@Fe3O4 and FePt NCs were not stable in phosphate-based buffers, and therefore, the corresponding graphs are not included in (a) and (b).
Stability of Cationic Au Nanocrystals
Although the cationic Au NCs studied here exhibited substantial buffer stability, some subtle differences were noted depending on the original surfactant and incoming ligand NCs that were initially hydrophobic and rendered cationic with TMABr showed the best stability in buffer and high ionic strength solutions (Figure 5e). Even large particles with diameters up to 11.9 nm were stable for at least six months as evidenced by the lack of precipitation and color change. In contrast, Au NCs originally capped by citrate or CTAC and functionalized with TMABr were not stable in Tris-HCl buffer or concentrated NaCl solutions, though they were stable for several months in PBS, HEPES, and 50 mM NaCl as indicated by DLS, color, and the absence of precipitation (Figure 5f,g). Cationic Au NCs produced from citrate or CTAC-capped NCs tended to have lower zeta potentials compared to those produced from oleylamine-capped Au NCs, which may explain the observed differences in stability.
Au NCs functionalized with the H-CALNR-NH2 peptide were stable in the two organic buffers and in 50 mM NaCl for several weeks and at least for 1 week in 500 mM NaCl (Figure 5h). However, these peptide-capped Au NCs were not stable in phosphate-containing buffers.
Aqueous and Buffer Stability of Large Cationic Nanocrystals
The cationic ligands investigated in this study were also investigated with larger Fe3O4, Au NCs, and Au NRs. It is notable that even such large NCs were colloidally stable in water for months. Colloidal stability in buffer and salt solutions was lower than that of the smaller NCs, but some stability was still observed in certain cases.
Water-soluble, 18.0 nm Fe3O4 NCs functionalized with DA-Arg do not aggregate as indicated by TEM (Figure 6a). They are stable in aqueous solution for at least six months and have zeta potentials of ca. +35 ± 3 mV, similar to the smaller NCs. Colloidal stability was observed in 10 mM HEPES buffer and 50 mM NaCl solution (Figure 6d), as well, though this stability was only short-term (hour scale).
All large cationic and spherical Au NCs and Au NRs appeared to be stable in aqueous solution for months as indicated by their deep cherry pink color and the absence of aggregates in TEM images (Figures 6b,c). They were also stable for several hours in 50 mM NaCl and the two organic buffers (Figure 6e).
Potential Applications of Cationic Nanocrystals
We envision two directions for practical applications of cationic NCs. First, electrostatic interactions with anionic NCs might be exploited to engineer solid-state devices where each component (cationic and anionic) has a desired spatial arrangement and may display a different functional property (semiconductor, plasmonic, or magnetic). The second opportunity for cationic NCs is in cellular imaging, exploiting the differential affinity of these NCs for certain negatively charged organelles and membranes. Below, we describe preliminary results illustrating these directions.
3D-Uniform Mixtures of Nanocrystals by Induced Homogenous Aggregation
Cationic and anionic NCs can be colloidally stable in the same solvents. Mixing of such solutions leads to uncontrolled or partially controlled aggregation, or self-assembly into organized structures, driven primarily by electrostatic attraction, as it has been shown previously for mixtures of metallic Au and Ag NCs.65,91 Such self-assembly affords composite materials with highly uniform mixtures of NCs. We show here that induced homogeneous aggregation can be extended to plasmonic, semiconductor, and magnetic NCs in desired particle number ratios. As one example, positively charged 19.5 nm Au–TMABr NCs and negatively charged 8.8 nm CdSe/CdS–MPA QDs were mixed in aqueous solution and incubated for approximately 10 min. The resulting structures have a strikingly uniform distribution, with each larger Au NC surrounded by a halo of QDs (Figures 7a and S3). Analogous structures composed of Au and Fe3O4 NCs were also formed (Figure S4). Such halo type structures can be formed only when one component (either cationic or anionic NCs) is in excess. Mixing similar amounts of the two components results in NC precipitation. However, we found that the NC distribution in settled aggregates and in the supernatant of unsettled dispersions is still very uniform. Therefore, various ratios of cationic and anionic NCs can be used for induced homogeneous aggregation. This property can be exploited to develop novel solid nanocrystalline materials for applications where mixtures of different NCs are desirable, but highly uniform distributions are required, such as in thermoelectrics,92,93 or when NCs are used as dopants in nanostructured assemblies.94
Figure 7.

Three applications of cationic NCs are presented here. (a) Induced homogenous aggregation: mixing positively and negatively charged NCs in optimized ratios leads to homogeneous flower-like structures. The main TEM scale bar is 50 nm, and the inset scale bar is 25 nm. (b) Alternating charge layer-by-layer deposition: the peak photoluminescence intensity of the glass substrates progressively increases after each coating (5 layers total). The inset shows a glass substrate containing 3 layers under UV light. (c) Cellular imaging in DIC and fluorescence modes: HeLa cells incubated with CdSe/CdS–TMABr for 30 min (left) and CdSe/CdS–TMABr and HS-PEG550 for 10 min (right). Scale bars are 50 μm.
2D-Uniform Layered Mixtures of Nanocrystals by Alternating Charge Layer-by-Layer Deposition
Thin films composed of oppositely charged NCs can be created through sequential deposition driven primarily by electrostatic attraction. High morphological quality in such films is guaranteed by two factors: (i) charge alteration enabling strong electrostatic attraction between NCs and uniform coverage of the incoming layer; (ii) mild conditions without the need for additional treatment, which is often rather destructive,95−97 or extra passivating layers of polymer.39 To illustrate this approach, we used cationic CdSe/CdS–TMABr QDs and anionic CdSe/CdS–S2– QDs to create highly luminescent layer-by-layer thin films deposited on glass substrates (Figure 7b). Scanning electron microscopy (SEM) revealed the films to be highly homogeneous and crack-free, even at the highest magnification tested (5 μm, Figure S5). Atomic force microscopy (AFM) confirmed that the films became progressively thicker with each additional layer, limited only by the diameter of the QDs (Figure S6). The emission profile of the films appeared unchanged compared to the corresponding colloidal dispersion although a redshift of approximately 2 nm was observed per added layer. We believe this approach could be used to create films with many more layers than tested here.
Cellular Imaging with Cationic Nanocrystals
Employing cationic NCs as cellular staining agents offers the opportunity to specifically target negatively charged cellular structures. For example, earlier work demonstrated that cationic NCs bind to lipid bilayers of cells due to the negative membrane potential and the negatively charged surface moieties.98 Upon adsorption onto the cell membrane, the NCs can enter via multiple pathways including endocytosis and energy-independent translocation, giving access to negatively charged interior structures such as the cell nucleus or ribosomes.98,99 Motivated by this precedent, we tested two highly stable cationic CdSe/CdS QD samples (QDs capped with TMABr and QDs capped with a mixed layer of TMABr and HS-PEG550) and neutral HS-PEG550-capped QDs for cellular staining. The TMABr-capped CdSe/CdS QDs clearly show an affinity for the cell membrane as indicated by the bright outline of the cells (Figure 7c). In contrast, QDs capped with mixed TMABr/HS-PEG550 ligands appear to stain the entire cell body allowing for easier visualization compared to the DIC image. By comparison, control experiments with neutral QDs capped with HS-PEG550 yielded subpar results, indicating that the positive charge from TMABr plays an important role in the staining process (Figure S7). These promising preliminary results demonstrate that buffer-stable cationic NCs can be used as site-specific cellular staining agents.
Conclusions
In summary, we have developed a collection of small (<2.5 nm) cationic ligands that confer colloidal stability in aqueous solution for various NCs. The coordinating group of these ligands was chosen on the basis of compatibility with the NC surface in accordance with the principles of hard–soft acid–base theory. Guanidium and quaternary amine groups, which retain their charge at high pH, were used to impart positive surface charge to the newly functionalized NCs. The generality of this approach was confirmed for a variety of NCs, including semiconductor, magnetic, and plasmonic materials of various shapes and ranging in size from 1.5 to 20 nm. Small (<12 nm) cationic NCs proved stable for months in water and for days to months, depending on NC/ligand pair, in buffer and salt solutions. Larger NCs, including 13 × 49 nm Au nanorods, showed short-term colloidal stability in some of the tested buffer solutions but are stable for months in water and several polar organic solvents.
The stability of these NCs in high ionic strength solutions and their positive surface charge allowed us to explore two possible applications. First, cationic NCs were found to be potentially useful as selective staining agents for cell imaging with characteristics that depend on the ligand layer composition. Second, combining highly stable cationic and anionic NCs yielded nanocrystalline solids with controllable and uniform 2D and 3D distributions of two distinct types of NCs. Engineering of such nanocrystalline solids is of interest for thermoelectric and photovoltaic applications, where highly uniform distributions of component NCs possessing different functionalities is appealing.
Acknowledgments
We acknowledge financial support from the European Union through the FP7 (ERC Starting Grant NANOSOLID, GA No. 306733; Marie Curie Fellowship IIF-GA-2012-330524), the Swiss National Science Foundation (SNF Ambizione Energy grant, Grant No. PZENP2_154287), and the ETH Zurich. We would like to thank Dr. Marc Walter for the SEM images and Matthias J. Grotevent for the AFM measurements. We are also grateful to Professor J.-C. Leroux for the use of the fluorescence microscope in his lab.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemmater.7b03504.
Materials and details of NCs and peptide ligands syntheses; FTIR, SEM, and AFM images; cellular imaging with neutral QDs (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Na H. B.; Song I. C.; Hyeon T. Inorganic Nanoparticles for MRI Contrast Agents. Adv. Mater. 2009, 21 (21), 2133–2148. 10.1002/adma.200802366. [DOI] [Google Scholar]
- Kamat P. V. Quantum Dot Solar Cells. The Next Big Thing in Photovoltaics. J. Phys. Chem. Lett. 2013, 4 (6), 908–918. 10.1021/jz400052e. [DOI] [PubMed] [Google Scholar]
- Ibáñez M.; Korkosz R. J.; Luo Z.; Riba P.; Cadavid D.; Ortega S.; Cabot A.; Kanatzidis M. G. Electron Doping in Bottom-Up Engineered Thermoelectric Nanomaterials through HCl-Mediated Ligand Displacement. J. Am. Chem. Soc. 2015, 137 (12), 4046–4049. 10.1021/jacs.5b00091. [DOI] [PubMed] [Google Scholar]
- Li J.-F.; Liu W.-S.; Zhao L.-D.; Zhou M. High-performance nanostructured thermoelectric materials. NPG Asia Mater. 2010, 2, 152–158. 10.1038/asiamat.2010.138. [DOI] [Google Scholar]
- Bao G.; Mitragotri S.; Tong S. Multifunctional Nanoparticles for Drug Delivery and Molecular Imaging. Annu. Rev. Biomed. Eng. 2013, 15 (1), 253–282. 10.1146/annurev-bioeng-071812-152409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biju V. Chemical modifications and bioconjugate reactions of nanomaterials for sensing, imaging, drug delivery and therapy. Chem. Soc. Rev. 2014, 43 (3), 744–764. 10.1039/C3CS60273G. [DOI] [PubMed] [Google Scholar]
- Hühn J.; Carrillo-Carrion C.; Soliman M. G.; Pfeiffer C.; Valdeperez D.; Masood A.; Chakraborty I.; Zhu L.; Gallego M.; Yue Z.; et al. Selected Standard Protocols for the Synthesis, Phase Transfer, and Characterization of Inorganic Colloidal Nanoparticles. Chem. Mater. 2017, 29 (1), 399–461. 10.1021/acs.chemmater.6b04738. [DOI] [Google Scholar]
- Murray C. B.; Norris D. J.; Bawendi M. G. Synthesis and characterization of nearly monodisperse CdE (E = sulfur, selenium, tellurium) semiconductor nanocrystallites. J. Am. Chem. Soc. 1993, 115 (19), 8706–8715. 10.1021/ja00072a025. [DOI] [Google Scholar]
- Park J.; Joo J.; Kwon S. G.; Jang Y.; Hyeon T. Synthesis of monodisperse spherical nanocrystals. Angew. Chem., Int. Ed. 2007, 46 (25), 4630–4660. 10.1002/anie.200603148. [DOI] [PubMed] [Google Scholar]
- Turkevich J.; Stevenson P. C.; Hillier J. A study of the nucleation and growth processes in the synthesis of colloidal gold. Discuss. Faraday Soc. 1951, 11 (0), 55–75. 10.1039/df9511100055. [DOI] [Google Scholar]
- Jana N. R.; Gearheart L.; Murphy C. J. Seed-Mediated Growth Approach for Shape-Controlled Synthesis of Spheroidal and Rod-like Gold Nanoparticles Using a Surfactant Template. Adv. Mater. 2001, 13 (18), 1389–1393. . [DOI] [Google Scholar]
- Piella J.; Bastús N. G.; Puntes V. Size-Controlled Synthesis of Sub-10-nanometer Citrate-Stabilized Gold Nanoparticles and Related Optical Properties. Chem. Mater. 2016, 28 (4), 1066–1075. 10.1021/acs.chemmater.5b04406. [DOI] [Google Scholar]
- Gaponik N.; Talapin D. V.; Rogach A. L.; Hoppe K.; Shevchenko E. V.; Kornowski A.; Eychmüller A.; Weller H. Thiol-Capping of CdTe Nanocrystals: An Alternative to Organometallic Synthetic Routes. J. Phys. Chem. B 2002, 106 (29), 7177–7185. 10.1021/jp025541k. [DOI] [Google Scholar]
- Pichaandi J.; Abel K. A.; Johnson N. J. J.; van Veggel F. C. J. M. Long-Term Colloidal Stability and Photoluminescence Retention of Lead-Based Quantum Dots in Saline Buffers and Biological Media through Surface Modification. Chem. Mater. 2013, 25 (10), 2035–2044. 10.1021/cm304091r. [DOI] [Google Scholar]
- Li W.; Hinton C. H.; Lee S. S.; Wu J.; Fortner J. D. Surface engineering superparamagnetic nanoparticles for aqueous applications: design and characterization of tailored organic bilayers. Environ. Sci.: Nano 2016, 3 (1), 85–93. 10.1039/C5EN00089K. [DOI] [Google Scholar]
- Dubertret B.; Skourides P.; Norris D. J.; Noireaux V.; Brivanlou A. H.; Libchaber A. Vivo Imaging of Quantum Dots Encapsulated in Phospholipid Micelles. Science 2002, 298 (5599), 1759–1762. 10.1126/science.1077194. [DOI] [PubMed] [Google Scholar]
- Yu W. W.; Chang E.; Falkner J. C.; Zhang J.; Al-Somali A. M.; Sayes C. M.; Johns J.; Drezek R.; Colvin V. L. Forming Biocompatible and Nonaggregated Nanocrystals in Water Using Amphiphilic Polymers. J. Am. Chem. Soc. 2007, 129 (10), 2871–2879. 10.1021/ja067184n. [DOI] [PubMed] [Google Scholar]
- Smith A. M.; Duan H.; Rhyner M. N.; Ruan G.; Nie S. A systematic examination of surface coatings on the optical and chemical properties of semiconductor quantum dots. Phys. Chem. Chem. Phys. 2006, 8 (33), 3895–3903. 10.1039/b606572b. [DOI] [PubMed] [Google Scholar]
- Carey G. H.; Yuan M.; Comin R.; Voznyy O.; Sargent E. H. Cleavable Ligands Enable Uniform Close Packing in Colloidal Quantum Dot Solids. ACS Appl. Mater. Interfaces 2015, 7 (39), 21995–22000. 10.1021/acsami.5b06890. [DOI] [PubMed] [Google Scholar]
- Turo M. J.; Macdonald J. E. Crystal-Bound vs Surface-Bound Thiols on Nanocrystals. ACS Nano 2014, 8 (10), 10205–10213. 10.1021/nn5032164. [DOI] [PubMed] [Google Scholar]
- Turo M. J.; Shen X.; Brandon N. K.; Castillo S.; Fall A. M.; Pantelides S. T.; Macdonald J. E. Dual-mode crystal-bound and X-type passivation of quantum dots. Chem. Commun. 2016, 52 (82), 12214–12217. 10.1039/C6CC05951A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerion D.; Pinaud F.; Williams S. C.; Parak W. J.; Zanchet D.; Weiss S.; Alivisatos A. P. Synthesis and Properties of Biocompatible Water-Soluble Silica-Coated CdSe/ZnS Semiconductor Quantum Dots. J. Phys. Chem. B 2001, 105 (37), 8861–8871. 10.1021/jp0105488. [DOI] [Google Scholar]
- Liu Y.; Li Y.; Li X.-M.; He T. Kinetics of (3-Aminopropyl)triethoxylsilane (APTES) Silanization of Superparamagnetic Iron Oxide Nanoparticles. Langmuir 2013, 29 (49), 15275–15282. 10.1021/la403269u. [DOI] [PubMed] [Google Scholar]
- Parak W. J.; Pellegrino T.; Plank C. Labelling of cells with quantum dots. Nanotechnology 2005, 16 (2), R9. 10.1088/0957-4484/16/2/R01. [DOI] [PubMed] [Google Scholar]
- Cho T. J.; MacCuspie R. I.; Gigault J.; Gorham J. M.; Elliott J. T.; Hackley V. A. Highly Stable Positively Charged Dendron-Encapsulated Gold Nanoparticles. Langmuir 2014, 30 (13), 3883–3893. 10.1021/la5002013. [DOI] [PubMed] [Google Scholar]
- Martin A. L.; Bernas L. M.; Rutt B. K.; Foster P. J.; Gillies E. R. Enhanced Cell Uptake of Superparamagnetic Iron Oxide Nanoparticles Functionalized with Dendritic Guanidines. Bioconjugate Chem. 2008, 19 (12), 2375–2384. 10.1021/bc800209u. [DOI] [PubMed] [Google Scholar]
- Medintz I. L.; Uyeda H. T.; Goldman E. R.; Mattoussi H. Quantum dot bioconjugates for imaging, labelling and sensing. Nat. Mater. 2005, 4 (6), 435–446. 10.1038/nmat1390. [DOI] [PubMed] [Google Scholar]
- Palui G.; Aldeek F.; Wang W.; Mattoussi H. Strategies for interfacing inorganic nanocrystals with biological systems based on polymer-coating. Chem. Soc. Rev. 2015, 44 (1), 193–227. 10.1039/C4CS00124A. [DOI] [PubMed] [Google Scholar]
- Uyeda H. T.; Medintz I. L.; Jaiswal J. K.; Simon S. M.; Mattoussi H. Synthesis of Compact Multidentate Ligands to Prepare Stable Hydrophilic Quantum Dot Fluorophores. J. Am. Chem. Soc. 2005, 127 (11), 3870–3878. 10.1021/ja044031w. [DOI] [PubMed] [Google Scholar]
- Pellegrino T.; Manna L.; Kudera S.; Liedl T.; Koktysh D.; Rogach A. L.; Keller S.; Rädler J.; Natile G.; Parak W. J. Hydrophobic Nanocrystals Coated with an Amphiphilic Polymer Shell: A General Route to Water Soluble Nanocrystals. Nano Lett. 2004, 4 (4), 703–707. 10.1021/nl035172j. [DOI] [Google Scholar]
- Parak W. J.; Gerion D.; Pellegrino T.; Zanchet D.; Micheel C.; Williams S. C.; Boudreau R.; Le Gros M. A.; Larabell C. A.; Alivisatos A. P. Biological applications of colloidal nanocrystals. Nanotechnology 2003, 14 (7), R15. 10.1088/0957-4484/14/7/201. [DOI] [Google Scholar]
- Sperling R. A.; Parak W. J. Surface modification, functionalization and bioconjugation of colloidal inorganic nanoparticles. Philos. Trans. R. Soc., A 2010, 368 (1915), 1333–1383. 10.1098/rsta.2009.0273. [DOI] [PubMed] [Google Scholar]
- Zhao G.; Tong L.; Cao P.; Nitz M.; Winnik M. A. Functional PEG–PAMAM-Tetraphosphonate Capped NaLnF4 Nanoparticles and their Colloidal Stability in Phosphate Buffer. Langmuir 2014, 30 (23), 6980–6989. 10.1021/la501142v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart M. H.; Susumu K.; Mei B. C.; Medintz I. L.; Delehanty J. B.; Blanco-Canosa J. B.; Dawson P. E.; Mattoussi H. Multidentate Poly(ethylene glycol) Ligands Provide Colloidal Stability to Semiconductor and Metallic Nanocrystals in Extreme Conditions. J. Am. Chem. Soc. 2010, 132 (28), 9804–9813. 10.1021/ja102898d. [DOI] [PubMed] [Google Scholar]
- Mei B. C.; Susumu K.; Medintz I. L.; Mattoussi H. Polyethylene glycol-based bidentate ligands to enhance quantum dot and gold nanoparticle stability in biological media. Nat. Protoc. 2009, 4 (3), 412–423. 10.1038/nprot.2008.243. [DOI] [PubMed] [Google Scholar]
- Liu W.; Howarth M.; Greytak A. B.; Zheng Y.; Nocera D. G.; Ting A. Y.; Bawendi M. G. Compact Biocompatible Quantum Dots Functionalized for Cellular Imaging. J. Am. Chem. Soc. 2008, 130 (4), 1274–1284. 10.1021/ja076069p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slocik J. M.; Stone M. O.; Naik R. R. Synthesis of Gold Nanoparticles Using Multifunctional Peptides. Small 2005, 1 (11), 1048–1052. 10.1002/smll.200500172. [DOI] [PubMed] [Google Scholar]
- Rio-Echevarria I. M.; Tavano R.; Causin V.; Papini E.; Mancin F.; Moretto A. Water-Soluble Peptide-Coated Nanoparticles: Control of the Helix Structure and Enhanced Differential Binding to Immune Cells. J. Am. Chem. Soc. 2011, 133 (1), 8–11. 10.1021/ja107588q. [DOI] [PubMed] [Google Scholar]
- Kovalenko M. V.; Bodnarchuk M. I.; Zaumseil J.; Lee J.-S.; Talapin D. V. Expanding the Chemical Versatility of Colloidal Nanocrystals Capped with Molecular Metal Chalcogenide Ligands. J. Am. Chem. Soc. 2010, 132 (29), 10085–10092. 10.1021/ja1024832. [DOI] [PubMed] [Google Scholar]
- Huang J.; Liu W.; Dolzhnikov D. S.; Protesescu L.; Kovalenko M. V.; Koo B.; Chattopadhyay S.; Shenchenko E. V.; Talapin D. V. Surface Functionalization of Semiconductor and Oxide Nanocrystals with Small Inorganic Oxoanions (PO43–, MoO42–) and Polyoxometalate Ligands. ACS Nano 2014, 8 (9), 9388–9402. 10.1021/nn503458y. [DOI] [PubMed] [Google Scholar]
- Dirin D. N.; Dreyfuss S.; Bodnarchuk M. I.; Nedelcu G.; Papagiorgis P.; Itskos G.; Kovalenko M. V. Lead Halide Perovskites and Other Metal Halide Complexes As Inorganic Capping Ligands for Colloidal Nanocrystals. J. Am. Chem. Soc. 2014, 136 (18), 6550–6553. 10.1021/ja5006288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakunin S.; Dirin D. N.; Protesescu L.; Sytnyk M.; Tollabimazraehno S.; Humer M.; Hackl F.; Fromherz T.; Bodnarchuk M. I.; Kovalenko M. V.; et al. High Infrared Photoconductivity in Films of Arsenic-Sulfide-Encapsulated Lead-Sulfide Nanocrystals. ACS Nano 2014, 8 (12), 12883–12894. 10.1021/nn5067478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalenko M. V.; Scheele M.; Talapin D. V. Colloidal Nanocrystals with Molecular Metal Chalcogenide Surface Ligands. Science 2009, 324 (5933), 1417–1420. 10.1126/science.1170524. [DOI] [PubMed] [Google Scholar]
- Speirs M. J.; Dirin D. N.; Abdu-Aguye M.; Balazs D. M.; Kovalenko M. V.; Loi M. A. Temperature dependent behaviour of lead sulfide quantum dot solar cells and films. Energy Environ. Sci. 2016, 9 (9), 2916–2924. 10.1039/C6EE01577H. [DOI] [Google Scholar]
- Balazs D. M.; Dirin D. N.; Fang H.-H.; Protesescu L.; ten Brink G. H.; Kooi B. J.; Kovalenko M. V.; Loi M. A. Counterion-Mediated Ligand Exchange for PbS Colloidal Quantum Dot Superlattices. ACS Nano 2015, 9 (12), 11951–11959. 10.1021/acsnano.5b04547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.-Y.; Harris M. T. Surface modification of magnetic nanoparticles capped by oleic acids: Characterization and colloidal stability in polar solvents. J. Colloid Interface Sci. 2006, 293 (2), 401–408. 10.1016/j.jcis.2005.06.062. [DOI] [PubMed] [Google Scholar]
- Ying E.; Li D.; Guo S.; Dong S.; Wang J. Synthesis and Bio-Imaging Application of Highly Luminescent Mercaptosuccinic Acid-Coated CdTe Nanocrystals. PLoS One 2008, 3 (5), e2222. 10.1371/journal.pone.0002222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagaria H. G.; Ada E. T.; Shamsuzzoha M.; Nikles D. E.; Johnson D. T. Understanding Mercapto Ligand Exchange on the Surface of FePt Nanoparticles. Langmuir 2006, 22 (18), 7732–7737. 10.1021/la0601399. [DOI] [PubMed] [Google Scholar]
- Pong B.-K.; Trout B. L.; Lee J.-Y. Modified Ligand-Exchange for Efficient Solubilization of CdSe/ZnS Quantum Dots in Water: A Procedure Guided by Computational Studies. Langmuir 2008, 24 (10), 5270–5276. 10.1021/la703431j. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Qin Y.; Palchoudhury S.; Bao Y. Water-Soluble Iron Oxide Nanoparticles with High Stability and Selective Surface Functionality. Langmuir 2011, 27 (14), 8990–8997. 10.1021/la201652h. [DOI] [PubMed] [Google Scholar]
- Jensen S. C.; Homan S. B.; Weiss E. A. Photocatalytic Conversion of Nitrobenzene to Aniline through Sequential Proton-Coupled One-Electron Transfers from a Cadmium Sulfide Quantum Dot. J. Am. Chem. Soc. 2016, 138 (5), 1591–1600. 10.1021/jacs.5b11353. [DOI] [PubMed] [Google Scholar]
- Dixit S. K.; Goicochea N. L.; Daniel M.-C.; Murali A.; Bronstein L.; De M.; Stein B.; Rotello V. M.; Kao C. C.; Dragnea B. Quantum Dot Encapsulation in Viral Capsids. Nano Lett. 2006, 6 (9), 1993–1999. 10.1021/nl061165u. [DOI] [PubMed] [Google Scholar]
- Kukushkin V. I.; Van’kov A. B.; Kukushkin I. V. Long-range manifestation of surface-enhanced Raman scattering. JETP Lett. 2013, 98 (2), 64–69. 10.1134/S0021364013150113. [DOI] [Google Scholar]
- Liu F. M.; Köllensperger P. A.; Green M.; Cass A. E. G.; Cohen L. F. A note on distance dependence in surface enhanced Raman spectroscopy. Chem. Phys. Lett. 2006, 430 (1–3), 173–176. 10.1016/j.cplett.2006.08.091. [DOI] [Google Scholar]
- Radziuk D.; Moehwald H. Prospects for plasmonic hot spots in single molecule SERS towards the chemical imaging of live cells. Phys. Chem. Chem. Phys. 2015, 17 (33), 21072–21093. 10.1039/C4CP04946B. [DOI] [PubMed] [Google Scholar]
- Aslan K.; Pérez-Luna V. H. Surface Modification of Colloidal Gold by Chemisorption of Alkanethiols in the Presence of a Nonionic Surfactant. Langmuir 2002, 18 (16), 6059–6065. 10.1021/la025795x. [DOI] [Google Scholar]
- Hamon C.; Bizien T.; Artzner F.; Even-Hernandez P.; Marchi V. Replacement of CTAB with peptidic ligands at the surface of gold nanorods and their self-assembling properties. J. Colloid Interface Sci. 2014, 424, 90–97. 10.1016/j.jcis.2014.03.002. [DOI] [PubMed] [Google Scholar]
- Pan Y.; Long M. J. C.; Li X.; Shi J.; Hedstrom L.; Xu B. Glutathione (GSH)-decorated magnetic nanoparticles for binding glutathione-S-transferase (GST) fusion protein and manipulating live cells. Chem. Sci. 2011, 2 (5), 945–948. 10.1039/c1sc00030f. [DOI] [Google Scholar]
- Pons T.; Uyeda H. T.; Medintz I. L.; Mattoussi H. Hydrodynamic Dimensions, Electrophoretic Mobility, and Stability of Hydrophilic Quantum Dots. J. Phys. Chem. B 2006, 110 (41), 20308–20316. 10.1021/jp065041h. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Purich D. L.; Wu C.; Wu Y.; Chen T.; Cui C.; Zhang L.; Cansiz S.; Hou W.; Wang Y.; et al. Ionic Functionalization of Hydrophobic Colloidal Nanoparticles To Form Ionic Nanoparticles with Enzymelike Properties. J. Am. Chem. Soc. 2015, 137 (47), 14952–14958. 10.1021/jacs.5b08533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woehrle G. H.; Brown L. O.; Hutchison J. E. Thiol-Functionalized, 1.5-nm Gold Nanoparticles through Ligand Exchange Reactions: Scope and Mechanism of Ligand Exchange. J. Am. Chem. Soc. 2005, 127 (7), 2172–2183. 10.1021/ja0457718. [DOI] [PubMed] [Google Scholar]
- Vigderman L.; Manna P.; Zubarev E. R. Quantitative Replacement of Cetyl Trimethylammonium Bromide by Cationic Thiol Ligands on the Surface of Gold Nanorods and Their Extremely Large Uptake by Cancer Cells. Angew. Chem., Int. Ed. 2012, 51 (3), 636–641. 10.1002/anie.201107304. [DOI] [PubMed] [Google Scholar]
- Kontogeorgis G. M.; Kiil S.. Introduction to Applied Colloid and Surface Chemistry; Wiley: Chichester, UK, 2016; p 219. [Google Scholar]
- Kalsin A. M.; Pinchuk A. O.; Smoukov S. K.; Paszewski M.; Schatz G. C.; Grzybowski B. A. Electrostatic Aggregation and Formation of Core–Shell Suprastructures in Binary Mixtures of Charged Metal Nanoparticles. Nano Lett. 2006, 6 (9), 1896–1903. 10.1021/nl060967m. [DOI] [PubMed] [Google Scholar]
- Kalsin A. M.; Kowalczyk B.; Smoukov S. K.; Klajn R.; Grzybowski B. A. Ionic-like Behavior of Oppositely Charged Nanoparticles. J. Am. Chem. Soc. 2006, 128 (47), 15046–15047. 10.1021/ja0642966. [DOI] [PubMed] [Google Scholar]
- Kalsin A. M.; Fialkowski M.; Paszewski M.; Smoukov S. K.; Bishop K. J. M.; Grzybowski B. A. Electrostatic Self-Assembly of Binary Nanoparticle Crystals with a Diamond-Like Lattice. Science 2006, 312 (5772), 420. 10.1126/science.1125124. [DOI] [PubMed] [Google Scholar]
- Elci S. G.; Jiang Y.; Yan B.; Kim S. T.; Saha K.; Moyano D. F.; Yesilbag Tonga G.; Jackson L. C.; Rotello V. M.; Vachet R. W. Surface Charge Controls the Suborgan Biodistributions of Gold Nanoparticles. ACS Nano 2016, 10 (5), 5536–5542. 10.1021/acsnano.6b02086. [DOI] [PubMed] [Google Scholar]
- Gessner A.; Lieske A.; Paulke B. R.; Müller R. H. Influence of surface charge density on protein adsorption on polymeric nanoparticles: analysis by two-dimensional electrophoresis. Eur. J. Pharm. Biopharm. 2002, 54 (2), 165–170. 10.1016/S0939-6411(02)00081-4. [DOI] [PubMed] [Google Scholar]
- Lundqvist M.; Stigler J.; Elia G.; Lynch I.; Cedervall T.; Dawson K. A. Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (38), 14265–14270. 10.1073/pnas.0805135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh W.-K.; Kim S.; Choi M.; Kim C.; Jeong Y. S.; Cho B.-R.; Hahn J.-S.; Jang J. Cellular Uptake, Cytotoxicity, and Innate Immune Response of Silica–Titania Hollow Nanoparticles Based on Size and Surface Functionality. ACS Nano 2010, 4 (9), 5301–5313. 10.1021/nn100561e. [DOI] [PubMed] [Google Scholar]
- Wang C.; Sun A.; Qiao Y.; Zhang P.; Ma L.; Su M. Cationic surface modification of gold nanoparticles for enhanced cellular uptake and X-ray radiation therapy. J. Mater. Chem. B 2015, 3 (37), 7372–7376. 10.1039/C5TB00766F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. T.; Saha K.; Kim C.; Rotello V. M. The Role of Surface Functionality in Determining Nanoparticle Cytotoxicity. Acc. Chem. Res. 2013, 46 (3), 681–691. 10.1021/ar3000647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N. B.; Vercellotti G. M.; White J. G.; Fegan A.; Wagner C. R.; Bischof J. C. Blood–Nanoparticle Interactions and in Vivo Biodistribution: Impact of Surface PEG and Ligand Properties. Mol. Mol. Pharmaceutics 2012, 9 (8), 2146–2155. 10.1021/mp200626j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townson J. L.; Lin Y.-S.; Agola J. O.; Carnes E. C.; Leong H. S.; Lewis J. D.; Haynes C. L.; Brinker C. J. Re-examining the Size/Charge Paradigm: Differing in Vivo Characteristics of Size- and Charge-Matched Mesoporous Silica Nanoparticles. J. Am. Chem. Soc. 2013, 135 (43), 16030–16033. 10.1021/ja4082414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong R.; Fischer N. O.; Emrick T.; Rotello V. M. Surface PEGylation and Ligand Exchange Chemistry of FePt Nanoparticles for Biological Applications. Chem. Mater. 2005, 17 (18), 4617–4621. 10.1021/cm0507819. [DOI] [Google Scholar]
- Wijaya A.; Hamad-Schifferli K. Ligand Customization and DNA Functionalization of Gold Nanorods via Round-Trip Phase Transfer Ligand Exchange. Langmuir 2008, 24 (18), 9966–9969. 10.1021/la8019205. [DOI] [PubMed] [Google Scholar]
- Hassinen J.; Liljeström V.; Kostiainen M. A.; Ras R. H. A. Rapid Cationization of Gold Nanoparticles by Two-Step Phase Transfer. Angew. Chem., Int. Ed. 2015, 54 (27), 7990–7993. 10.1002/anie.201503655. [DOI] [PubMed] [Google Scholar]
- Mehtala J. G.; Zemlyanov D. Y.; Max J. P.; Kadasala N.; Zhao S.; Wei A. Citrate-Stabilized Gold Nanorods. Langmuir 2014, 30 (46), 13727–13730. 10.1021/la5029542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen A. K. L.; Hutton G. A.; Masters A. F.; Maschmeyer T. The interplay of catechol ligands with nanoparticulate iron oxides. Dalton Trans. 2012, 41 (9), 2545–2559. 10.1039/c2dt11864e. [DOI] [PubMed] [Google Scholar]
- Schladt T. D.; Schneider K.; Schild H.; Tremel W. Synthesis and bio-functionalization of magnetic nanoparticles for medical diagnosis and treatment. Dalton Trans. 2011, 40 (24), 6315–6343. 10.1039/c0dt00689k. [DOI] [PubMed] [Google Scholar]
- Amstad E.; Gillich T.; Bilecka I.; Textor M.; Reimhult E. Ultrastable Iron Oxide Nanoparticle Colloidal Suspensions Using Dispersants with Catechol-Derived Anchor Groups. Nano Lett. 2009, 9 (12), 4042–4048. 10.1021/nl902212q. [DOI] [PubMed] [Google Scholar]
- Amstad E.; Zurcher S.; Mashaghi A.; Wong J. Y.; Textor M.; Reimhult E. Surface Functionalization of Single Superparamagnetic Iron Oxide Nanoparticles for Targeted Magnetic Resonance Imaging. Small 2009, 5 (11), 1334–1342. 10.1002/smll.200801328. [DOI] [PubMed] [Google Scholar]
- Xu C.; Xu K.; Gu H.; Zheng R.; Liu H.; Zhang X.; Guo Z.; Xu B. Dopamine as A Robust Anchor to Immobilize Functional Molecules on the Iron Oxide Shell of Magnetic Nanoparticles. J. Am. Chem. Soc. 2004, 126 (32), 9938–9939. 10.1021/ja0464802. [DOI] [PubMed] [Google Scholar]
- Corona-Avendaño S.; Alarcón-Angeles G.; Rosquete-Pina G. A.; Rojas-Hernández A.; Gutierrez A.; Ramírez-Silva M. T.; Romero-Romo M.; Palomar-Pardavé M. New Insights on the Nature of the Chemical Species Involved during the Process of Dopamine Deprotonation in Aqueous Solution: Theoretical and Experimental Study. J. Phys. Chem. B 2007, 111 (7), 1640–1647. 10.1021/jp0637227. [DOI] [PubMed] [Google Scholar]
- Korpany K. V.; Habib F.; Murugesu M.; Blum A. S. Stable water-soluble iron oxide nanoparticles using Tiron. Mater. Chem. Phys. 2013, 138 (1), 29–37. 10.1016/j.matchemphys.2012.10.015. [DOI] [Google Scholar]
- Nag A.; Kovalenko M. V.; Lee J.-S.; Liu W.; Spokoyny B.; Talapin D. V. Metal-free Inorganic Ligands for Colloidal Nanocrystals: S2–, HS–, Se2–, HSe–, Te2–, HTe–, TeS32–, OH–, and NH2– as Surface Ligands. J. Am. Chem. Soc. 2011, 133 (27), 10612–10620. 10.1021/ja2029415. [DOI] [PubMed] [Google Scholar]
- Haynes W. M.CRC handbook of chemistry and physics: a ready-reference book of chemical and physical data; CRC Press: Boca Raton, FL, 2011. [Google Scholar]
- Lévy R.; Thanh N. T. K.; Doty R. C.; Hussain I.; Nichols R. J.; Schiffrin D. J.; Brust M.; Fernig D. G. Rational and Combinatorial Design of Peptide Capping Ligands for Gold Nanoparticles. J. Am. Chem. Soc. 2004, 126 (32), 10076–10084. 10.1021/ja0487269. [DOI] [PubMed] [Google Scholar]
- Oyelere A. K.; Chen P. C.; Huang X.; El-Sayed I. H.; El-Sayed M. A. Peptide-Conjugated Gold Nanorods for Nuclear Targeting. Bioconjugate Chem. 2007, 18 (5), 1490–1497. 10.1021/bc070132i. [DOI] [PubMed] [Google Scholar]
- Alkilany A. M.; Nagaria P. K.; Hexel C. R.; Shaw T. J.; Murphy C. J.; Wyatt M. D. Cellular Uptake and Cytotoxicity of Gold Nanorods: Molecular Origin of Cytotoxicity and Surface Effects. Small 2009, 5 (6), 701–708. 10.1002/smll.200801546. [DOI] [PubMed] [Google Scholar]
- Kolny J.; Kornowski A.; Weller H. Self-Organization of Cadmium Sulfide and Gold Nanoparticles by Electrostatic Interaction. Nano Lett. 2002, 2 (4), 361–364. 10.1021/nl0156843. [DOI] [Google Scholar]
- Ibáñez M.; Luo Z.; Genc A.; Piveteau L.; Ortega S.; Cadavid D.; Dobrozhan O.; Liu Y.; Nachtegaal M.; Zebarjadi M.; et al. High-performance thermoelectric nanocomposites from nanocrystal building blocks. Nat. Commun. 2016, 7, 10766. 10.1038/ncomms10766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadavid D.; Ibáñez M.; Gorsse S.; López A. M.; Cirera A.; Morante J. R.; Cabot A. Bottom-up processing of thermoelectric nanocomposites from colloidal nanocrystal building blocks: the case of Ag2Te–PbTe. J. Nanopart. Res. 2012, 14 (12), 1–10. 10.1007/s11051-012-1328-0.22448125 [DOI] [Google Scholar]
- Urban J. J.; Talapin D. V.; Shevchenko E. V.; Kagan C. R.; Murray C. B. Synergism in binary nanocrystal superlattices leads to enhanced p-type conductivity in self-assembled PbTe/Ag2Te thin films. Nat. Mater. 2007, 6 (2), 115–121. 10.1038/nmat1826. [DOI] [PubMed] [Google Scholar]
- Jin H.; Choi S.; Velu R.; Kim S.; Lee H. J. Preparation of Multilayered CdSe Quantum Dot Sensitizers by Electrostatic Layer-by-Layer Assembly and a Series of Post-treatments toward Efficient Quantum Dot-Sensitized Mesoporous TiO2 Solar Cells. Langmuir 2012, 28 (12), 5417–5426. 10.1021/la202892h. [DOI] [PubMed] [Google Scholar]
- Jin H.; Choi S.; Xing G.; Lee J.-H.; Kwon Y.; Chong W. K.; Sum T. C.; Jang H. M.; Kim S. SnS44–, SbS43–, and AsS33– Metal Chalcogenide Surface Ligands: Couplings to Quantum Dots, Electron Transfers, and All-Inorganic Multilayered Quantum Dot Sensitized Solar Cells. J. Am. Chem. Soc. 2015, 137 (43), 13827–13835. 10.1021/jacs.5b05787. [DOI] [PubMed] [Google Scholar]
- Law M.; Beard M. C.; Choi S.; Luther J. M.; Hanna M. C.; Nozik A. J. Determining the Internal Quantum Efficiency of PbSe Nanocrystal Solar Cells with the Aid of an Optical Model. Nano Lett. 2008, 8 (11), 3904–3910. 10.1021/nl802353x. [DOI] [PubMed] [Google Scholar]
- Cho E. C.; Au L.; Zhang Q.; Xia Y. The Effects of Size, Shape, and Surface Functional Group of Gold Nanostructures on Their Adsorption and Internalization by Cells. Small 2010, 6 (4), 517–522. 10.1002/smll.200901622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nativo P.; Prior I. A.; Brust M. Uptake and Intracellular Fate of Surface-Modified Gold Nanoparticles. ACS Nano 2008, 2 (8), 1639–1644. 10.1021/nn800330a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.