

Abstract
Many adverse drug reactions are thought to be caused by electrophilically reactive drug metabolites that conjugate to nucleophilic sites within DNA and proteins, causing cancer or toxic immune responses. Quinone species, including quinone-imines, quinone-methides, and imine-methides, are electrophilic Michael acceptors that are often highly reactive and comprise over 40% of all known reactive metabolites. Quinone metabolites are created by cytochromes P450 and peroxidases. For example, cytochromes P450 oxidize acetaminophen to N-acetyl-p-benzoquinone imine, which is electrophilically reactive and covalently binds to nucleophilic sites within proteins. This reactive quinone metabolite elicits a toxic immune response when acetaminophen exceeds a safe dose. Using a deep learning approach, this study reports the first published method for predicting quinone formation: the formation of a quinone species by metabolic oxidation. We model both one- and two-step quinone formation, enabling accurate quinone formation predictions in nonobvious cases. We predict atom pairs that form quinones with an AUC accuracy of 97.6%, and we identify molecules that form quinones with 88.2% AUC. By modeling the formation of quinones, one of the most common types of reactive metabolites, our method provides a rapid screening tool for a key drug toxicity risk. The XenoSite quinone formation model is available at http://swami.wustl.edu/xenosite/p/quinone.
Graphical abstract
INTRODUCTION
Unanticipated toxicity frequently causes drug candidates to be discontinued late in development.1 Furthermore, idiosyncratic adverse drug reactions (IADRs) are often detected only after approval, causing significant morbidity and mortality.2,3 Around 75% of IADR cases result in liver transplants or death.4,5 There is considerable evidence that many varieties of drug toxicity, including IADRs, may be driven by a common mechanism: drug-metabolizing enzymes bioactivate drugs into electrophilically reactive metabolites, which covalently bind to sites within DNA or proteins.6–9 Frequently, DNA adducts are mutagenic and may interfere with transcription, replication, or regulation, causing gene dysfunction or initiating cancer.10–12 Likewise, protein adducts can disturb important biological activities or engender adverse immune responses.12–14 This study focuses on quinones, a major class of Michael acceptors, which are soft electrophiles15 and the most common type of reactive metabolites.16 Quinone species, such as quinone-imines, quinone-methides, or imine-methides, are commonly highly electrophilically reactive.17 Over 40% of all known reactive metabolites are quinones.16 Quinone formation, the formation of quinones by metabolic oxidation, is performed by cytochromes P45018–20 and peroxidases.9,21–23 Quinone formation generally occurs in one or two metabolic steps, where the final step is usually the coreduction of a cytochrome P450’s catalytic iron by the substrate, thereby dehydrogenating and oxidizing the substrate into a quinone species.24 For example, in a single step both cytochromes P45025 and peroxidases26 convert acetaminophen to the toxic metabolite N-acetyl-p-benzoquinone imine, which covalently binds to proteins (Figure 1). In contrast, lumiracoxib requires two steps to form a quinone. First, cytochromes P450 hydroxylate lumiracoxib, the product of which then forms a quinone after further metabolism by cytochromes P450 or peroxidases.18,27,28
Figure 1.
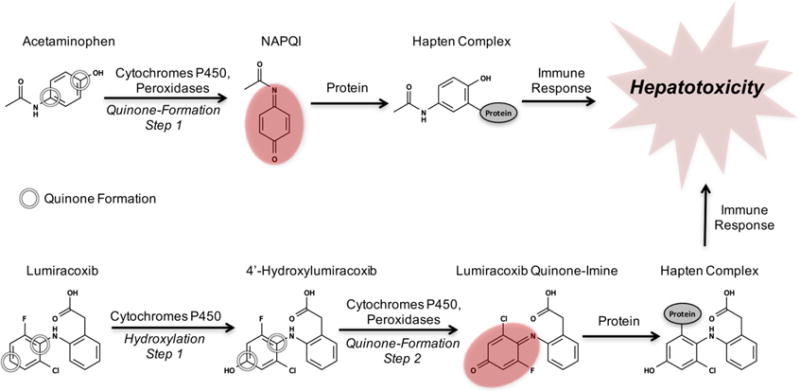
Examples of one- and two-step quinone formation: the formation of a quinone by metabolic oxidation. The quinone (indicated by red ovals) metabolites of acetaminophen and lumiracoxib cause hepatotoxicity due to the formation of a hapten complex with off-target proteins that triggers toxic immune responses.18,27,28 Acetaminophen’s conversion to the reactive quinone N-acetyl-p-benzoquinone imine (NAPQI) illustrates one-step quinone formation, and lumiracoxib’s conversion to lumiracoxib quinone-imine via the intermediate 4′-hydroxylumiracoxib demonstrates two-step quinone formation.18,27,28 The quinone formation model encompasses both one- and two-step quinone formation.
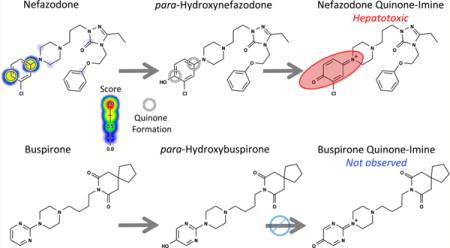
Detecting or anticipating quinone formation is critical for avoiding drug candidates early in drug development that can form reactive metabolites. Experimental methods for detecting quinone formation, most commonly incubation with glutathione, are well developed.29 However, these techniques require time and resources, which can be significant, especially when considering thousands of candidates during the initial screening phase of drug development. Moreover, some methods detect metabolic events that are inconclusive for quinone formation, such as aromatic hydroxylation, because the product may not form a subsequent quinone in the in vitro screens, which are often tuned to only generate one-step metabolites. For example, buspirone undergoes aromatic hydroxylation but does not form a quinone (Figure 2). In contrast, after hydroxylation at a structurally similar motif, the analogue nefazodone does proceed to form a hepatotoxic quinone.30,31 An accurate computational method for predicting both one-and two-step quinone formation pathways would be complementary to experimental screens and might identify otherwise undetected quinone formation risk.
Figure 2.
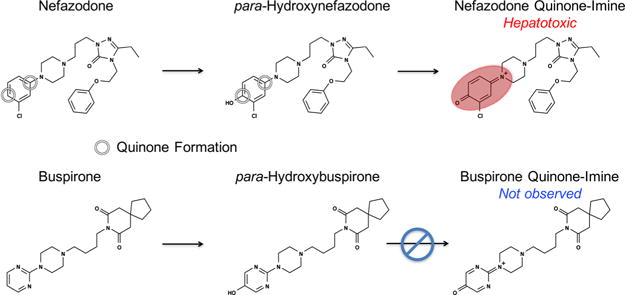
Both quinone-forming and nonquinone-forming molecules can undergo comparable initial metabolic events. Nefazodone first undergoes aromatic hydroxylation to form hydroxy-nefazodone, which is then oxidized to form the reactive nefazodone quinone, thereby causing hepatotoxicity in some patients.30,31 In contrast, the analogue buspirone does not form a quinone, despite undergoing a comparable initial hydroxylation event.31 A key goal of our model was to accurately identify molecules that form quinones.
Several studies demonstrated that computational methods can predict sites of metabolism.32–38 However, these methods do not predict the metabolic structures produced by these reactions, and cytochromes P450 can alter the same site in different ways, each with different consequences to toxicity.39,40 In contrast, we recently published a computational model that went beyond site of metabolism prediction to specifically predict the formation of epoxides, the second most common type of reactive metabolites after Michael acceptors such as quinones, which demonstrated the feasibility and utility of modeling the formation of specific reactive metabolites.41
In this study, we built the quinone formation model (Figure 3), which succeeded at two crucial tasks. First, the model accurately predicted the specific atom pairs within molecules that form quinones: their quinone-forming pairs (QPs). Knowledge of QPs can guide structural modifications to reduce the chances of reactive metabolite formation. Second, the model distinguished quinone-forming and nonquinone-forming molecules. Molecule quinone formation predictions can be used to quickly screen for molecules likely to form reactive metabolites, a key toxicity risk.
Figure 3.
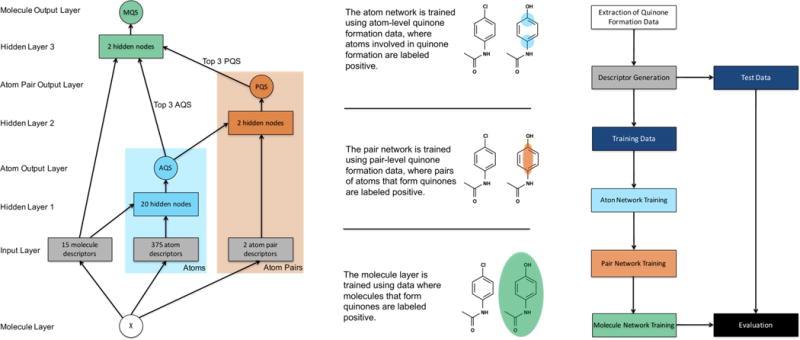
Structure of the quinone formation model. The network on the left depicts how information propagated through the model, which contained one molecule layer, one input layer, three hidden layers, and three output layers. From the structure of an input molecule, several molecule-level, atom-level, and pair-level descriptors were calculated at the input layer. The molecule- and atom-level descriptors flowed into the first hidden layer, which outputted an atom quinone formation score (AQS) for each atom in the input molecule. In this study, we defined a quinone-forming pair (QP) as the exact pair of ring carbons that forms a quinone. To predict QP, the atom-level scores of both atoms and the pair-level descriptors were presented to the second hidden layer, which computed a pair-level quinone formation score (PQS). Finally, the top three atom-level scores, the top three pair-level scores, and all molecule-level descriptors were submitted to the third hidden layer, which calculated a molecule quinone formation score (MQS). On the right, atom-level data are illustrated on the bottom (with atoms of quinone formation circled), pair-level data are illustrated in the middle (with a pair of atoms circled), and molecule-level data are illustrated on top (with the quinone-forming molecule circled). The molecule input node is a chemical structure, and all other circles are predictions ranging from 0 to 1. Blocks are vectors of real numbers. Right, a flowchart visualizes the overall experimental method. First, quinone formation data were extracted from a literature-derived database. Next, chemical descriptors were generated from the structures of these molecules. The data were then split into a training set and a test set. The training data were modeled by three steps of sequential training, on the atom-, pair-, and molecule-levels. Finally, the trained model was evaluated on the test data. Ten-fold cross-validation was used, a standard procedure in machine learning for estimating generalization accuracy. For each cross-validation fold, one-tenth of the data was set aside as the test set and the remaining data used as the training set.
MATERIALS AND METHODS
Quinone Formation Training Data
From the literature-derived Accelrys Metabolite Database (AMD), we extracted an extensive data set of chemically diverse metabolic reactions that form quinones. Overall, 576 reactions were extracted, each observed in humans, human cells, or human microsomes. These reactions consisted of 377 single-step quinone formation reactions and 199 two-step quinone formation reactions. An automated algorithm used the structure of each quinone product to label the QP on its metabolic parent, for one-step quinone formation, and its metabolic grandparent, for two-step quinone formation. We use “metabolic parent” to refer to the starting molecule of a one-step quinone formation reaction and “metabolic grandparent” to refer to the starting molecule of a two-step quinone formation reaction. Metabolic parents form quinones directly, whereas metabolic grandparents form a quinone through an intermediate structure. Both metabolic parents and grandparents were included in the training data. The results of the labeling algorithm were manually inspected and corrected as necessary.
For this study, we defined a QP as the pair of ring carbons that forms a quinone. In depictions, the atoms of these QPs are indicated by circles. When a pair of atoms was topologically equivalent to an observed QP, it was itself labeled as a QP. Duplicate starting molecules were merged into a lone training molecule with all observed QPs marked. The final data set included 359 quinone-forming molecules, each with its QPs labeled. Primarily, these QPs were found in six-membered rings (Table S1). Excluding atoms shared between fused rings of different sizes, such as the fused six- and seven-member rings of nevirapine in Figure 4, 90.2% of the QP atoms were in six-membered rings, compared to 8.6% and 1.2% for five- and seven-membered rings, respectively.
Figure 4.
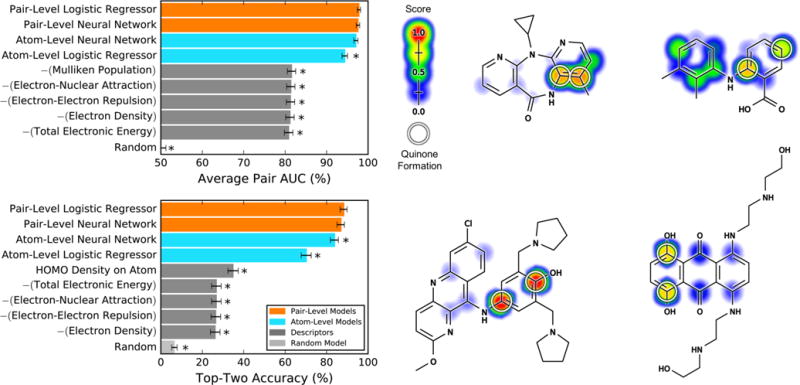
Pair-level model, computing pair quinone formation scores (PQS), accurately predicted quinone-forming pairs (QP). Top left, average pair area under the curve (AUC) was calculated for 359 molecules extracted from the Accelrys Metabolite Database and labeled with their QPs. Within these quinone-forming molecules, the average pair AUC metric quantifies how frequently QP were ranked higher than other atom pairs.41,44,45 Bottom left, the top-two metric was calculated, which counts a molecule correctly predicted if any of its QP received the first- or second-highest prediction.32,34,41,52,53 By both metrics, the cross-validated pair-level scores outperformed atom quinone formation scores (AQS), computed by a control model that did not include training on the pair-level, instead mapping the atom-level to the pair-level by multiplying the scores of both atoms together. The pair-level model performance also exceeded that of quantum chemical descriptors, the five best of which are included in each panel. The baseline of each metric, indicated by the random model, was computed by calculating performance over randomized predictions. For each metric, asterisks indicate results significantly worse than the best-performing method, as determined by a paired t-test.54 Examples from the data set are visualized with their scores (ranging from 0 to 0.98) indicated by the colored shading, assigning each atom the probabilistic OR of all pair-level quinone formation scores including the atom. Top center, nevirapine;89,90 top right, mefenamic acid;91 bottom center, pyronaridine;92 and bottom right, mitoxantrone.93 The atoms of experimentally observed QPs are circled.
Predicting which molecules will form quinones is a key goal of a useful model. Therefore, the training data set also included molecules that do not form quinones. To assemble these molecules, first all metabolically studied molecules in human-relevant experiments were extracted from the AMD. Second, all molecules that contained a six-membered aromatic ring were selected. Third, all molecules that formed quinones were filtered out, including quinones that formed in multiple steps. This procedure yielded a pool of 11884 molecules that have been studied in humans, human cells, or human microsomes but have not been reported to form quinones. From this pool, 359 (the same number of quinone-forming molecules) randomly selected nonquinone-forming molecules were added to the training set, for a total of 718 training molecules.
External Non-Quinone-Forming Test Sets
After removing the 359 nonquinone-forming molecules added to the training data, 11525 nonquinone-forming molecules remained. From this pool, external test sets were similarly extracted by randomly selecting 359 molecules for each test set. We repeated this process 20 times, thereby constructing 20 external test sets with 359 molecules apiece. All of these molecules were withheld from model training and were only evaluated by the final model.
Because of the terms of our AMD license, we were not able to share the structures of the training and test molecules. Instead, all reaction and molecule registry numbers were included in the Supporting Information, consistent with the June 2015 AMD release.
Topological Descriptors
To predict quinone formation, the first step of our method assigned each atom a vector of numbers, topological descriptors, which described chemical properties of that atom. Each atom was assigned 390 numbers, including 375 atom descriptors and 15 molecule descriptors. These descriptors were computed by in-house Python software using the structure of each molecule as input.42 We used topological descriptors, previous versions of which have been used for models of metabolism,32,43 reactivity,44,45 and epoxidation.41 For this study, we started with our most recent set of topological descriptors,44 which we supplemented with several new atom descriptors for ortho, meta, and para substituents. A second descriptor generation step assigned two descriptors to each pair of atoms: the connectivity distance between the atoms, and whether this distance was odd or even. A comprehensive listing of descriptors is available in the Supporting Information (Tables S3–S7), which also specifies the new topological descriptors developed for this study.
Combined Pair- and Molecule-Level Quinone Formation Model
We built a model using a deep convolutional neural network with one molecule layer, one input layer, three hidden layers, and three output layers (Figure 3). The top output layer computed molecule quinone formation scores (MQS), the middle output layer computed pair quinone formation scores (PQS), and the bottom output layer computed atom quinone formation scores (AQS). Respectively, the molecule-, pair-, and atom-level scores represented the probability that a molecule, pair, or atom formed a quinone, each score ranging from zero to one. The network was trained in three stages.
First, the atom-level network was trained to produce accurate atom-level scores. In this training step, each ring carbon within a molecule was considered a possible atom of quinone formation. Each atom was assigned a vector of numbers that described chemical properties of that atom. The data set was a matrix, with one row per atom, and one column per descriptor. Experimentally observed atoms of quinone formation were labeled with a 1 in a final binary target vector. Using gradient descent on the cross-entropy error, we trained the network weights so that atoms of quinone formation received higher scores than other atoms.
Second, the pair-level network was trained to compute scores for each pair of atoms. In this training step, each row of the data matrix was a pair of ring carbons, and each column was a descriptor. Descriptors included the atom-level score of each atom and both pair-level descriptors. For each atom pair, the two associated atom-level scores were sorted by magnitude and transformed by the logit function into log-odds. The pair-level network was trained to assign experimentally observed QPs with higher scores than all other atom pairs.
Third, the molecule-level network was trained. Each row of this data matrix was a molecule, and each column was a descriptor. Descriptors included the top three atom-level scores, the top three pair-level scores, and all molecule-level descriptors. The weights of the network were trained to assign quinone-forming molecules with higher scores than those for nonquinone-forming molecules.
The final model included hidden layers at all three stages. Several alternative models were considered, including a logistic regressor at all three stages, an AND function that mapped the atom-level to the pair-level, and a max layer that used a molecule’s maximum PQS as its molecule-level score. For all three stages, adding a hidden layer offered either higher classification performance or a better scaled prediction than alternative methods.
Quantum Chemical Descriptors
Currently, there are no other methods for explicitly predicting quinone formation. However, there are several computational tools available for predicting sites of metabolism, which frequently use quantum chemical calculations that correlate to the activation energies of cytochromes P450.34,35,46–49 Because of the absence of other methods for predicting quinone formation, we computed several quantum chemical descriptors to establish baseline performances (Tables S8 and S9). These descriptors were calculated by MOPAC, a quantum chemistry package that performs self-consistent field computations, with the COSMO implicit solvent model and the semiempirical method PM7.50,51 On both the pair- and molecule-level, we compared the performances of quantum chemical descriptors to those of the quinone formation predictions from the model, which only used topological descriptors. Atom-level quantum chemical descriptors were mapped to the pair-level by multiplying the descriptor values of both atoms together.
RESULTS AND DISCUSSION
The following sections analyze the performance and applications of the quinone formation model. First, we quantified the power of pair quinone formation scores (PQS) to predict quinone-forming atom pairs within quinone-forming molecules. Second, we evaluated the ability of molecule quinone formation scores (MQS) to separate quinone-forming and nonquinone-forming molecules. Third, we investigated whether MQS can separate drugs containing the same structural alert. Fourth, we screened for drugs that are predicted to form quinones but are not currently known to do so. Finally, we studied how the model can potentially direct rational drug modifications to prevent quinone formation.
Accuracy at Predicting Quinone-Forming Pairs
To minimize reaction metabolite formation during drug design, identifying a molecule’s QP is critical. Additionally, knowledge of the QP, the exact pair of carbons involved in quinone formation, provides a precise hypothesis about the source of a molecule’s toxicity. Information about QPs also suggests where a molecule could be modified to prevent quinone formation, thereby minimizing a key toxicity risk. The quinone formation model is the first published computational method that specifically predicts QPs across a wide range of molecules.
To predict QPs, the model was trained in two stages. First, the model computed an atom quinone formation score (AQS) for each ring carbon within a test molecule. The atom-level scores ranged from zero to one, reflecting the probability than an atom was involved in quinone formation. The weights of the network were trained such that experimentally known atoms of quinone formation receive high scores, and all other atoms received low scores.
The atom-level scores were intended to predict whether a single atom is part of a quinone formation reaction. However, because quinone formation always operates on a pair of atoms, the atom-level scores did not directly predict quinone formation. Therefore, a second training step maps the atom score to the pair level, producing a pair-level score. Within each molecule, this step considered all pairs of ring carbons as possible QPs. For each pair of atoms, this training step took as input two atom level scores (corresponding to each atom), as well as pair-level descriptors. The weights of this stage of the network were trained to assign QPs with high pair-level scores and all other pairs with low scores.
For both the atom-level and pair-level training steps, 10-fold cross-validation was used, a standard procedure in machine learning for estimating generalization accuracy. Within quinone-forming molecules, accurate predictions should differentiate QPs from all other pairs. We used two metrics to quantify the accuracy of cross-validated atom- and pair-level scores. First, we calculated the “average pair AUC” by computing the area under the receiver operating characteristic curve (AUC) for each molecule and averaging the AUCs across all molecules to measure performance for the entire data set.41,44,45 Second, we calculated “top-two” performance, which considers a molecule correctly predicted if any of its QPs are predicted in the first or second rank positions, a standard metric for site of metabolism predictions.32,34,41,52,53
To assess the performance of the atom-level model, we translated the atom-level scores to the pair-level by multiplying the scores of both atoms together, thereby calculating the probability that both atoms were involved in quinone formation. By this approach, the atom-level scores computed by a neural network yielded an average pair AUC performance of 97.1% and a top-two performance of 83.8% (Figure 4). In contrast, a logistic regressor computed significantly less accurate predictions, with performances of 94.4% (average pair AUC) and 70.2% (top-two). On the basis of this result, we selected the neural network for the atom-level training stage.
For the second training stage, on the pair-level, we similarly tested both a neural network and a logistic regressor. The neural network predicted QPs with performances of 97.6% and 86.9%, for average pair AUC and top-two, respectively. These did not exceed the performances of the logistic regressor: 97.8% (average pair AUC) and 88.3% (top-two). However, both the pair-level neural network and the pair-level logistic regressor significantly improved performance compared to only training on the atom-level, especially by the top-two metric. For example, comparing the top-two performances of the pair-level logistic regressor and the atom-level neural network by a paired t-test54 yielded a p-value of 0.037. Consequently, we retained the pair-level training and model.
However, at the pair level, choosing between the neural network and the logistic regressor was more difficult because they had statistically indistinguishable classification performances. Paired t-tests54 indicated that the neural network and the logistic regressor were statistically equivalent for both average pair AUC (p-value 0.339) and top-two (p-value 0.286). To break the tie, we constructed reliability plots, which quantify how well predictions correlate to probabilities (Figure 5).41,55 These plots revealed that the neural network is a better scaled prediction than the logistic regressor, with the lowest root-mean-square error to a perfectly scaled prediction. Well-scaled predictions are interpretable as probabilities, so we decided to use the neural network. Nevertheless, the choice between the neural network and the logistic regressor is arguable because the logistic regressor is a simpler model structure. While we settled on the neural network for this component of the model, we believe a logistic regressor could likely be utilized with comparable results.
Figure 5.
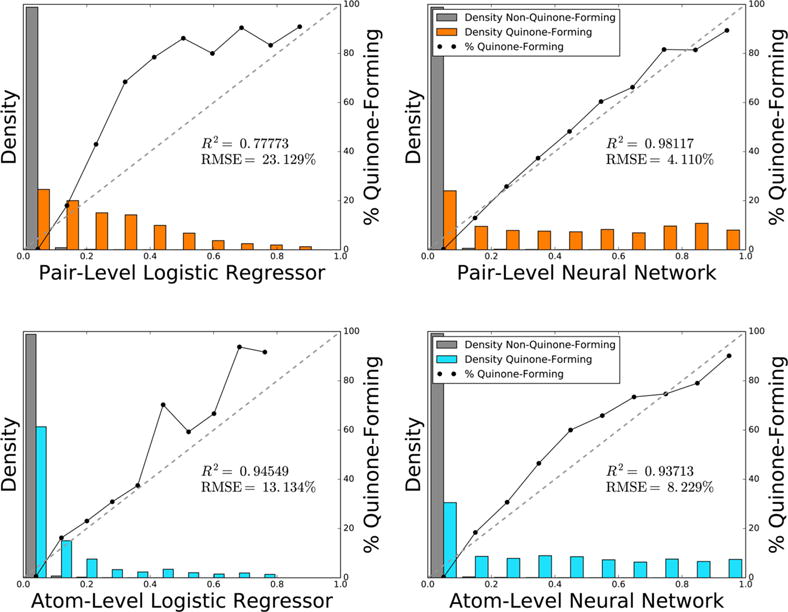
Pair-level neural network computes the best scaled prediction of quinone-forming pairs and corresponds closely with a probability. Across 78341 atom pairs within 718 quinone-forming and nonquinone-forming molecules, the bar graphs plot the normalized distributions of the pair-level logistic regressor, the pair-level neural network, the atom-level logistic regressor, and the atom-level neural network. Using non-normalized frequencies, the solid lines in each bin plot the percentage of atom pairs that form quinones. Atom-level scores were mapped to the pair-level by multiplying the scores of both atoms together. On the pair-level, the logistic regressor and the neural network used the outputs of the atom-level neural network as inputs, along with pair-level descriptors. Each diagonal dashed line indicates a hypothetical perfectly scaled prediction. Of the four methods, the pair-level neural network provided the best scaled prediction, with the lowest root-mean-square error of a perfectly scaled prediction, and the highest correlation to the best-fit line. Therefore, the pair-level neural network most accurately reflected the probability that an atom pair will form a quinone.
The majority of the QPs in the data were found in six-membered rings (Table S1), which raised concerns that we would not accurately predict quinone formation on five-membered rings due to insufficient data. With this in mind, we investigated whether QPs in five-membered rings were also accurately predicted by calculating accuracy only on those molecules. Across the 36 molecules where a five-membered ring formed a quinone, pair-level scores predicted QPs with performances of 96.7% and 86.1%, for average pair AUC and top-two, respectively. These performances matched the accuracies across the whole data set of 97.6% (average pair AUC) and 86.9% (top-two). We accurately predicted QPs in five-membered rings despite limited training data, suggesting that the underlying principles that guide quinone formation on six-membered rings also applied to quinone formation on five-membered rings.
Currently, there are no other models published for predicting quinone formation to which this model can be compared. Instead, to provide a baseline we calculated several quantum chemical descriptors (Tables S8 and S9), which correlate to the activation energies of cytochromes P450 and are often used by sites of metabolism predictors.34,35,46–49 Each quantum chemical descriptor was considered as a model to predict QPs. To map each descriptor to the pair-level, the descriptor value of both atoms was multiplied together. None of the quantum chemical descriptors approached the accuracy of the pair-level score. For example, the best descriptor by the top-two metric was the density of the highest occupied molecular orbital (HOMO) on the test atom, which only had a top-two performance of 34.8%, far below the pair-level model’s performance of 86.9%. By considering many chemical attributes together using machine learning, the model predicted QPs more accurately than any quantum chemical descriptor.
Accuracy at Identifying Quinone-Forming Molecules
We also evaluated how well the model separated quinone-forming from nonquinone-forming molecules. A model that accurately predicts molecule quinone formation could be used to quickly screen drug candidates for a key toxicity risk.
In this assessment, we trained the model to differentiate molecules that formed quinones from those that did not. In addition to quinone-forming molecules, the training data contained molecules that could form quinones, defined as containing a six-membered aromatic ring, and that were metabolically studied in human-relevant experiments in the Accelrys Metabolite Database (AMD) but did not form quinones. Following pair-level training, we investigated several methods of discriminating between quinone-forming and nonquinone-forming molecules (Figure 6). To measure performance at this objective, we calculated the AUC across the full data set (molecule AUC).
Figure 6.
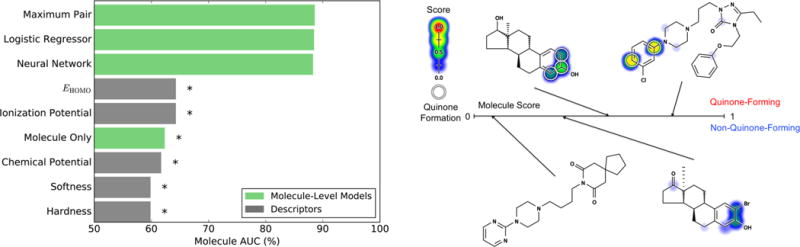
Molecule quinone formation scores (MQS) accurately identified quinone-forming molecules. Left, the ability of several methods to identify molecules that form quinones was computed, including taking each molecule’s maximum pair quinone formation score (PQS), training a logistic regressor or a neural network on the top three pair-level scores, the top three atom-level scores, and all molecule descriptors, and training a control model in the form of a neural network that only used as input molecule descriptors. The data set set included 359 quinone-forming molecules and 359 nonquinone-forming molecules. To quantify performance, the AUC was computed across quinone-forming and nonquinone-forming molecules (Molecule AUC), using cross-validated predictions. Asterisks indicate results significantly worse than the best-performing method, as determined by a false positive rate paired t-test.54 Right, closely related example pairs of molecules that do and do not form quinones are visualized. Cross-validated scores (ranging from 0 to 0.78) are indicated by the colored shading, assigning each atom the probabilistic OR of all pair-level quinone formation scores including the atom. The number line denotes each molecule’s cross-validated score, computed by the neural network. From left to right, top to bottom: estradiol (MQS: 0.60),94 nefazodone (MQS: 0.83),30,31 buspirone (MQS: 0.09),30,31 and a 2α-substituted androstenedione analogue (MQS: 0.42).95 The atoms of experimentally observed quinone-forming pairs are circled.
First, we tried the most straightforward method of predicting molecule quinone formation: taking each molecule’s maximum cross-validated pair-level scores. This procedure distinguished quinone-forming and nonquinone-forming molecules with a molecule AUC of 88.5%. Second, we added a training step that submitted the top three pair-level scores, the top three atom-level scores, and all molecule descriptors to a neural network or a logistic regressor. Using 10-fold cross-validation, neither the neural network nor the logistic regressor outperformed the maximum pair-level score, with molecule AUCs of 88.2% (p-value 0.16) and 88.4% (p-value 0.31), respectively, with significance evaluated by a false positive rate paired t-test.54
However, the neural network supplied a better scaled prediction than both the logistic regressor and the maximum pair quinone formation score, as indicated by the reliability plots of Figure 7. Furthermore, compared to both of the other methods, the neural network predicted more quinone-forming molecules with high scores and nonquinone-forming molecules with low scores. On the other hand, both the logistic regressor or the maximum PQS were simpler models with equivalent molecule AUC performance. Still, because of its well-scaled predictions, we decided to use the neural network, but we expect that the other two methods would provide similar results.
Figure 7.
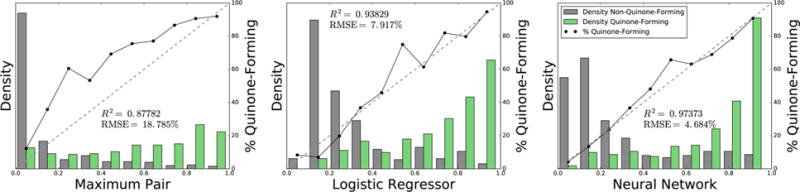
Neural network produces a well-scaled probabilistic prediction of molecule quinone formation. Across 718 quinone-forming and nonquinone-forming molecules, the bar graphs plot the normalized distributions of the maximum pair-level score, the molecule-level logistic regressor, and the molecule-level neural network. Using non-normalized frequencies, the solid lines in each bin plot the percentage of molecules that form quinones. Each diagonal dashed line indicates a hypothetical perfectly scaled prediction. The neural network provided the best scaled prediction, with the lowest root-mean-square error (RMSE) of a perfectly scaled prediction, and the highest correlation to the best-fit line. Therefore, the neural network most accurately reflected the probability that a molecule will form a quinone.
Compared to the performance of the pair-level model in identifying QPs (97.6% AUC), the molecule-level model had a considerably lower AUC of 88.2%. This drop in accuracy might be because the molecule-level data contained more noise than the pair-level data. When selecting “nonquinone-forming” molecules, we inferred that a molecule did not form a quinone if it was not reported to do so in the AMD. This supposition, while obligatory for molecule-level training, was not a robust indication that molecules do not form quinones because not every study measures quinone products. As a result, our data set construction procedure imprecisely labeled some quinone-forming molecules as nonquinone-forming. In contrast, the pair-level quinone formation data were much cleaner because they were extracted from experiments capable of reporting quinone formation. Our previous studies with the AMD have observed comparable drops in site-level (equivalent to the pair-level in this study) to molecule-level accuracy.41,44,45
Even so, the molecule-level scores classified quinone-forming molecules with 88.2% AUC, greatly exceeding that of any comparison quantum chemical descriptor and demonstrating that the model produces illuminating molecule-level predictions. The best quantum chemical descriptor was the energy of the highest occupied molecular orbital (EHOMO), which only had an AUC of 64.2%. The relatively high performance of the molecule-level model supports our assumption that many of the molecules labeled as nonquinone-forming indeed do not form quinones. If the quinone-forming and nonquinone-forming molecules were actually extracted from identical chemical populations, separating them in our cross-validated experiments, which simulated performance on external data, would be highly unlikely.
To further confirm that molecule-level performance was not a consequence of overfitting, we evaluated the model on several external test sets of nonquinone-forming molecules. Each external test set was predicted by the final trained model. Separation between each external nonquinone-forming test set and the quinone-forming training molecules was measured by the molecule AUC. Over the 20 test sets, the AUC was 85.9% ± 1.2%, which is comparable to the previously reported cross-validated AUC of 88.2% computed by training on both quinone-forming and nonquinone-forming molecules. The model successfully generalized to new data, assigning nonquinone-forming molecules it had never seen before with much lower scores than quinone-forming training molecules.
Comparison to Structural Alerts
We compared the model to “structural alerts,” which are motifs known to commonly form reactive metabolites. A library of structural alerts can be easily used to flag potentially problematic molecules during drug development.18,56 This is a widely used approach, but unfortunately structural alerts for quinone formation, such as anilines or phenols, are also found in many safe drugs because they are not bioactivated due to specific molecular context. We would hope that the model could identify which structural alerts are bioactivated to quinones and which are not.
To systematically evaluate whether the model distinguishes structural alerts, we focused on several motifs known to commonly form quinones. The exact patterns used are listed in the Supporting Information (Table S2). We included both motifs that form quinones in one step, such as hydroquinones and catechols, and motifs that form quinones in two steps, such as an oxygen or nitrogen ortho or para to a hydrogen. For each motif, we extracted all atom pairs from the training data set that matched the structure and recorded whether this pair formed a quinone. Next, we assigned each pair its cross-validated pair-level score and calculated the AUC across all pairs of that substructure, the pair AUC (Figure 8 and Table 1). For each structural alert, the model predicted quinone formation with performances significantly higher than random.
Figure 8.
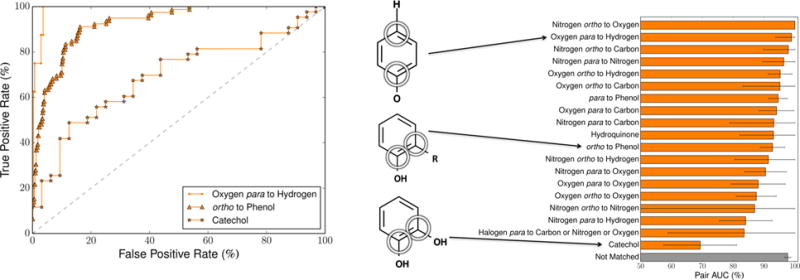
Pair-level scores predicted whether several structural alerts form quinones. We tested several motifs that are structural alerts for both one-and two-step quinone formation. For each motif, across the 718 training molecules, the cross-validated scores of all atom pairs matching that motif were extracted. Left, the receiver operating characteristic (ROC) curve is displayed across all the atom pairs of three example motifs (visualized in the center). The graph displays example ROC curves for the second best, typical, and worst performing alerts. The diagonal dashed line indicates baseline performance. The ROC curves of all the motifs are available in the Supporting Information (Figure S1). In the depiction of the ortho to phenol motif, the “R” represents any atom, including hydrogens. Right, the area under the ROC curve (AUC) was calculated across all the atom pairs of each motif. The error bars represent 95% two-sided confidence intervals.58
Table 1.
Pair-Level Scores Predicted Whether Structural Alerts Form Quinones
| motif | category | quinone-forming pairs | nonquinone-forming pairs | pair AUC (%) |
|---|---|---|---|---|
| nitrogen ortho to oxygen | one step | 3 | 31 | 100.0 |
| oxygen para to hydrogen | two step | 8 | 276 | 98.9 |
| nitrogen ortho to carbon | one step | 6 | 204 | 97.9 |
| nitrogen para to nitrogen | one step | 15 | 24 | 96.4 |
| oxygen ortho to hydrogen | two step | 57 | 981 | 95.2 |
| oxygen ortho to carbon | one step | 6 | 338 | 95.2 |
| para to phenol | one and two step | 108 | 325 | 94.6 |
| oxygen para to carbon | one step | 33 | 312 | 94.1 |
| nitrogen para to carbon | one step | 6 | 44 | 93.2 |
| hydroquinone | one step | 24 | 3 | 93.1 |
| ortho to phenol | one and two step | 79 | 813 | 92.8 |
| nitrogen ortho to hydrogen | two step | 12 | 483 | 91.4 |
| nitrogen para to oxygen | one step | 41 | 40 | 90.5 |
| oxygen para to oxygen | one step | 33 | 24 | 88.1 |
| oxygen ortho to oxygen | one step | 51 | 145 | 87.5 |
| nitrogen ortho to nitrogen | one step | 1 | 23 | 87.0 |
| nitrogen para to hydrogen | two step | 34 | 140 | 84.1 |
| halogen para to carbon or nitrogen or oxygen | two step | 4 | 105 | 83.6 |
| catechol | one step | 43 | 32 | 69.3 |
| not matched | 417 | 74398 | 97.7 |
For example, the catechol motif commonly forms an ortho-quinone in a single step (Figure 9).18,56,57 Catechols might be considered a motif that “obviously” forms quinones. However, of the 75 catechols in our data set, only 43 actually are known to form quinones.
Figure 9.
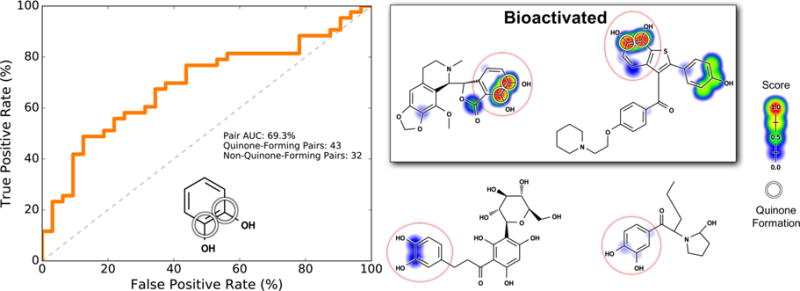
Pair-level scores predicted whether catechols form quinones. Of all structural alerts tested, the model performed the worst on catechols (Figure 8). Nevertheless, the model distinguished catechols with a statistically significant performance. This motif (indicated by the red circles) requires a single step to form a quinone. Across the 718 training molecules, the cross-validated scores of all atom pairs matching this motif were extracted. Left, the receiver operating characteristic (ROC) curve is displayed across all these atom pairs. The diagonal dashed line indicates baseline performance: 50% area under the ROC curve (AUC). Four example molecules are visualized. Scores (ranging from 0 to 0.97) are indicated by the colored shading, assigning each atom the probabilistic OR of all pair-level quinone formation scores including the atom. The atoms of experimentally observed quinone-forming pairs are circled. Top center, a noscapine metabolite96 and top right, a raloxifene metabolite,97 both of which are correctly predicted to form quinones at the catechol. Bottom center, aspalathin98 and bottom right, a methylenedioxypyrovalerone metabolite99 neither of which forms quinones at the catechol-para-to-hydrogen motifs, which are correctly assigned low scores.
The quinone formation model’s pair-level scores separated quinone-forming and nonquinone-forming catechols with a pair AUC of 69.3%. This was the lowest performance of all the structural alerts we tested, and there is certainly room for improvement. Nevertheless, as indicated by the 95% confidence intervals58 in Figure 9, we separated catechols with a performance significantly higher than baseline (AUC 50%). We find this an encouraging result because the method helps identify which catechols might not actually form quinones, unlike the structural alert approach that considers all catechols a bioactivation risk without evaluating specific molecular context.
We also found that pair-level scores are informative for two-step quinone formation structural alerts. For example, for the motif of a nitrogen para to a hydrogen to form a quinone, hydroxylation must first occur at the unsubstituted carbon, followed by quinone formation. The nitrogen-para-to-hydrogen motif can be found in some anilines or anilides, which are well-known structural alerts.18,56,59 The entire training data set contains 174 total nitrogen-para-to-hydrogen atom pairs, of which 34 are actually known to form quinones. Encouragingly, the quinone formation model’s pair-level scores accurately separated quinone-forming and nonquinone-forming nitrogen-para-to-hydrogen groups with a pair AUC of 84.1% (Figure 10).
Figure 10.
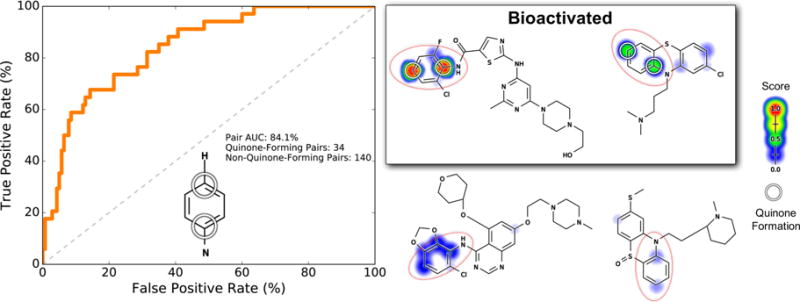
Pair-level scores predicted whether nitrogen-bound carbons para to a carbon with a hydrogen form quinones. This motif (indicated by the red circles) requires aromatic hydroxylation to form a quinone. Across the 718 training molecules, the cross-validated scores of all atoms pairs matching this motif were extracted. Left, the receiver operating characteristic (ROC) curve is displayed across all of these atom pairs. The diagonal dashed line indicates baseline performance: 50% area under the ROC curve (AUC). Four example molecules are visualized. Scores (ranging from 0 to 0.96) are indicated by the colored shading, assigning each atom the probabilistic OR of all pair-level quinone formation scores including that atom. The atoms of experimentally observed quinone-forming pairs are circled. Top center, a dasatinib analogue100 and top right, chlorpromazine,101 both of which are correctly predicted to form quinones at the nitrogen-para-to-hydrogen motif. Bottom center, saracatinib100 and bottom right, a thioridazine metabolite,102 neither of which forms quinones at the nitrogen-para-to-hydrogen motifs, are correctly assigned low scores.
The phenol motif is a broad structural alert found in a wide variety of drugs.18,56 In contrast to catechols (which require one step to form a quinone) and nitrogen-para-to-hydrogen (which require two steps), phenols can form quinones in one or two steps, depending on ring substituents. For example, phenols can form a quinone-methide in a single step if there is a para carbon or form a quinone in two steps via intermediate aromatic hydroxylation at the para position. Across our training data set, there are 433 carbons para to a phenol. For each atom, we extracted the atom-pair-level score of the carbon and the ipso-carbon of the corresponding phenol. Of these atom pairs, 108 form a para quinone.
The model accurately distinguished whether a quinone will form para to a phenol, with a pair AUC of 94.6% (Figure 11). Both one- and two-step quinone formation at the para position to a phenol were accurately predicted. For example, both oxymetazoline and an analogue of androstenedione contain a phenol-para-to-carbon motif, but only in oxymetazoline does the phenol-para-to-carbon motif form a quinone-methide.60,61 The model’s pair-level scores cleanly distinguished the phenol-para-to-carbon in both molecules. Similarly, both propofol and doxycycline contain a phenol-para-to-hydrogen, which can form a quinone in two steps with an intermediate hydroxylation. Propofol’s phenol-para-to-hydrogen received a much higher pair-level score than doxycycline, consistent with only propofol being known to form a quinone.62,63
Figure 11.
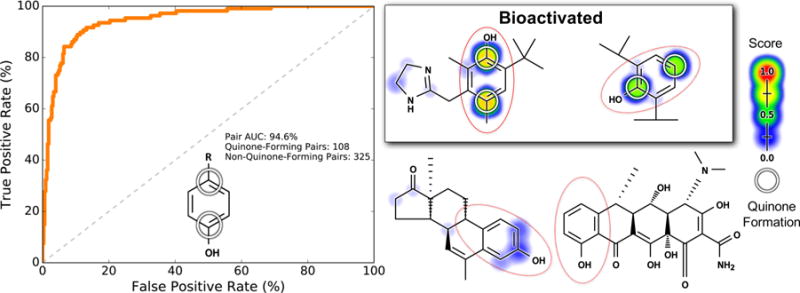
Pair-level scores predicted whether a quinone will form para to a phenol. This motif (indicated by the red circles) may require one or two metabolic steps to form a quinone. In the depiction of the phenol motif on the left, the “R” represents any atom, including hydrogens. Across the 718 training molecules, the cross-validated scores of all atom pairs matching this motif were extracted. Left, the receiver operating characteristic (ROC) curve is displayed across all these atom pairs. The diagonal dashed line indicates baseline performance: 50% area under the ROC curve (AUC). Four example molecules are visualized. Scores (ranging from 0 to 0.77) are indicated by the colored shading, assigning each atom the probabilistic OR of all pair-level quinone formation scores including that atom. The atoms of experimentally observed quinone-forming pairs are circled. Top center, oxymetazoline60 and top right, propofol,62 both of which are correctly predicted to form quinones at the para-to-phenol motif. Bottom center, an androstenedione analogue61 and bottom right, doxycycline,63 neither of which forms quinones at their para-to-phenol motifs, are correctly assigned low scores.
These results demonstrate that the model has better specificity than structural alerts. Specific substructures can determine the parts of molecules that have the capacity to form quinones, but they do not assess the likelihood of the quinone actually forming. The quinone formation model, however, can stratify molecules by their propensity of forming quinones, identifying which alerts are bioactivated and which are not.
Structural alerts are retrospective in nature. Consequently, they cannot detect quinone formation for motifs that have not previously been observed to form quinones. As a result, molecules that form quinones at atypical sites will likely be missed by structural alerts. In contrast, our model can potentially detect these unusual cases because it is built on a diverse data set that includes a wide variety of typical and atypical sites of quinone formation. To test this, we investigated the accuracy of the model at predicting quinone formation at sites that do not match structural alerts. After filtering out all atom pairs that matched any of the quinone structural alerts used (Table 1), we calculated the pair AUC over all remaining atom pairs. The model accurately predicted quinone formation at these nonobvious sites with a pair AUC of 97.7% (Figure 12). This indicates that the model is able to accurately predict quinone formation even for atom pairs that would not generally be considered to be at risk.
Figure 12.
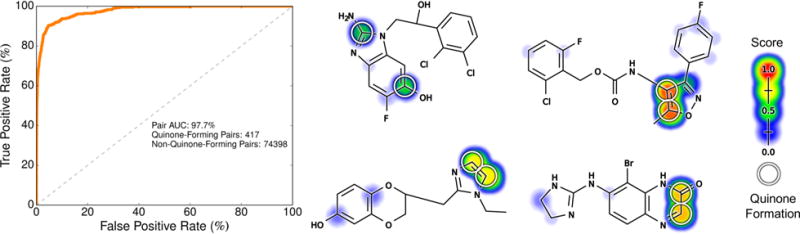
Pair-level scores predicted whether quinones form at motifs that do not match structural alerts. Across the 718 training molecules, the cross-validated scores of all atom pairs that did not match any structural alert were extracted. Left, the ROC curve is displayed across all these atom pairs. The diagonal dashed line indicates baseline performance: 50% area under the ROC curve (AUC). Four example molecules are visualized. Top center, an antimalarial drug candidate.103 Top right, a voltage-gated sodium channel inhibitor drug candidate.104 Bottom center, an imiloxan metabolite.105 Bottom right, a brimonidine metabolite.106 The atoms of experimentally observed quinone forming pairs are circled. Scores (ranging from 0 to 0.92) are indicated by the colored shading, assigning each atom the probabilistic OR of all pair-level quinone formation scores including the atom.
Case Studies
We considered several case studies to investigate the model’s utility. One potential application is to screen for molecules that are likely to form quinones but that are currently not known to do so. To explore this application, we predicted the quinone formation of drugs that were not reported to form quinones in the AMD. After downloading a database of FDA-approved and withdrawn drugs, the molecules present in the training data set were removed.64 Next, each drug was submitted to the trained quinone formation model, thereby assigning a molecule quinone formation score to each drug.
To define a cutoff above which a drug was considered predicted to form a quinone, we used the optimal point on the cross-validated molecule ROC curve from the training data (Figure S4). This optimal point, corresponding to a molecule score of 0.515, is the cutoff that offers the best trade-off between sensitivity and specificity. We selected all drugs that were assigned a higher score than the optimal cutoff. This revealed 288 drugs that are predicted to form quinones but that are currently thought to be nonquinone-forming across all the relevant literature in the AMD. All 288 drugs and their molecule quinone formation scores are available in the Supporting Information. Here, we highlight three example drugs (Figure 13). Each drug carries a risk of idiosyncratic toxicity, the causes of which are currently obscure.
Figure 13.
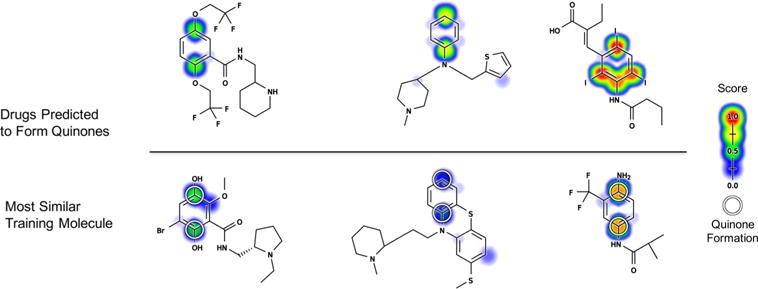
Examples of drugs associated with idiosyncratic toxicity that received high molecule quinone formation scores (MQS) but are not known to form quinones in our data set. Each drug predicted to form a quinone is visualized above its most similar quinone-forming molecule from the training data. Similarity was quantified using path based fingerprints and MinMax similarity, a modification of Tanimoto appropriate for count fingerprints.107 Top, flecainide (MQS: 0.81), thenalidine (MQS: 0.79), and bunamiodyl (MQS: 0.96). Bottom, a remoxipride metabolite (MQS: 0.58),108 thioridazine (MQS 0.36),101 and a flutamide metabolite (MQS 0.88).109 The atoms of experimentally observed quinone forming pairs are circled. Scores (ranging from 0 to 0.99) are indicated by colored shading, assigning each atom the probabilistic OR of all pair-level quinone formation scores including the atom. For the training examples, cross-validated scores are reported.
First, the antiarrhythmic flecainide is associated with idiosyncratic hepatotoxicity, the origin of which is currently unknown.18,59,65 There are no known reactive metabolites of flecainide, and previous studies have suggested that flecainide is harmlessly metabolized to a phenol via oxygen-dealkylation, followed by glucuronidation.18,59,66 However, our model suggests that the phenol metabolite may actually form a quinone.
Second, the antihistamine thenalidine is associated with idiosyncratic cases of agranulocytosis, a potentially deadly condition that entails a severely lowered blood white cell count that leaves a patient vulnerable to critical infection.67–70 Indeed, three years after thenalidine was introduced to the U.S. market, it was withdrawn in 1958 due to fatal cases of agranulocytosis.71–74 The mechanism of thenalidine’s idiosyncratic toxicity was never uncovered, and it is has no known reactive metabolites. An obvious hypothesis is that thenalidine’s thiophene ring, a well-known structural alert, could undergo bioactivation through epoxidation or sulfur-oxidation.18,75 However, the quinone model’s predictions yield a second, more surprising hypothesis: thenalidine may undergo hydroxylation at the para position on its aniline ring, followed by formation of a potentially highly reactive quinone-imine.
Third, bunamiodyl carries a risk of severe nephropathy.71,76 It was introduced in 1958 as a cholecystographic contrast medium but was withdrawn in 1964 after around 100 patients died due to renal failure.71,74,76,77 While bunamiodyl is not known to produce reactive metabolites, our model predicts that it undergoes dehalogenation followed by formation of an ortho or para quinone-imine.
As seen for flecainide, thenalidine, and bunamiodyl, the model can yield an explicit, testable hypothesis about the mechanisms of a molecule’s toxicity. Experimentally validating these quinone formation predictions is beyond the scope of this study but is planned for future work.
In another application, we see that the model’s scores accurately reflect the impact of rational drug modifications. Often, small modification to a molecule can prevent reactive metabolite formation while retaining drug efficacy. For example, the antimalarial drug amodiaquine forms a reactive quinone-imine metabolite that causes the hepatotoxicity and agranulocytosis sometimes associated with amodiaquine treatment, severely limiting its use.18,78–80 Amodiaquine has an analogue that retains pharmacological efficacy while preventing reactive metabolite formation.18,81,82 The quinone formation model detects the effect of this subtle change, cleanly distinguishing amodiaquine from its safe analogue (Figure 14). The quinone formation model also correctly predicts amodiaquine’s QP, demonstrating how the model could help guide rational drug modifications that require specific knowledge about where on a molecule a reactive metabolite forms.
Figure 14.
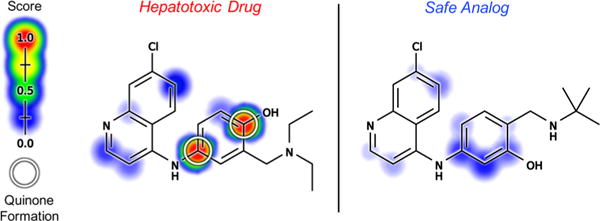
Model reflects the impact of structural modification to prevent quinone formation. The hepatotoxicity and agranulocytosis that have significantly curtailed the use of the antimalarial drug amodiaquine (left) have been traced to the formation of a reactive quinone-imine metabolite.18,78–80 An amodiaquine analogue (right) has been identified that retains pharmacological efficacy while preventing reactive metabolite formation.18,81,82 Molecule quinone formation scores (MQS) cleanly distinguished amodiaquine (MQS: 0.93) from this safe analogue (MQS: 0.18). Additionally, pair-level quinone formation scores (PQS) correctly predicted amodiaquine’s quinone formation pair, the atoms of which are circled. Scores (ranging from 0 to 0.98) are indicated by the colored shading, assigning each atom the probabilistic OR of all pair-level quinone formation scores including the atom. Cross-validated scores are reported for amodiaquine.
MODEL LIMITATIONS
Quinone formation is only one piece of the toxicity puzzle, and this model is only one step toward effective toxicity management. For example, while the model predicts quinone formation, it does not predict the reactivity of those quinones. Subtle changes in ring substituents can have a large impact on quinone reactivity and toxicity.83 Consequently, in future work we plan to incorporate the quinone formation model with already-developed reactivity models.44,45
Moreover, the quinone formation model does not correct for alternate metabolic pathways that may detoxify molecules before they can form quinones. Because of these metabolic alternatives, quinone formation is sometimes observed in vitro but does not occur in vivo.18 Furthermore, after quinones form, they can be further metabolized by reduction, which, for example, can produce hydroquinones that can then be conjugated and eliminated.84 In the long run, integration of the quinone formation model with models of reduction and detoxification, such as uridine 5′-diphospho-glucuronosyltransferase conjugation,85 could offer more nuanced predictions by weighing quinone formation against other metabolic routes.
Likewise, quinone formation that occurs in several metabolic steps may not be detected by the current model, which focused on one- and two-step quinone formation. As we continue to develop more complex systems of metabolite structure prediction, we plan to expand the quinone formation model by explicitly modeling intermediate structures, potentially extending its utility beyond two metabolic steps. Additionally, the current method only uses topological descriptors. As evidenced by the quinone formation model’s accuracy, these descriptors are tightly correlated with electronic structure, but performance on rare subclasses or new molecules could be affected by not explicitly including quantum chemical descriptors. In the future, we will consider expanding our method to include quantum chemistry, such as descriptors relating to the half-reactions between the substrate and Compound I within cytochromes P450.
Finally, there is no guarantee that the model’s applicability domain extends beyond its training domain of metabolically studied drug-like molecules. New areas of chemical space, possibly only currently explored in proprietary data within pharmaceutical companies, may not be well suited to the current topological-descriptor-based model trained on literature-derived data. However, this study’s methodology could be easily applied to new data, thereby expanding the utility of the model.
CONCLUSIONS
This study constructed a novel method that forecasts the formation of reactive quinone metabolites. The QP-trained quinone formation model predicted with 97.6% AUC the QPs within quinone-forming molecules. These pair-level predictions suggest where drug candidates could be modified to make them safer. The model also separated quinone-forming and non-quinone-forming molecules with 88.2% AUC. Furthermore, the model distinguished molecules containing the same quinone-formation structural alert and detected the impact of rational drug modification to prevent quinone formation. Molecule-level quinone formation predictions can be used to flag problematic molecules in the early stages of drug development. However, eventually both reactivity and metabolism must be modeled to accurately predict reactive metabolite formation. While we have recently made progress on modeling the reactivity of diverse chemicals,44,45 previous studies of metabolism have primarily focused on predicting sites of metabolism, rather than actual metabolite structures.32,37,43,47,85–88 This study explicitly predicted the formation of quinones, one of the most common types of reactive metabolites, thereby supplying an essential piece of a unified model of reactivity and metabolism.
Supplementary Material
Acknowledgments
We thank Na Le Dang and Matthew Matlock for invaluable constructive feedback. We are also grateful to the developers of the open-source chemoinformatics tools Open Babel and RDKit.
Funding
Research reported in this publication was supported by the National Library Of Medicine of the National Institutes of Health under award numbers R01LM012222 and R01LM012482. Computations were performed using the facilities of the Washington University Center for High Performance Computing, which were partially funded by National Institutes of Health grants numbers 1S10RR022984-01A1 and 1S10OD018091-01. We also thank both the Department of Immunology and Pathology at the Washington University School of Medicine and the Washington University Center for Biological Systems Engineering for their generous support of this work.
ABBREVIATIONS
- AMD
Accelrys metabolite database
- AQS
atom quinone formation scores
- AUC
area under the receiver operating characteristic curve
- HOMO
highest occupied molecular orbital
- IADRs
idiosyncratic adverse drug reactions
- MQS
molecule quinone formation scores
- PQS
pair quinone formation scores
- QPs
quinone-forming pairs
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemrestox.6b00385.
Frequencies of different ring sizes in which atoms of quinone formation were found; the exact patterns used to define the structural alerts investigated in Table 1 and Figures 8–12; ROC curves for all motifs; atom-, atom-pair-, and molecule-level topological descriptors used to train the model; quantum chemical descriptors used to establish baseline performances; model’s reliance on individual descriptors; cross-validated molecule ROC curve from the training data; and comparison of two approaches for mapping atom-pair-level predictions to the atom-level for visualization (PDF)
Files in the MotifPairScores directory corresponding to the motifs listed in Table S2; list of drugs predicted to form quinones; and the reaction and molecule registry numbers, labeled as RXNREGNO and MOLREGNO of the quinone formation data sets drawn from the June 2015 AMD release (ZIP)
ORCID
Tyler B. Hughes: 0000-0001-6221-9014
Notes
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare no competing financial interest.
References
- 1.McKim JM., Jr Building a tiered approach to in vitro predictive toxicity screening: a focus on assays with in vivo relevance. Comb Chem High Throughput Screening. 2010;13:188. doi: 10.2174/138620710790596736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guengerich FP. Mechanisms of drug toxicity and relevance to pharmaceutical development. Drug Metab Pharmacokinet. 2011;26:3–14. doi: 10.2133/dmpk.dmpk-10-rv-062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discovery. 2004;3:711–716. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- 4.Lee WM. Drug-induced hepatotoxicity. N Engl J Med. 2003;349:474–485. doi: 10.1056/NEJMra021844. [DOI] [PubMed] [Google Scholar]
- 5.Ostapowicz G, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 2002;137:947–954. doi: 10.7326/0003-4819-137-12-200212170-00007. [DOI] [PubMed] [Google Scholar]
- 6.Stachulski AV, et al. The Generation, Detection, and Effects of Reactive Drug Metabolites. Med Res Rev. 2013;33:985–1080. doi: 10.1002/med.21273. [DOI] [PubMed] [Google Scholar]
- 7.Pichler WJ, Naisbitt DJ, Park BK. Immune pathomechanism of drug hypersensitivity reactions. J Allergy Clin Immunol. 2011;127:S74–S81. doi: 10.1016/j.jaci.2010.11.048. [DOI] [PubMed] [Google Scholar]
- 8.Adams DH, Ju C, Ramaiah SK, Uetrecht J, Jaeschke H. Mechanisms of immune-mediated liver injury. Toxicol Sci. 2010;115:307–321. doi: 10.1093/toxsci/kfq009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shenton JM, Chen J, Uetrecht JP. Animal models of idiosyncratic drug reactions. Chem-Biol Interact. 2004;150:53–70. doi: 10.1016/j.cbi.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 10.Lutz WK. In vivo covalent binding of organic chemicals to DNA as a quantitative indicator in the process of chemical carcinogenesis. Mutat Res, Rev Genet Toxicol. 1979;65:289–356. doi: 10.1016/0165-1110(79)90006-x. [DOI] [PubMed] [Google Scholar]
- 11.Gelboin HV. Benzo [alpha] pyrene metabolism, activation and carcinogenesis: role and regulation of mixed-function oxidases and related enzymes. Physiol Rev. 1980;60:1107–1166. doi: 10.1152/physrev.1980.60.4.1107. [DOI] [PubMed] [Google Scholar]
- 12.Srivastava A, Maggs J, Antoine D, Williams D, Smith D, Park B. Adverse Drug Reactions. Springer; Berlin, Germany: 2010. pp. 165–194. [Google Scholar]
- 13.Knowles SR, Uetrecht J, Shear NH. Idiosyncratic drug reactions: the reactive metabolite syndromes. Lancet. 2000;356:1587–1591. doi: 10.1016/S0140-6736(00)03137-8. [DOI] [PubMed] [Google Scholar]
- 14.Numata K, Kubo M, Watanabe H, Takagi K, Mizuta H, Okada S, Kunkel SL, Ito T, Matsukawa A. Overexpression of suppressor of cytokine signaling-3 in T cells exacerbates acetaminophen-induced hepatotoxicity. J Immunol. 2007;178:3777–3785. doi: 10.4049/jimmunol.178.6.3777. [DOI] [PubMed] [Google Scholar]
- 15.Schultz TW, Yarbrough JW, Hunter RS, Aptula AO. Verification of the structural alerts for Michael acceptors. Chem Res Toxicol. 2007;20:1359–1363. doi: 10.1021/tx700212u. [DOI] [PubMed] [Google Scholar]
- 16.Testa B, Pedretti A, Vistoli G. Reactions and enzymes in the metabolism of drugs and other xenobiotics. Drug Discovery Today. 2012;17:549–560. doi: 10.1016/j.drudis.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 17.Schwöbel J, Wondrousch D, Koleva Y, Madden J, Cronin M, Schüürmann G. Prediction of michael-type acceptor reactivity toward glutathione. Chem Res Toxicol. 2010;23:1576–1585. doi: 10.1021/tx100172x. [DOI] [PubMed] [Google Scholar]
- 18.Stepan AF, Walker DP, Bauman J, Price DA, Baillie TA, Kalgutkar AS, Aleo MD. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: a perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem Res Toxicol. 2011;24:1345–1410. doi: 10.1021/tx200168d. [DOI] [PubMed] [Google Scholar]
- 19.Monks TJ, Jones DC. The metabolism and toxicity of quinones, quinonimines, quinone methides, and quinone-thioethers. Curr Drug Metab. 2002;3:425–438. doi: 10.2174/1389200023337388. [DOI] [PubMed] [Google Scholar]
- 20.Dahlin DC, Miwa GT, Lu A, Nelson SD. N-acetyl-p-benzoquinone imine: a cytochrome P-450-mediated oxidation product of acetaminophen. Proc Natl Acad Sci U S A. 1984;81:1327–1331. doi: 10.1073/pnas.81.5.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Awad HM, Boersma MG, Vervoort J, Rietjens IM. Peroxidase-catalyzed formation of quercetin quinone methide–glutathione adducts. Arch Biochem Biophys. 2000;378:224–233. doi: 10.1006/abbi.2000.1832. [DOI] [PubMed] [Google Scholar]
- 22.Meunier G, Meunier B. Peroxidase-catalyzed O-demethylation reactions. Quinone-imine formation from 9-methoxyellipticine derivatives. J Biol Chem. 1985;260:10576–10582. [PubMed] [Google Scholar]
- 23.Basketter D, Dooms-Goossens A, Karlberg AT, Lepoittevin JP. The chemistry of contact allergy: why is a molecule allergenic? Contact Dermatitis. 1995;32:65–73. doi: 10.1111/j.1600-0536.1995.tb00748.x. [DOI] [PubMed] [Google Scholar]
- 24.Anzenbacher P, Anzenbacherova E. Cytochromes P450 and metabolism of xenobiotics. Cell Mol Life Sci. 2001;58:737–747. doi: 10.1007/PL00000897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Albano E, Rundgren M, Harvison P, Nelson S, Moldeus P. Mechanisms of N-acetyl-p-benzoquinone imine cytotoxicity. Mol Pharmacol. 1985;28:306–311. [PubMed] [Google Scholar]
- 26.Ross D, Moldeus P. Generation of reactive species and fate of thiols during peroxidase-catalyzed metabolic activation of aromatic amines and phenols. Environ Health Perspect. 1985;64:253. doi: 10.1289/ehp.8564253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang P, Dalvie D, Smith E, Renner M. Bioactivation of lumiracoxib by peroxidases and human liver microsomes: identification of multiple quinone imine intermediates and GSH adducts. Chem Res Toxicol. 2009;22:106–117. doi: 10.1021/tx8002356. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Slatter JG, Zhang Z, Li Y, Doss GA, Braun MP, Stearns RA, Dean DC, Baillie TA, Tang W. In vitro metabolic activation of lumiracoxib in rat and human liver preparations. Drug Metab Dispos. 2008;36:469–473. doi: 10.1124/dmd.107.019018. [DOI] [PubMed] [Google Scholar]
- 29.Zhu M, Ma L, Zhang H, Humphreys WG. Detection and structural characterization of glutathione-trapped reactive metabolites using liquid chromatography-high-resolution mass spectrometry and mass defect filtering. Anal Chem. 2007;79:8333–8341. doi: 10.1021/ac071119u. [DOI] [PubMed] [Google Scholar]
- 30.Bauman JN, Frederick KS, Sawant A, Walsky RL, Cox LM, Obach RS, Kalgutkar AS. Comparison of the bioactivation potential of the antidepressant and hepatotoxin nefazodone with aripiprazole, a structural analog and marketed drug. Drug Metab Dispos. 2008;36:1016–1029. doi: 10.1124/dmd.108.020545. [DOI] [PubMed] [Google Scholar]
- 31.Kalgutkar AS, Vaz AD, Lame ME, Henne KR, Soglia J, Zhao SX, Abramov YA, Lombardo F, Collin C, Hendsch ZS, Hop CECA. Bioactivation of the nontricyclic antidepressant nefazodone to a reactive quinone-imine species in human liver microsomes and recombinant cytochrome P450 3A4. Drug Metab Dispos. 2005;33:243–253. doi: 10.1124/dmd.104.001735. [DOI] [PubMed] [Google Scholar]
- 32.Zaretzki J, Matlock M, Swamidass SJ. XenoSite: Accurately predicting CYP-mediated sites of metabolism with neural networks. J Chem Inf Model. 2013;53:3373–3383. doi: 10.1021/ci400518g. [DOI] [PubMed] [Google Scholar]
- 33.Campagna-Slater V, Pottel J, Therrien E, Cantin LD, Moitessier N. Development of a Computational Tool to Rival Experts in the Prediction of Sites of Metabolism of Xenobiotics by P450s. J Chem Inf Model. 2012;52:2471. doi: 10.1021/ci3003073. [DOI] [PubMed] [Google Scholar]
- 34.Rydberg P, Gloriam DE, Zaretzki J, Breneman C, Olsen L. SMARTCyp: A 2D method for prediction of cytochrome P450-mediated drug metabolism. ACS Med Chem Lett. 2010;1:96–100. doi: 10.1021/ml100016x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zaretzki J, Rydberg P, Bergeron C, Bennett KP, Olsen L, Breneman CM. RS-Predictor Models Augmented with SMARTCyp Reactivities: Robust Metabolic Regioselectivity Predictions for Nine CYP Isozymes. J Chem Inf Model. 2012;52:1637–1659. doi: 10.1021/ci300009z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones JP, Mysinger M, Korzekwa KR. Computational models for cytochrome P450: a predictive electronic model for aromatic oxidation and hydrogen atom abstraction. Drug Metab Dispos. 2002;30:7–12. doi: 10.1124/dmd.30.1.7. [DOI] [PubMed] [Google Scholar]
- 37.Kim DN, Cho KH, Oh WS, Lee CJ, Lee SK, Jung J, No KT. EaMEAD: Activation energy prediction of cytochrome P450 mediated metabolism with effective atomic descriptors. J Chem Inf Model. 2009;49:1643–1654. doi: 10.1021/ci900011g. [DOI] [PubMed] [Google Scholar]
- 38.Dixit VA, Deshpande S. Advances in Computational Prediction of Regioselective and Isoform-Specific Drug Metabolism Catalyzed by CYP450s. ChemistrySelect. 2016;1:6571–6597. [Google Scholar]
- 39.Guengerich FP. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem Res Toxicol. 2001;14:611–650. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- 40.Lamb DC, Waterman MR, Zhao B. Streptomyces cytochromes P450: applications in drug metabolism. Expert Opin Drug Metab Toxicol. 2013;9:1279–1294. doi: 10.1517/17425255.2013.806485. [DOI] [PubMed] [Google Scholar]
- 41.Hughes TB, Miller GP, Swamidass SJ. Modeling Epoxidation of Drug-like molecules with a Deep Machine Learning Network. ACS Cent Sci. 2015;1:168–180. doi: 10.1021/acscentsci.5b00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.O’Boyle NM, Morley C, Hutchison GR. Pybel: a Python wrapper for the OpenBabel cheminformatics toolkit. Chem Cent J. 2008;2:5. doi: 10.1186/1752-153X-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matlock MK, Hughes TB, Swamidass SJ. XenoSite server: a web-available site of metabolism prediction tool. Bioinformatics. 2015;31:1136–1137. doi: 10.1093/bioinformatics/btu761. [DOI] [PubMed] [Google Scholar]
- 44.Hughes TB, Dang NL, Miller GP, Swamidass SJ. Modeling Reactivity to Biological Macromolecules with a Deep Multitask Network. ACS Cent Sci. 2016;2:529–537. doi: 10.1021/acscentsci.6b00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hughes TB, Miller GP, Swamidass SJ. Site of Reactivity Models Predict Molecular Reactivity of Diverse Chemicals with Glutathione. Chem Res Toxicol. 2015;28:797–809. doi: 10.1021/acs.chemrestox.5b00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rydberg P, Gloriam DE, Olsen L. The SMARTCyp cytochrome P450 metabolism prediction server. Bio-informatics. 2010;26:2988–2989. doi: 10.1093/bioinformatics/btq584. [DOI] [PubMed] [Google Scholar]
- 47.Rydberg P, Olsen L. Predicting drug metabolism by cytochrome P450 2C9: comparison with the 2D6 and 3A4 isoforms. ChemMedChem. 2012;7:1202–1209. doi: 10.1002/cmdc.201200160. [DOI] [PubMed] [Google Scholar]
- 48.Zaretzki J, Bergeron C, Rydberg P, Huang T-W, Bennett KP, Breneman CM. RS-Predictor: A new tool for predicting sites of cytochrome P450-mediated metabolism applied to CYP 3A4. J Chem Inf Model. 2011;51:1667–1689. doi: 10.1021/ci2000488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zaretzki J, Bergeron C, Huang T-W, Rydberg P, Swamidass SJ, Breneman CM. RS-WebPredictor: a server for predicting CYP-mediated sites of metabolism on drug-like molecules. Bioinformatics. 2013;29:497–498. doi: 10.1093/bioinformatics/bts705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stewart JJ. MOPAC: a semiempirical molecular orbital program. J Comput-Aided Mol Des. 1990;4:1–103. doi: 10.1007/BF00128336. [DOI] [PubMed] [Google Scholar]
- 51.Hostaš J, Řezác J, Hobza P. On the performance of the semiempirical quantum mechanical PM6 and PM7 methods for noncovalent interactions. Chem Phys Lett. 2013;568:161–166. [Google Scholar]
- 52.Cruciani G, Baroni M, Benedetti P, Goracci L, Fortuna CG. Exposition and reactivity optimization to predict sites of metabolism in chemicals. Drug Discovery Today: Technol. 2013;10:e155–e165. doi: 10.1016/j.ddtec.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 53.Zamora I, Afzelius L, Cruciani G. Predicting drug metabolism: a site of metabolism prediction tool applied to the cytochrome P450 2C9. J Med Chem. 2003;46:2313–2324. doi: 10.1021/jm021104i. [DOI] [PubMed] [Google Scholar]
- 54.Swamidass SJ, Azencott CA, Daily K, Baldi P. A CROC stronger than ROC: measuring, visualizing and optimizing early retrieval. Bioinformatics. 2010;26:1348–1356. doi: 10.1093/bioinformatics/btq140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Charba JP, Klein WH. Skill in precipitation forecasting in the National Weather Service. Bull Am Meteorol Soc. 1980;61:1546–1555. [Google Scholar]
- 56.Kalgutkar A. Should the Incorporation of Structural Alerts be Restricted in Drug Design? An Analysis of Structure-Toxicity Trends with Aniline-Based Drugs. Curr Med Chem. 2015;22:438–464. doi: 10.2174/0929867321666141112122118. [DOI] [PubMed] [Google Scholar]
- 57.O’brien P. Molecular mechanisms of quinone cytotoxicity. Chem-Biol Interact. 1991;80:1–41. doi: 10.1016/0009-2797(91)90029-7. [DOI] [PubMed] [Google Scholar]
- 58.Cortes C, Mohri M. Confidence Intervals for the Area Under the ROC Curve. Adv Neural Inf Proc Syst. 2004:305–312. [Google Scholar]
- 59.Kalgutkar AS, Dalvie D. Predicting Toxicities of Reactive Metabolite-Positive Drug Candidates. Annu Rev Pharmacol Toxicol. 2015;55:35–54. doi: 10.1146/annurev-pharmtox-010814-124720. [DOI] [PubMed] [Google Scholar]
- 60.Mahajan MK, Uttamsingh V, Daniels JS, Gan LS, LeDuc BW, Williams DA. In vitro metabolism of oxymetazoline: evidence for bioactivation to a reactive metabolite. Drug Metab Dispos. 2011;39:693–702. doi: 10.1124/dmd.110.036004. [DOI] [PubMed] [Google Scholar]
- 61.Numazawa M, Yoshimura A. Biological aromatization of Δ 4, 6-and Δ 1, 4, 6-androgens and their 6-alkyl analogs, potent inhibitors of aromatase. J Steroid Biochem Mol Biol. 1999;70:189–196. doi: 10.1016/s0960-0760(99)00105-3. [DOI] [PubMed] [Google Scholar]
- 62.Favetta P, Guitton J, Degoute C, Van Daele L, Boulieu R. High-performance liquid chromatographic assay to detect hydroxylate and conjugate metabolites of propofol in human urine. J Chromatogr, Biomed Appl. 2000;742:25–35. doi: 10.1016/s0378-4347(00)00097-9. [DOI] [PubMed] [Google Scholar]
- 63.Nelis H, De Leenheerx A. Evidence for metabolic inertness of doxycycline. J Pharm Sci. 1981;70:226–228. doi: 10.1002/jps.2600700230. [DOI] [PubMed] [Google Scholar]
- 64.Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, Tzur D, Gautam B, Hassanali M. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36:D901–D906. doi: 10.1093/nar/gkm958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hopmann G, Surmann T. Cholestatic jaundice during flecainide therapy. Dtsch Med Wochenschr. 1984;109:1863. [PubMed] [Google Scholar]
- 66.McQuinn R, Quarfoth G, Johnson J, Banitt E, Pathre S, Chang S, Ober R, Conard G. Biotransformation and elimination of 14C-flecainide acetate in humans. Drug Metab Dispos. 1984;12:414–420. [PubMed] [Google Scholar]
- 67.Andrès E, Federici L, Weitten T, Vogel T, Alt M. Recognition and management of drug-induced blood cytopenias: the example of drug-induced acute neutropenia and agranulocytosis. Expert Opin Drug Saf. 2008;7:481–489. doi: 10.1517/14740338.7.4.481. [DOI] [PubMed] [Google Scholar]
- 68.Edwards I, Lindquist M, Wiholm BE, Napke E. Quality criteria for early signals of possible adverse drug reactions. Lancet. 1990;336:156–158. doi: 10.1016/0140-6736(90)91669-2. [DOI] [PubMed] [Google Scholar]
- 69.Stevenson J, Kennedy A. A fatal case of agranulocytosis due to thenalidine tartrate complicated by acute renal failure and mycelial abscesses of brain. Scott Med J. 1961;6:522–5. doi: 10.1177/003693306100601104. [DOI] [PubMed] [Google Scholar]
- 70.Arneborn P, Palmblad J. Drug-Induced Neutropenia–A Survey for Stockholm 1973–1978. Acta Med Scand. 1982;212:289–292. doi: 10.1111/j.0954-6820.1982.tb03216.x. [DOI] [PubMed] [Google Scholar]
- 71.Stephens MD. The Dawn of Drug Safety. George Mann; Hampshire, U.K.: 2010. [Google Scholar]
- 72.Adams DA, Perry S. Agranulocytosis associated with thenalidine (sandostene) tartrate therapy: report of three cases. J Am Med Assoc. 1958;167:1207–1210. doi: 10.1001/jama.1958.02990270013004. [DOI] [PubMed] [Google Scholar]
- 73.Zolov B. Agranulocytosis resulting from sandostene. J Maine Med Assoc. 1958;49:335–337. [PubMed] [Google Scholar]
- 74.Ninan B, Wertheimer AI. Withdrawing Drugs in the US Versus Other Countries. Innovations Pharm. 2012;3:6. [Google Scholar]
- 75.Gramec D, Peterlin Mašič L, Sollner Dolenc M. Bioactivation potential of thiophene-containing drugs. Chem Res Toxicol. 2014;27:1344–1358. doi: 10.1021/tx500134g. [DOI] [PubMed] [Google Scholar]
- 76.Orhan H. Extrahepatic targets and cellular reactivity of drug metabolites. Curr Med Chem. 2015;22:408–437. doi: 10.2174/0929867321666140826113716. [DOI] [PubMed] [Google Scholar]
- 77.Schreiner GE. Nephrotoxicity and Diagnostic Agents. J Am Med Assoc. 1966;196:413–415. [PubMed] [Google Scholar]
- 78.Churchill FC, Patchen LC, Campbell CC, Schwartz IK, Nguyen-Dinh P, Dickinson CM. Amodiaquine as a prodrug: importance of metabolite (s) in the antimalarial effect of amodiaquine in humans. Life Sci. 1985;36:53–62. doi: 10.1016/0024-3205(85)90285-1. [DOI] [PubMed] [Google Scholar]
- 79.Maggs J, Tingle M, Kitteringham N, Park B. Drug-protein conjugates–XIV: mechanisms of formation of protein-arylating intermediates from amodiaquine, a myelotoxin and hepatotoxin in man. Biochem Pharmacol. 1988;37:303–311. doi: 10.1016/0006-2952(88)90733-2. [DOI] [PubMed] [Google Scholar]
- 80.Christie G, Breckenridge A, Park B. Drug-protein conjugates–XVIII: Detection of antibodies towards the antimalarial amodiaquine and its quinone imine metabolite in man and the rat. Biochem Pharmacol. 1989;38:1451–1458. doi: 10.1016/0006-2952(89)90184-6. [DOI] [PubMed] [Google Scholar]
- 81.O’Neill PM, Harrison AC, Storr RC, Hawley SR, Ward SA, Park BK. The effect of fluorine substitution on the metabolism and antimalarial activity of amodiaquine. J Med Chem. 1994;37:1362–1370. doi: 10.1021/jm00035a017. [DOI] [PubMed] [Google Scholar]
- 82.O’Neill PM, et al. Synthesis, antimalarial activity, and preclinical pharmacology of a novel series of 4′-fluoro and 4′-chloro analogues of amodiaquine. Identification of a suitable “back-up” compound for N-tert-butyl isoquine. J Med Chem. 2009;52:1828–1844. doi: 10.1021/jm8012757. [DOI] [PubMed] [Google Scholar]
- 83.Chan K, Jensen N, O’Brien PJ. Structure– activity relationships for thiol reactivity and rat or human hepatocyte toxicity induced by substituted p-benzoquinone compounds. J Appl Toxicol. 2008;28:608–620. doi: 10.1002/jat.1312. [DOI] [PubMed] [Google Scholar]
- 84.Monks TJ, Hanzlik RP, Cohen GM, Ross D, Graham DG. Quinone chemistry and toxicity. Toxicol Appl Pharmacol. 1992;112:2–16. doi: 10.1016/0041-008x(92)90273-u. [DOI] [PubMed] [Google Scholar]
- 85.Dang NL, Hughes TB, Krishnamurthy V, Swamidass SJ. A simple model predicts UGT-mediated metabolism. Bioinformatics. 2016;32:3183–3189. doi: 10.1093/bioinformatics/btw350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zaretzki J, Boehm KM, Swamidass SJ. Improved Prediction of CYP-Mediated Metabolism with Chemical Fingerprints. J Chem Inf Model. 2015;55:972–982. doi: 10.1021/ci5005652. [DOI] [PubMed] [Google Scholar]
- 87.Zaretzki J, Browning MR, Hughes TB, Swamidass SJ. Extending P450 Site-of-Metabolism Models with Region-Resolution Data. Bioinformatics. 2015;31:1966–1973. doi: 10.1093/bioinformatics/btv100. [DOI] [PubMed] [Google Scholar]
- 88.Rudik A, Dmitriev A, Lagunin A, Filimonov D, Poroikov V. SOMP: web-server for in silico prediction of sites of metabolism for drug-like compounds. Bioinformatics. 2015;31:2046–2048. doi: 10.1093/bioinformatics/btv087. [DOI] [PubMed] [Google Scholar]
- 89.Sharma AM, Li Y, Novalen M, Hayes MA, Uetrecht J. Bioactivation of nevirapine to a reactive quinone methide: implications for liver injury. Chem Res Toxicol. 2012;25:1708–1719. doi: 10.1021/tx300172s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wen B, Chen Y, Fitch WL. Metabolic activation of nevirapine in human liver microsomes: dehydrogenation and inactivation of cytochrome P450 3A4. Drug Metab Dispos. 2009;37:1557–1562. doi: 10.1124/dmd.108.024851. [DOI] [PubMed] [Google Scholar]
- 91.Zheng J, Ma L, Xin B, Olah T, Humphreys WG, Zhu M. Screening and identification of GSH-trapped reactive metabolites using hybrid triple quadruple linear ion trap mass spectrometry. Chem Res Toxicol. 2007;20:757–766. doi: 10.1021/tx600277y. [DOI] [PubMed] [Google Scholar]
- 92.Lee J, Son J, Chung SJ, Lee ES, Kim DH. In vitro and in vivo metabolism of pyronaridine characterized by low-energy collision-induced dissociation mass spectrometry with electro-spray ionization. J Mass Spectrom. 2004;39:1036–1043. doi: 10.1002/jms.663. [DOI] [PubMed] [Google Scholar]
- 93.Salvatorelli E, Menna P, Paz OG, Chello M, Covino E, Singer JW, Minotti G. The novel anthracenedione, pixantrone, lacks redox activity and inhibits doxorubicinol formation in human myocardium: insight to explain the cardiac safety of pixantrone in doxorubicin-treated patients. J Pharmacol Exp Ther. 2013;344:467–478. doi: 10.1124/jpet.112.200568. [DOI] [PubMed] [Google Scholar]
- 94.Yue W, Santen R, Wang JP, Li Y, Verderame M, Bocchinfuso W, Korach K, Devanesan P, Todorovic R, Rogan E, Cavalier E. Genotoxic metabolites of estradiol in breast: potential mechanism of estradiol induced carcinogenesis. J Steroid Biochem Mol Biol. 2003;86:477–486. doi: 10.1016/s0960-0760(03)00377-7. [DOI] [PubMed] [Google Scholar]
- 95.Numazawa M, Handa W, Hasegawa C, Takahashi M. Structure-activity relationships of 2α-substituted androstenedione analogs as aromatase inhibitors and their aromatization reactions. J Steroid Biochem Mol Biol. 2005;97:353–359. doi: 10.1016/j.jsbmb.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 96.Fang ZZ, Yang L. Characterization of in vitro bioactivation of noscapine in Human Liver Microsomes. Drug Metab Rev. 2010:79–79. [Google Scholar]
- 97.Yu L, Liu H, Li W, Zhang F, Luckie C, Van Breemen RB, Thatcher GR, Bolton JL. Oxidation of raloxifene to quinoids: potential toxic pathways via a diquinone methide and o-quinones. Chem Res Toxicol. 2004;17:879–888. doi: 10.1021/tx0342722. [DOI] [PubMed] [Google Scholar]
- 98.Courts FL, Williamson G. The C-glycosyl flavonoid, aspalathin, is absorbed, methylated and glucuronidated intact in humans. Mol Nutr Food Res. 2009;53:1104–1111. doi: 10.1002/mnfr.200800569. [DOI] [PubMed] [Google Scholar]
- 99.Meyer MR, Du P, Schuster F, Maurer HH. Studies on the metabolism of the α-pyrrolidinophenone designer drug methylenedioxy-pyrovalerone (MDPV) in rat and human urine and human liver microsomes using GC–MS and LC–high-resolution MS and its detectability in urine by GC–MS. J Mass Spectrom. 2010;45:1426–1442. doi: 10.1002/jms.1859. [DOI] [PubMed] [Google Scholar]
- 100.Li X, He Y, Ruiz CH, Koenig M, Cameron MD. Characterization of dasatinib and its structural analogs as CYP3A4 mechanism-based inactivators and the proposed bioactivation pathways. Drug Metab Dispos. 2009;37:1242–1250. doi: 10.1124/dmd.108.025932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wen B, Zhou M. Metabolic activation of the phenothiazine antipsychotics chlorpromazine and thioridazine to electrophilic iminoquinone species in human liver microsomes and recombinant P450s. Chem-Biol Interact. 2009;181:220–226. doi: 10.1016/j.cbi.2009.05.014. [DOI] [PubMed] [Google Scholar]
- 102.Lin G, Hawes E, McKay G, Korchinski E, Midha K. Metabolism of piperidine-type phenothiazine antipsychotic agents. IV. Thioridazine in dog, man and rat. Xenobiotica. 1993;23:1059–1074. doi: 10.3109/00498259309059422. [DOI] [PubMed] [Google Scholar]
- 103.Srivastava A, Ramachandran S, Hameed SP, Ahuja V, Hosagrahara VP. Identification and mitigation of a reactive metabolite liability associated with aminoimidazoles. Chem Res Toxicol. 2014;27:1586–1597. doi: 10.1021/tx500212c. [DOI] [PubMed] [Google Scholar]
- 104.Bylund J, Macsari I, Besidski Y, Olofsson S, Petersson C, Arvidsson PI, Bueters T. Novel bioactivation mechanism of reactive metabolite formation from phenyl methylisoxazoles. Drug Metab Dispos. 2012;40:2185–2191. doi: 10.1124/dmd.112.047431. [DOI] [PubMed] [Google Scholar]
- 105.Fitch WL, Chen Y, Liu L, Paehler A, Young M. Application of modern drug metabolism structure determination tools and assays to the in vitro metabolism of imiloxan. Drug Metab Lett. 2010;4:77–87. doi: 10.2174/187231210791292753. [DOI] [PubMed] [Google Scholar]
- 106.Ni J, Rowe J, Heidelbaugh T, Sinha S, Acheampong A. Characterization of benzimidazole and other oxidative and conjugative metabolites of brimonidine in vitro and in rats in vivo using on-line H/D exchange LC-MS/MS and stable-isotope tracer techniques. Xenobiotica. 2007;37:205–220. doi: 10.1080/00498250601047897. [DOI] [PubMed] [Google Scholar]
- 107.Ralaivola L, Swamidass SJ, Saigo H, Baldi P. Graph kernels for chemical informatics. Neural Networks. 2005;18:1093–1110. doi: 10.1016/j.neunet.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 108.Erve JC, Svensson MA, von Euler-Chelpin H, Klasson-Wehler E. Characterization of glutathione conjugates of the remoxipride hydroquinone metabolite NCQ-344 formed in vitro and detection following oxidation by human neutrophils. Chem Res Toxicol. 2004;17:564–571. doi: 10.1021/tx034238n. [DOI] [PubMed] [Google Scholar]
- 109.Li F, Chordia MD, Huang T, Macdonald TL. In vitro nimesulide studies toward understanding idiosyncratic hepatotoxicity: diiminoquinone formation and conjugation. Chem Res Toxicol. 2009;22:72–80. doi: 10.1021/tx800152r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.