

Abstract
Fast, inexpensive and noninvasive identification of Alzheimer’s disease (AD) before clinical symptoms emerge would augment our ability to intervene early in the disease. Individuals with fully-penetrant genetic mutations causing autosomal dominant Alzheimer’s disease (ADAD) are essentially certain to develop the disease, providing a unique opportunity to examine biomarkers during the preclinical stage. Using a generalization task that has previously shown to be sensitive to medial temporal lobe pathology, we compared preclinical individuals carrying ADAD mutations to non-carrying kin to determine whether generalization (the ability to transfer previous learning to novel but familiar recombinations) is vulnerable early, before overt cognitive decline. As predicted, results revealed that preclinical ADAD mutation carriers made significantly more errors during generalization than non-carrying kin, despite no differences between groups during learning or retention. This impairment correlated with left hippocampal volume, particularly in mutation carriers. Such identification of generalization deficits in early ADAD may provide an easily implementable and potentially linguistically and culturally neutral way to identify and track cognition in ADAD.
Keywords: Alzheimer’s disease, associative learning, generalization, presenilin-1, amyloid precursor protein, mutation, hippocampus, early onset, preclinical
In the preclinical phase of Alzheimer’s disease (AD), which can last years or decades, underlying pathology of the medial temporal lobe (MTL) region accumulates before reaching a “tipping point” where cognitive and clinical symptoms emerge and affect daily life (Sperling, 2011). In vivo markers of pathology are useful for early detection, allowing therapeutic intervention before cognitive symptoms emerge. Early identification of incipient symptoms is of particular interest in cases of late-onset AD, the most common form of the disease whose incidence increases after age 65 to involve over 40% of persons age 85 and above (Evans et al., 1989). However, future development of overt dementia is often difficult to predict. The study of relatively young persons with autosomal dominant Alzheimer’s disease (ADAD, typically with age of onset between the 30’s and 50’s), in whom the future development of dementia can be predicted with essentially 100% certainty, can provide a model of the more common late-onset AD.
Several biomarkers show great promise for early identification of ADAD, including markers of beta-amyloid deposition, tau accumulation, changes in brain structure (e.g., hippocampal or MTL atrophy), and other biochemical changes (e.g., synaptic damage, oxidative stress) (e.g., see Ringman et al., 2008). These measures provide the foundation for understanding the underlying neurobiological mechanisms of ADAD, and can now be used as a validating standard for exams that are more appropriate for the masses – that is, easy-to-administer, inexpensive, fast and non-invasive cognitive screening tools. This paper investigates one promising measure: the simple, computer-based Acquired Equivalence task.
In this task, individuals learn a series of antecedent-consequent pairs using feedback during an acquisition phase, and then are challenged to transfer previous learning to new situations without feedback in a generalization phase, when familiar stimuli are presented in novel pairings. Early computational models predicted that dysfunction of MTL circuits would produce generalization failures when task demands change, despite intact learning of associations (Gluck & Myers, 1993). More recent patient work, including individuals with MTL damage such as post-traumatic stress disorder or hypoxia, has validated that generalization is a sensitive and selective behavioral marker of hippocampal dysfunction (Bodi, Csibri, Myers, Gluck, & Keri, 2009; Levy-Gigi et al., 2012; Myers et al., 2008a). Recent work has also implicated generalization deficits in mild stages of AD (Bodi et al., 2009). Similar generalization tasks have shown poor generalization among non-demented elderly with hippocampal atrophy visible on structural imaging (Myers et al., 2002; Myers et al., 2003) and that generalization performance could predict cognitive outcome (normal, MCI, probable AD) two years later among non-demented elders who showed no objective evidence of a disease state that could affect neural or cognitive function (Myers, Kluger, Golomb, Gluck, & Ferris, 2008b).
The present study extends this approach to test generally healthy, preclinical, young adult carriers of fully-penetrant autosomal dominant mutations in genes coding for presenilin 1 (PSEN1), presenilin 2 (PSEN2), or amyloid precursor protein (APP) who will develop AD later in life (Kennedy et al., 1993; Sherrington et al., 1996). Although mutation carriers account for only a small percentage of AD cases, they provide a unique opportunity to sensitively and accurately evaluate the earliest cognitive manifestations of the disorder. Though minor qualitative and quantitative differences exist between ADAD and late-onset AD, overall the neuropathological changes, especially in the MTL, are similar (Ringman et al., 2016). For example, presymptomatic ADAD carriers showed increased hippocampus activity relative to non-carrier controls in a face-name associative encoding task that mirrored brain activity in AD patients (Quiroz et al., 2010). Similarly, relative to non-carrier kin, ADAD mutation carriers have reduced fractional anisotropy in the fornix, a major efferent of the hippocampus (Ringman et al., 2007) and show decreased MTL volumes as they approach the expected age of disease diagnosis (Lee et al., 2013). Such MTL atrophy in ADAD mutation carriers is consistent with early AD (Apostolova et al., 2011; Cash et al., 2013).
Aggregate data from the Dominantly Inherited Alzheimer Network (DIAN) confirm the development of cerebrospinal fluid changes, fibrillar amyloid deposition, regional hypometabolism, and measurable structural brain changes in ADAD carriers as long as 15 – 20 years prior to the anticipated time of overt clinical symptoms (Bateman et al., 2012). Similarly, using quantitative neuropsychological testing, cognitive deficits have been identified 5 – 12 years before functional decline and typically parallel those seen early in late-onset AD, with deficits in memory and executive function often seen first (Aguirre-Acevedo et al., 2016; Almkvist et al., 2017; Ringman et al., 2005). However, conventional neuropsychological assessments typically involve paper-and-pencil tasks that are difficult to apply across diverse languages, cultures, and educational levels, and so tests with minimal language components have been proposed. For example, one computerized task involving short-term retention for the binding of features in a visual stimulus differentiated asymptomatic ADAD mutation carriers from healthy controls better than standard neuropsychological tests and did so in persons approximately 9 years prior to the onset of MCI (Parra et al., 2010). A flash-card version of this task has been developed which might be of wider utility, for example, in primary care settings (Della Sala, Kozlova, Stamate, & Parra, 2016). Here, we evaluated whether our visual matching Acquired Equivalence task, which has previously demonstrated sensitivity to MTL pathology and early cognitive decline in non-demented older adults, could differentiate performance among generally healthy, young, preclinical ADAD mutation carriers and their non-carrying kin before other overt cognitive decline. Arguably, this task is more useful for early detection than existent tasks because it takes approximately 15 minutes to complete, runs automatically on a standard laptop computer, is relatively engaging (similar to a short video game) and has been translated and successfully used in populations speaking Arabic, French, Chinese, Italian, Hungarian, and Hebrew.
Based on previous research showing that generalization in our Acquired Equivalence task depends on processing in the MTL (Herzallah et al., 2010; Myers et al., 2008a; Myers et al., 2003), we predicted that young ADAD mutation carriers would show significantly worse generalization than their non-carrying kin despite equivalent learning. Further, we examined the neural correlates of this deficit. Specifically, prior studies have found hippocampal volume reduction in non-demented elderly correlates with poor generalization on this task (Myers et al., 2003) as well as similar tasks (Myers et al., 2002); accordingly, we predicted that worse generalization among mutation carriers in the current study would likewise be associated with smaller hippocampal volumes. We also examined a second MTL region, entorhinal cortex thickness, because it has been implicated in generalization in animal models of acquired equivalence (Coutureau et al., 2002), and because entorhinal atrophy accompanies (or even precedes) hippocampal atrophy in sporadic AD (Bobinski et al., 1999; de Toledo-Morrell, Goncharova, Dickerson, Wilson, & Bennett, 2000). Finally, as a control region, we considered the frontal lobe; we predicted that generalization would not be associated with middle frontal gyrus thickness, based on results from a similar task showing that generalization does not involve the frontal cortex (Chase et al., 2008). Our expectation of a relationship between hippocampal volume and generalization would be consistent with the idea that MTL dysfunction appears early in the progression of preclinical ADAD and that generalization tests might have utility as behavioral markers of this dysfunction, before other cognitive and clinical symptoms emerge.
Methods
Participants
Subjects were a subset of participants in a study of ADAD being performed at UCLA. Participants were either native English- or Spanish-speaking. Participants older than 55 years were excluded to better age-match with non-carrying kin. All participants were non-demented and scored <1 on the Clinical Dementia Rating Scale (CDR), a structured interview of the subject and informant in which subjects are rated: 0 (asymptomatic), 0.5 (equivocal impairment), 1 (mild), 2 (moderate), or 3 (severe dementia) (Morris, 1997).
Participants included 34 non-demented carriers of ADAD mutations and 11 non-carrying kin; see demographic information in Table 1. Participants came from 27 different families with pathogenic mutations in the PSEN1, PSEN2, or APP genes. Of the 45 participants, 13 were at-risk for a common APP mutation (V717I) (Mullan et al., 1993) and 12 for a common PSEN1 mutation (A431E) (Murrell et al., 2006). One subject was at-risk for a PSEN2 mutation (N141I) and the rest of the subjects were at-risk for different PSEN1 mutations (G206A (n = 5), L235V (n = 4), R269H (n = 3), A260V, E184D, E280A, H163R, S212Y, C410Y, G378E (n = 1 each).
Table 1.
Demographics and neuropsychological performance
| Mutation carriers (n = 34) | Non-carrying kin (n = 11) | t-value/chi-square (n.s.) | |
|---|---|---|---|
| Chronological age in years | 35.6 (10.1) | 32.4 (8.9) | −1.14 |
| # female (%) | 23 (68%) | 7 (64%) | .06 |
| Years of education | 11.65 (4.35) | 13.45 (4.13) | 1.21 |
| # whose language of testing was Spanish (%) | 14 (41%) | 2 (18%) | 1.92 |
| Mutation in family (PSEN, APP)* | 24/10 | 8/3 | .02 |
| # positive for APOE ε4 allele (%) | 4 (12%) | 4 (36%) | 3.44 |
| Adjusted Age (Age relative to median age of dementia diagnosis in family) | −15.2 (8.5)† | −19.3 (9.5) | −1.45 |
| MMSE score | 27.8 (3.2) | 28.2 (1.8) | .47 |
| CDR, global score | .24 (.25) | .18 (.25) | .16 |
| (18 CDR=0, 16 CDR=0.5) | (7 CDR=0, 4 CDR=0.5) | .39 | |
| Mean CASI score | 92.81 (6.7) | 92.86 (6.0) | .02 |
Notes. All scores are given as mean (s.d.). MMSE = Mini Mental State Examination; CDR = Clinical Dementia Rating. CASI = Cognitive Abilities Screening Instrument
Further breakdown by specific mutation is not shown to protect subject confidentiality
Data unavailable for two individuals.
n.s. indicates that there were no significant group differences in demographics or neuropsychological peformance (all p’s > .08).
The UCLA Institutional Review Board approved all experimental procedures and research was conducted in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki. The protocol included storing and sharing of data and biospecimens with collaborators and their use in future research studies. All participants provided written informed consent before initiation of any experimental procedures.
Clinical Assessments
Subjects underwent clinical assessments in their preferred language. Assessment included the Mini-Mental Status Examination (MMSE) (Folstein, Folstein, & McHugh, 1975) and the Cognitive Abilities Screening Instrument (CASI), which have both English and Spanish versions and have been used in cross-cultural studies of cognitive aging (Teng et al., 1994).
Each subject’s age in relation to his or her estimated age of dementia onset was calculated. As the age of onset of symptoms is fairly consistent within a family and mutation but more variable between families, an “adjusted age” can be calculated that estimates how many years from disease manifestation a given subject is (Ryman et al., 2014). In our experience, the age of clinical diagnosis of dementia is a more reproducible measure; therefore we calculated an adjusted age for each subject (regardless of mutation status) as his or her chronological age minus the median age of dementia diagnosis in his or her family. For example, someone who is 32 and comes from a family in which median age of dementia diagnosis is 40 would have an adjusted age of “−8,” indicating approximately 8 years until diagnosable dementia. Even though non-carrying kin do not carry the mutation, we still calculated an adjusted age for each subject in the same way as mutation carriers for comparison purposes.
Genetic Testing
Extraction of DNA and genotyping of apolipoprotein E were performed using standard techniques. ApoE SNP genotyping was carried out by real-time PCR on an Applied Biosystems 7900HT Real Time PCR machine (Applied Biosystems, Foster City, CA), using Taqman SNP Genotyping Assays (#C___3084793_20 and C____904973_10 for rs429358 and rs7412, respectively). SDS version 2.3 software was used to analyze the raw data and to call the genotype. The presence or absence of the specific mutation each subject was known to be at-risk for were assessed using standard Sanger sequencing, according to published protocols and primers.
Acquired Equivalence Task
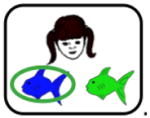
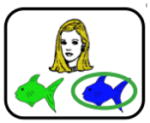
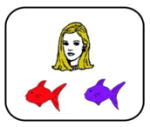
Participants were tested in a quiet room, using software programmed and presented using SuperCard (Allegiant Technologies, San Diego, CA). Instructions were translated into Spanish for Spanish-speaking participants but the task itself is otherwise essentially non-verbal. Methods follow those described previously (Myers et al., 2003). In brief, on each trial, the participant sees both a cartoon face (either a brown-haired man, a blonde-haired woman, a blonde-haired boy or a brown-haired girl, labeled in Table 2 as A1, A2, B1 or B2) and a pair of colored fish (red, green, blue or purple, labeled in Table 2 as X1, X2, Y1 or Y2). Assignment and mapping of faces and fish is randomized across subjects. Each face shares one binary-valued feature with another face: gender (male, female), age (child, adult) and hair color (blonde, brown). The participant is asked to indicate which fish belongs to each face. Correct responses (right vs. left) varied randomly. There is a 1 second pause between trials.
Table 2.
Acquired Equivalence Task and sample screen displays
| Training Phase (Feedback) | Test Phase* (No feedback) | ||
|---|---|---|---|
| Stage 1: Shaping | Stage 2: Equivalence Training | Stage 3: New Consequents | Generalization and Retention |
|
|

|
|
| A1→X1 | A1→X1 | A1→X1 | A1→X1 |
| A2→X1 | A2→X1 | A2→X1 | |
| A1→X2 | A1→X2 | ||
| A2→X2 | |||
| B1→Y1 | B1→Y1 | B1→Y1 | B1→Y1 |
| B2→Y1 | B2→Y1 | B2→Y1 | |
| B1→Y2 | B1→Y2 | ||
| B2→Y2 | |||
Note. During stage 1, participants learn the first 2 associations between difference faces (A1, B1) and fishes (X1, Y1). During stage 2, different faces (A2, B2) are associated with the same fishes (equivalence training), whereas during stage 3, new consequents (X2, Y2) are added. At each stage of the task, participants continued to receive maintenance trials with previously learned fish-face pairs. During the testing phase, participants are tested for retention of the associations learned in stages 1–3 and also on generalization to new pairings of faces and fishes (i.e., A2→X2, B2→Y2). Retention and generalization pairs are interleaved randomly during the test phase. Screen shots of the task (shown above) represent examples of the bolded items; however note that assignment of specific faces and fishes to stimuli (A1, A2, X1, X2, etc.) is randomized across subjects.
The task has two distinct phases: training and test. During training, participants receive feedback to guide learning (e.g., the selected fish is circled and “Correct” or “Incorrect” is displayed for 1 second). Training has three stages, though the start of a new stage is not signaled. In the “shaping” stage, participants learn to pair two faces with specific colored fish (e.g., A1-X1, B1-Y1). This stage continues until the participant makes 4 consecutive correct responses or for a maximum of 20 trials. Next, in the “equivalence training” stage, participants learn to pair new faces with the same colored fish (e.g., A2-X1, B2-Y1). In the process, participants typically learn that some faces (e.g. A1, A2) are equivalent because they map onto the same consequent (e.g., X1). This stage terminates after 8 consecutive correct responses or a maximum of 32 trials. Finally, in a “new consequents” stage, participants learn to pair the original faces with a new colored fish (e.g., A1-X2); this stage terminates after 12 consecutive correct responses or a maximum of 60 trials. At each stage, maintenance trials with previously-trained pairs are interleaved with the new pairs.
A test phase follows in which participants no longer receive feedback. To measure retention, participants are tested on their recall of the trained fish-face associations in random order (12 trials per block). Intermixed within retention trials are 2 novel fish-face pairings representing generalization (presented once each with the fish in either left-right order). Although these pairings were never explicitly trained, participants should show successful generalization, defined as predicting the same outcome (fish) for faces that were trained to be equivalent. For example, because A1 was trained to be functionally equivalent to A2 (in that both predict X1), and because A1 was also paired with X2, participants should generalize that A2 is also paired with X2. Test includes three blocks (36 retention and 12 generalization trials); trial order within a block is randomized across subjects.
MRI Scanning and Volumetric Analyses
Subjects underwent structural MRI scanning using a 3D T1 weighted MPRAGE sequence using the following parameters: 192 slices at 1mm slice thickness, voxel size= 1×1×1mm3, TR/TE=1620/3ms, TI=950ms, TE=3ms, the scan time of 6 minutes. Volumetric segmentation of T1-weighted MPRAGE MRI volumes was completed automatically using FreeSurfer software version 5.3.0 (http://surfer.nmr.mgh.harvard.edu.). The automated cortical reconstruction procedure assigns a neuroanatomical label to each voxel in a MRI volume based on a combination of intensity mapping and probabilistic spatial atlases. FreeSurfer volumetric processing has been validated as an automated method to obtain subcortical volumes (Thompson et al., 1997, but see Wenger et al., 2014). Full description of processing methods have been described previously (Fischl et al., 2002). ROI volumes for cortical and subcortical structures are calculated during this process by multiplying the number of voxels in an ROI by the single voxel volume. To account for individual differences in total brain volume, a residual normalization approach was used to control for total intracranial volume. Therefore, the studentized residual for each region of interest was used in the partial correlation analyses with task performance.
Our brain regions of interest included the bilateral hippocampus, entorhinal cortex, and middle frontal gyrus. The hippocampus was selected based on (1) the memory construct measured by our task as predicted by Gluck and Myers’ (1993) computational model of cortico-hippocampal function in generalization and (2) findings relating poorer generalization to hippocampal atrophy, as measured by neuroimaging (Myers et al., 2002; Myers et al., 2003). The entorhinal cortex was selected based on animal models (Coutureau et al., 2002) and patients with broader MTL dysfunction (e.g., hypoxia (Myers et al., 2008a), post-traumatic stress disorder (Levy-Gigi et al., 2012), and AD (Bodi et al., 2009)). We selected the middle frontal gyrus as a control region based on evidence that generalization deficits are unrelated to frontal functioning and volume (e.g., Chase et al., 2008; Farkas et al., 2008). No other brain areas were examined.
Statistical analysis
Mixed-design ANCOVA, using SPSS 20, compared mutation carriers and non-carrying kin on their cognitive performance during training (acquisition) and test (retention and generalization), followed by planned one-way ANOVAs and Fisher’s Least Significant Difference (LSD) post-hoc tests. Partial correlations examined relationships between generalization performance and our three brain regions of interest (hippocampal volume, entorhinal cortex thickness, middle frontal gyrus thickness), separately for the left and right regions. Analyses were performed for all subjects and, where appropriate, separately for all mutation carriers and non-carriers, and for carriers of APP and PSEN mutations. We controlled for chronological age and education in all analyses. These covariates were selected because separate analyses using chronological age and education as independent variables revealed significant differences in task performance (p’s < .05). The level of significance was set at α= 0.05.
Results
Subjects
All demographic and neuropsychological results are presented in Table 1. Genetic testing confirmed which participants were carriers or non-carrier kin. Participants were all younger or middle-aged adults (mutation carriers: M = 35.6 years, SD = 10.1, range: 19 to 48 years; non-carrying kin: M = 32.4 years, SD = 8.9, range: 19 to 53 years) and were, on average, 16 years from expected age of dementia onset (mutation carriers: M = −15.0 years, SD = 8.3, range: −35 to −4 years; non-carrying kin: M = −19.1 years, SD = 9.4, range: −34 to −1 years). As shown in Table 1, groups did not differ significantly in chronological age, gender, education, language of testing, mutation in family, prevalence of the ε4 allele of APOE, adjusted age to typical familial dementia onset, or neuropsychological performance (all p’s > .08).
Training Phase
All participants reached criterion, by completing each training stage in fewer than the maximum allowed trials. On average, mutation carriers required 54 ± 30 trials to reach criterion across training while non-carriers required 53 ± 24 trials.
A Group (mutation carriers, non-carrying kin) x Training Stage (1–3) mixed-design ANCOVA on mean number of total errors, with chronological age and education as covariates, revealed no significant main effects of Group, F(1, 41) = .27, p = .61, η2= .02, or Training Stage, F(2, 82) = 1.04, p = .36, η2= .03, as well as no interaction, F(2, 82) = .12, p = .89, η2= .01 (see Figure 1).
Figure 1. Performance on the training phase of the Acquired Equivalence task.

Adjusted mean error scores for Training Phase. Covariates include chronological age and education. Error bars represent the standard error of the mean.
Testing Phase
A mixed ANCOVA on mean proportion of errors, with Group and Trial Type (Generalization, Retention) as independent variables and chronological age, education and learning performance (i.e., number of total training trials) as covariates (see Figure 2). We controlled for learning here because generalization (at least partially) depends on representational changes that occur over the course of training (Shohamy, Myers, Kalanithi, & Gluck, 2008) and indeed, learning performance did correlate with generalization, r(43)= .58, p < .001. A main effect of Group, F(1, 40) = 6.91, p =.012, η2= .15, revealed that non-carrying kin made fewer errors overall than mutation carriers. There was no main effect of Trial Type, F(1, 40) = 1.68, p =.20, η2= .04, indicating that errors were approximately equal across groups for retention and generalization. However, the critical interaction was significant, F(1, 40) = 5.29, p = .027, η2= .12. Follow-up tests revealed no group differences on retention (p = .25); however, as predicted, mutation carriers showed worse generalization (M = 33.8% errors) than non-carrying kin (M = 12.6% errors) (p = .02).
Figure 2. Performance on the test phase of the Acquired Equivalence task.

Adjusted proportion error scores for Test Phase: Retention (old-pairs) and Generalization (new-pairs). Covariates include chronological age, education and learning performance (i.e., number of total training trials). Error bars represent the standard error of the mean.
Because we were interested in cognitive changes during preclinical stages of AD, we reran all analyses excluding individuals who had a total MMSE score less than 25 (n=4, all mutation carriers). Patterns of significance were unchanged with these individuals removed. Similarly, because a CDR score of 0.5 may be considered already in a symptomatic/clinical stage, we added CDR scores as an independent variable. This produced no significant main effects or interactions; all patterns of significance remained otherwise unchanged, indicating no differences in task performance between the subgroups with CDR=0 and CDR=0.5.
Finally, to examine the utility of our behavioral task across cultures and ethnic groups, we examined whether language of testing (English, Spanish) had any influence on generalization. Results showed no significant main effect of language, F(1, 41) = 1.63, p > .05.
MRI Volumetric Analyses
Only 41 participants had available T1 MRI data (30 mutation carriers, 11 non-carrying kin); four scans were either not obtained due to claustrophobia in the scanner or were not available for analysis. In this subset, we observed no group differences in the left or right hippocampal volume (p’s > .49), entorhinal cortex thickness (p’s > .43), or middle frontal gyrus cortical thickness (p’s > .24).
Partial correlations, controlling for chronological age and education, examined the relationship between mean proportion of generalization errors and our brain regions of interest, separately for the left and right regions. In the left hippocampus, a negative correlation across all participants, r(37) = −0.34, p = 0.034, showed that those with more generalization errors had smaller left hippocampal volumes. In a subgroup analysis stratified by mutation status, the association between generalization and hippocampal volumes persisted in the gene mutation carriers only, r(30) = −0.43, p = 0.014 (see Figure 3A), but not in non-carrying kin, r(7)=−0.01, p = 0.98. This association was present both for carriers of APP mutations, r(5) = −0.89, p = 0.008, and PSEN mutations, r(19) = −0.47, p = 0.031, despite the smaller numbers in both of these groups. No significant associations were found with the right hippocampus and generalization (see Figure 3B, all p’s > .21).
Figure 3. Partial correlation for Generalization (Proportion Error) and Left (A) and Right (B) Hippocampal Volume.


Standardized residuals are reported, using chronological age and education as covariates.
To test the specificity of this relationship between hippocampal volume and generalization, we used partial correlations to demonstrate that bilateral hippocampal volume did not generally correlate with task performance (i.e., acquisition and/or retention). That is, neither the right or left hippocampus correlated with acquisition (p’s > .14) or retention (p’s > .22). Furthermore, in contrast to the results linking the hippocampus with generalization, partial correlations revealed that generalization was not associated with entorhinal cortex thickness (right: r(37) = .05, p = .731; left: r(37) = .004, p = .983). Similarly, partial correlations revealed no significant associations between generalization and frontal middle gyrus: left (r(37) = −.10, p = .53) and right (r(37) = .10, p = .55).
Discussion
This study examined whether young, preclinical individuals carrying ADAD mutations show deficits in generalization, before other overt cognitive decline. Otherwise healthy ADAD mutation carriers, who were on average 16 years younger than the expected age of AD diagnosis, exhibited poor generalization relative to their non-carrying kin. This deficit is consistent with MTL dysfunction and emerged despite intact learning and retention of stimulus pairs. Our pattern of results supports previous observations in a group of non-demented, healthy older adults with hippocampal atrophy (consistent with preclinical sporadic AD) who also showed spared learning and retention but impaired generalization compared to non-atrophied controls (Myers et al., 2008b). Furthermore, in the present study, we observed worse generalization among those mutation carriers with smaller left hippocampal volumes. The specificity of the relationship between hippocampal volume and generalization was underscored by findings that hippocampus volume did not relate to learning or retention, and that generalization was not associated with middle frontal gyrus or entorhinal cortex thickness. Taken together, our findings suggest that computer-based generalization tests may be a sensitive measure of the preclinical effects of ADAD.
While the non-carrying kin sample was small relative to the carriers, their low error rate on generalization (M = 13%) matched what has been previously reported among healthy adult controls (typically between 10 to 15%) in studies that used this task (Herzallah et al., 2010; Levy-Gigi et al., 2012; Mattyassy et al., 2012). More importantly, the larger generalization error rate for young, preclinical mutation carriers (mean = 34%) is similar to what has been reported in sporadic AD patients (~50%; Bodi et al., 2009), individuals with impaired hippocampal function due to alcohol dependence (~25%; Mattyassy et al., 2012), older adults with confirmed hippocampal atrophy (~40%; Myers et al., 2003), and amnesic patients with bilateral hippocampal damage (~45%; Myers et al., 2008a). Thus, by demonstrating poor generalization among younger adult ADAD mutation carriers, we reveal cognitive deficits that likely reflect functional changes in the MTL that accompany preclinical AD. The observed relationship of more pronounced generalization deficits in ADAD patients who had smaller left hippocampal volumes confirms this idea. While our task may have utility as a screening tool to detect risk for developing ADAD or in tracking response to therapeutic interventions in early pre-clinical disease, substantial variability in generalization performance among mutation carriers (despite the fact that these individuals will develop ADAD with near certainty) suggests that more work is needed to examine the factors that lead to impaired generalization in ADAD.
The current study overcomes some limits of previous work in interpreting generalization deficits as an indicator of later AD diagnosis. For example, reports using our same task found at-chance generalization in both non-demented individuals who had documented hippocampal atrophy consistent with early AD (Myers et al., 2003) as well as in older adults greater than 75 years of age (Simon & Gluck, 2013) when hippocampal declines may accelerate (Zhang et al., 2010); however, hippocampal atrophy and advanced age are imperfect predictors of future development of AD. Similarly, a different task showed that generalization at baseline, together with delayed paragraph recall, could predict two-year outcome for which non-demented elderly adults would be diagnosed with mild cognitive impairment (MCI) (Myers et al., 2008b). Yet, in some cases, individuals who had been diagnosed with MCI were reclassified as cognitively normal 2 years later, demonstrating the difficulty in assessing later development of AD from a single evaluation in preclinical populations (see Bruscoli & Lovestone, 2004). Our findings, in contrast, examine individuals within whom rates of progression to AD are not as variable; ADAD mutation carriers will develop AD with essentially 100% certainty.
The selective generalization impairment among ADAD mutation carriers does not reflect impaired learning; mutation carriers and non-carrying kin produced similar error responses during learning and required the same number of trials to reach criterion. In fact, generalization was impaired among mutation carriers even when controlling for learning performance. This is consistent with literature showing that learning, but not generalization, is intact among AD patients (Kennedy et al., 1993; Sherrington et al., 1996) whereas the reverse is true in patients with disruption to the striatal system only (Bodi et al., 2009). Poor generalization is also not due to forgetting of the trained pairs that support flexible transfer because mutation carriers and non-carrying kin had nearly equivalent error rates at retention. It is also unlikely that generalization deficits reflect prefrontal dysfunction that may accompany AD (Klunk et al., 2007; Ringman et al., 2011), as we observed no relationships between generalization and the middle frontal gyrus in our groups. This finding is consistent with previous work; generalization abilities do not correlate with tests of frontal lobe functioning, such as Wisconsin Card Sorting, n-back working memory, Trail-Making, and Controlled Oral Word Associations (Chase et al., 2008; Head, Snyder, Girton, Morris, & Buckner, 2005), and generalization deficits were not reported in patients with frontal damage (Farkas et al., 2008).
It is not clear why only the left hippocampus would show a significant relationship with generalization errors in ADAD. To our knowledge, there is no clear evidence for hemispheric lateralization in hippocampal atrophy as conveyed by genetic carriage. Previous studies have suggested that hippocampal volume loss occurs more with some ADAD mutations (e.g. those in APP) than others (e.g. in PSEN1) (Scahill et al., 2013). However, despite our small sample size, we found similar correlations between generalization performance and left hippocampal volume among carriers of APP and PSEN mutations, providing further evidence that the left hippocampus underlies this ability and our findings do not reflect an idiosyncrasy of a specific mutation type. Given that the task could be viewed as a visual matching task, one might have predicted that the right hippocampus would be more involved in generalization (Smith & Milner, 1981). However, our task uses visual stimuli that can be verbalized (i.e., the participant can describe each of the stimuli in a verbal way, such as “blonde woman”), which may recruit left hemisphere processes (Brown, Roth, Saykin, & Beverly-Gibson, 2007; Heilbronner, 1992). That said, our findings align with two recent meta-analyses: one revealed that atrophy of the left MTL is, on average, slightly more severe, which may make it more sensitive to early AD pathology (Shi, Liu, Zhou, Yu, & Jiang, 2009), and another found that reduced left hippocampal volume is the most consistent neurostructural biomarker in predicting development of AD in MCI patients (Ferreira, Diniz, Forlenza, Busatto, & Zanetti, 2011). Similarly, our results complement data from different associative learning tasks that required participants to generalize previously acquired information. For example, one study found that activity in the left posterior hippocampal region alone correlated with the ability to transfer knowledge to novel settings (Kumaran, Summerfield, Hassabis, & Maguire, 2009). Moreover, the left hippocampus has been implicated in context-dependent episodic memory (Burgess, Maguire, & O’Keefe, 2002). Nonetheless, future studies need to elucidate laterality issues of the hippocampus in supporting generalization in ADAD given that the precise division of labor between the right and left hippocampi is not yet clear.
Moreover, future work will need to disentangle the specificity of generalization deficits to the left hippocampus as compared to the MTL more broadly. We did not necessarily expect an absence of association between entorhinal cortex and generalization, especially given evidence that entorhinal atrophy and hippocampal atrophy are often correlated in sporadic AD (Bobinski et al., 1999; de Toledo-Morrell et al., 2000) and that entorhinal cortex has been implicated in generalization in animal models of acquired equivalence (Coutureau et al., 2002). Our results could reflect a true lack of involvement of entorhinal cortex in generalization. Alternatively, the association may be difficult to detect due to decreased precision in measuring entorhinal cortex thickness or that this structure may change less predictably with neuronal loss in ADAD than sporadic AD (Ringman et al., 2014). Further, the small size of the entorhinal cortex may hinder the ability to detect volumetric changes even if functional changes are occurring, or our small sample may limit our power to detect significant brain-behavior correlations in this region. Additional studies, especially involving those involving cases of sporadic AD, may clarify whether hippocampal volume measurements are simply a better marker of general MTL atrophy in ADAD or whether the hippocampus is more functionally involved in generalization than other MTL structures.
The present study is limited by a relatively small sample size, the heterogeneity of the different mutations included, and uncertainty regarding the degree to which results with ADAD mutation carriers can be applied to sporadic late-onset AD. Furthermore, some previous work suggests the automated segmentation algorithm used by Freesurfer may overestimate hippocampal volumes when compared to manual tracings (Morey et al., 2009; Wenger et al., 2014). That said, our participants were relatively young, and therefore had fewer comorbid illnesses (e.g., hypertension) that might contribute to preclinical cognitive decline. In addition, our kin control group limits some environmental or biological factors that might otherwise confound the results.
Our study included both native English and Spanish speakers, suggesting our task has utility as a “linguistically-neutral” assessment tool to assess cognitive impairment in early AD. Similar to Parra et al. (2010), this linguistic neutrality likely reflects that our task uses primarily visual vs. verbal stimuli, in contrast to other commonly used tests of hippocampal-dependent memory, such as explicit verbal recall. Thus, our quick, easily implementable, inexpensive task may provide a culturally neutral way to identify and track cognition in AD in a way that complements standard neuropsychological tests. Such early prediction of AD is critical, given that existing pharmacological interventions for AD are typically aimed to slow advancement of the disease, rather than reverse or stop its progress.
Highlights.
A generalization task detected cognitive deficits in preclinical ADAD adults
ADAD mutation carriers performed worse on generalization than non-carrying kin
Generalization was related to left hippocampal volumes in ADAD mutation carriers
Generalization tests may predict AD onset in ADAD mutation carriers and sporadic AD
Acknowledgments
This research was supported by NIA/NIH grant R03AG044610-01A1 (PIs: Petok & Gluck), NIH/NIA grant R56-AG053961 (PI: Gluck), as well as Easton Consortium for Alzheimer’s Disease Drug Discovery and Biomarker Development, NIA K08 AG-22228 (PI: Ringman), the Dominantly Inherited Alzheimer Network (DIAN) U01 AG032438 (PI: Morris), the UCLA Alzheimer’s Disease Research Center Grant, P50 AG16570 (PI: Ringman), the USC Alzheimer’s Disease Center Research Grant P50 AG-005142, (PI: H. Chui) and the UCLA Clinical Translational Research Institute 1UL1-RR033176 (PI: Dubinett). The authors want to thank Lisa Haber-Chalom for help with data collection, as well as Sylvia Larson and Angelica Boeve for help with manuscript preparation. Preliminary findings from this project were presented at the American Academy of Neurology in San Diego, CA in 2013.
Footnotes
Author Contributions
J. Ringman, C. E. Myers and M. Gluck developed the study concept and study design. C. E. Myers provided experimental software. Testing and data collection were performed by authors D. Wharton, L. Medina, and M. Casado under the supervision of J. Ringman. J. Petok performed the behavioral data analysis and interpretation under the supervision of J. Ringman, C. E. Myers and M. Gluck. J. Pa and Z. Hobel provided the neuroimaging analysis. J. Petok drafted the manuscript, and J. Ringman, C. E. Myers and M. Gluck provided critical revisions. All authors approved the final version of the manuscript for submission.
Conflicts of Interest
The authors declare that they have no conflict of interest and no relevant disclosures. Dr. Ringman has received personal compensation for serving on a scientific advisory boards for Takeda Pharmaceuticals and StemCells, Inc, consulting fees from Innosense, LLC, and research support from Janssen, Pfizer, Accera, Bristol Myers Squibb, and Wyeth Pharmaceuticals.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguirre-Acevedo DC, Lopera F, Henao E, Tirado V, Munoz C, Giraldo M, … Jaimes F. Cognitive Decline in a Colombian Kindred With Autosomal Dominant Alzheimer Disease: A Retrospective Cohort Study. JAMA Neurol. 2016;73:431–438. doi: 10.1001/jamaneurol.2015.4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almkvist O, Rodriguez-Vieitez E, Thordardottir S, Amberla K, Axelman K, Basun H, … Graff C. Predicting Cognitive Decline across Four Decades in Mutation Carriers and Non-carriers in Autosomal-Dominant Alzheimer’s Disease. J Int Neuropsychol Soc. 2017;23:195–203. doi: 10.1017/S1355617716001028. [DOI] [PubMed] [Google Scholar]
- Apostolova LG, Hwang KS, Medina LD, Green AE, Braskie MN, Dutton RA, … Ringman JM. Cortical and hippocampal atrophy in patients with autosomal dominant familial Alzheimer’s disease. Dementia and Geriatric Cognitive Disorders. 2011;32:118–125. doi: 10.1159/000330471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, Xiong C, Benzinger TLF, AM, Goate A, Fox NC, Marcus DS, … Klunk WEM, Martins E, Masters RN, Mayeux CL, Ringman R, Rossor JM, Schofield MN, Sperling PR, Salloway RA, Morris S, JC Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. The New England Journal of Medicine. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobinski M, de Leon MJ, Convit A, De Santi S, Wegiel J, Tarshish CY, … Wisniewski HM. MRI of entorhinal cortex in mild Alzheimer’s disease. Lancet. 1999;353:38–40. doi: 10.1016/s0140-6736(05)74869-8. [DOI] [PubMed] [Google Scholar]
- Bodi N, Csibri E, Myers CE, Gluck MA, Keri S. Associative learning, acquired equivalence, and flexible generalization of knowledge in mild Alzheimer disease. Cognitive and Behavioral Neurology. 2009;22:89–94. doi: 10.1097/WNN.0b013e318192ccf000146965-200906000-00003. [pii] [DOI] [PubMed] [Google Scholar]
- Brown FC, Roth RM, Saykin AJ, Beverly-Gibson G. A new measure of visual location learning and memory: development and psychometric properties for the Brown Location Test (BLT) Clin Neuropsychol. 2007;21:811–825. doi: 10.1080/13854040600878777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruscoli M, Lovestone S. Is MCI really just early dementia? A systematic review of conversion studies. Internatioanal Psychogeriatrics. 2004;16:129–140. doi: 10.1017/s1041610204000092. [DOI] [PubMed] [Google Scholar]
- Burgess N, Maguire EA, O’Keefe J. The human hippocampus and spatial and episodic memory. Neuron. 2002;35:625–641. doi: 10.1016/s0896-6273(02)00830-9. [DOI] [PubMed] [Google Scholar]
- Cash DM, Ridgway GR, Liang Y, Ryan NS, Kinnunen KM, Yeatman T, … Fox NC. The pattern of atrophy in familial Alzheimer disease: Volumetric MRI results from the DIAN study. Neurology. 2013;81:1425–1433. doi: 10.1212/WNL.0b013e3182a841c6. doi: http://dx.doi.org/10.1212/WNL.0b013e3182a841c6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase HW, Clark L, Myers CE, Gluck MA, Sahakian BJ, Bullmore ET, Robbins TW. The role of the orbitofrontal cortex in human discrimination learning. Neuropsychologia. 2008;46:1326–1337. doi: 10.1016/j.neuropsychologia.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Coutureau E, Killcross AS, Good M, Marshall VJ, Ward-Robinson J, Honey RC. Acquired equivalence and distinctiveness of cues: II. Neural manipulations and their implications. J Exp Psychol Anim Behav Process. 2002;28:388–396. [PubMed] [Google Scholar]
- de Toledo-Morrell L, Goncharova I, Dickerson B, Wilson RS, Bennett DA. From healthy aging to early Alzheimer’s disease: in vivo detection of entorhinal cortex atrophy. Ann N Y Acad Sci. 2000;911:240–253. doi: 10.1111/j.1749-6632.2000.tb06730.x. [DOI] [PubMed] [Google Scholar]
- Della Sala S, Kozlova I, Stamate A, Parra MA. A transcultural cognitive marker of Alzheimer’s Disease. Int J Geriatr Psychiatry. 2016 doi: 10.1002/gps.4610. [DOI] [PubMed] [Google Scholar]
- Evans DA, Funkenstein HH, Albert MS, Scherr PA, Cook NR, Chown MJ, … Taylor JO. Prevalence of Alzheimer’s disease in a community population of older persons. Higher than previously reported. JAMA. 1989;262:2551–2556. [PubMed] [Google Scholar]
- Farkas M, Polgar P, Kelemen O, Rethelyi J, Bitter I, Myers CE, … Keri S. Associative learning in deficit and nondeficit schizophrenia. Neuroreport. 2008;19:55–58. doi: 10.1097/WNR.0b013e3282f2dff6. [DOI] [PubMed] [Google Scholar]
- Ferreira LK, Diniz BS, Forlenza OV, Busatto GF, Zanetti MV. Neurostructural predictors of Alzheimer’s disease: a meta-analysis of VBM studies. Neurobiol Aging. 2011;32:1733–1741. doi: 10.1016/j.neurobiolaging.2009.11.008. [DOI] [PubMed] [Google Scholar]
- Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, … Dale AM. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33:341–355. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. Journal of Psychiatric Research. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Gluck MA, Myers CE. Hippocampal mediation of stimulus representation: a computational theory. Hippocampus. 1993;3:491–516. doi: 10.1002/hipo.450030410. [DOI] [PubMed] [Google Scholar]
- Head D, Snyder AZ, Girton LE, Morris JC, Buckner RL. Frontal-hippocampal double dissociation between normal aging and Alzheimer’s disease. Cerebral Cortex. 2005;15:732–739. doi: 10.1093/cercor/bhh174. [DOI] [PubMed] [Google Scholar]
- Heilbronner RL. The search for a “pure” visual memory test: Pursuit of perfection? The Clinical Neuropsychologist. 1992;6:105–112. doi: http://dx.doi.org/10.1080/13854049208404124. [Google Scholar]
- Herzallah MM, Moustafa AA, Misk AJ, Al-Dweib LH, Abdelrazeq SA, Myers CE, Gluck MA. Depression impairs learning whereas anticholinergics impair transfer generalization in Parkinson patients tested on dopaminergic medications. Cognitive and Behavioral Neurology. 2010;23:98–105. doi: 10.1097/WNN.0b013e3181df3048. [DOI] [PubMed] [Google Scholar]
- Kennedy AM, Newman S, McCaddon A, Ball J, Roques P, Mullan M, et al. Familial Alzheimer’s disease. A pedigree with a mis-sense mutation in the amyloid precursor protein gene (amyloid precursor protein 717 valine-->glycine) Brain. 1993;116(Pt 2):309–324. doi: 10.1093/brain/116.2.309. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Price JC, Mathis CA, Tsopelas ND, Lopresti BJ, Ziolko SK, … DeKosky ST. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. Journal of Neuroscience. 2007;27:6174–6184. doi: 10.1523/JNEUROSCI.0730-07.2007. doi: http://dx.doi.org/10.1523/JNEUROSCI.0730-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaran D, Summerfield JJ, Hassabis D, Maguire EA. Tracking the emergence of conceptual knowledge during human decision making. Neuron. 2009;63:889–901. doi: 10.1016/j.neuron.2009.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GJ, Lu PH, Medina LD, Rodriguez-Agudelo Y, Melchor S, Coppola G, … Ringman JM. Regional brain volume differences in symptomatic and presymptomatic carriers of familial Alzheimer’s disease mutations. Journal of Neurology, Neurosurgery & Psychiatry. 2013;84:154–162. doi: 10.1136/jnnp-2011-302087. doi: http://dx.doi.org/10.1136/jnnp-2011-302087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy-Gigi E, Keri S, Myers CE, Lencovsky Z, Sharvit-Benbaji H, Orr SP, … Gluck MA. Individuals with posttraumatic stress disorder show a selective deficit in generalization of associative learning. Neuropsychology. 2012;26:758–767. doi: 10.1037/a0029361. [DOI] [PubMed] [Google Scholar]
- Mattyassy A, Keri S, Myers CE, Levy-Gigi E, Gluck MA, Kelemen O. Impaired generalization of associative learning in patients with alcohol dependence after intermediate-term abstinence. Alcohol and Alcoholism. 2012;47:533–537. doi: 10.1093/alcalc/ags050. [DOI] [PubMed] [Google Scholar]
- Morey RA, Petty CM, Xu Y, Hayes JP, Wagner HR, 2nd, Lewis DV, … McCarthy G. A comparison of automated segmentation and manual tracing for quantifying hippocampal and amygdala volumes. Neuroimage. 2009;45:855–866. doi: 10.1016/j.neuroimage.2008.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC. Clinical dementia rating: a reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. International Psychogeriatrics. 1997;9(Suppl 1):173–176. doi: 10.1017/s1041610297004870. discussion 177–178. [DOI] [PubMed] [Google Scholar]
- Mullan MJ, Tsuji S, Miki T, Katsuya T, Naruse S, Kaneko K, … Roses A. Clinical comparison of Alzheimer’s disease in pedigrees with the codon 717 Val→Ile mutation in the amyloid precursor protein gene. Neurobiology of Aging. 1993;14:407–419. doi: 10.1016/0197-4580(93)90099-w. doi: http://dx.doi.org/10.1016/0197-4580(93)90099-W. [DOI] [PubMed] [Google Scholar]
- Murrell J, Ghetti B, Cochran E, Macias-Islas MA, Medina L, Varpetian A, … Ringman JM. The A431E mutation in PSEN1 causing Familial Alzheimer’s Disease originating in Jalisco State, Mexico: an additional fifteen families. Neurogenetics. 2006;7:277–279. doi: 10.1007/s10048-006-0053-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CE, Hopkins RO, DeLuca J, Moore NB, Wolansky LJ, Sumner JM, Gluck MA. Learning and generalization deficits in patients with memory impairments due to anterior communicating artery aneurysm rupture or hypoxic brain injury. Neuropsychology. 2008a;22:681–686. doi: 10.1037/0894-4105.22.5.681. [DOI] [PubMed] [Google Scholar]
- Myers CE, Kluger A, Golomb J, Ferris S, de Leon MJ, Schnirman G, Gluck MA. Hippocampal atrophy disrupts transfer generalization in nondemented elderly. Journal of Geriatric Psychiatry and Neurology. 2002;15:82–90. doi: 10.1177/089198870201500206. [DOI] [PubMed] [Google Scholar]
- Myers CE, Kluger A, Golomb J, Gluck MA, Ferris S. Learning and generalization tasks predict short-term cognitive outcome in nondemented elderly. Journal of Geriatric Psychiatry and Neurology. 2008b;21:93–103. doi: 10.1177/0891988708316858. [DOI] [PubMed] [Google Scholar]
- Myers CE, Shohamy D, Gluck MA, Grossman S, Kluger A, Ferris S, … Schwartz R. Dissociating hippocampal versus basal ganglia contributions to learning and transfer. Joural of Cognitive Neuroscience. 2003;15:185–193. doi: 10.1162/089892903321208123. [DOI] [PubMed] [Google Scholar]
- Parra MA, Abrahams S, Logie RH, Mendez LG, Lopera F, Della Sala S. Visual short-term memory binding deficits in familial Alzheimer’s disease. Brain. 2010;133:2702–2713. doi: 10.1093/brain/awq148. [DOI] [PubMed] [Google Scholar]
- Quiroz YT, Budson AE, Celone K, Ruiz A, Newmark R, Castrillón G, … Stern CE. Hippocampal hyperactivation in presymptomatic familial Alzheimer’s disease. Annals of Neurology. 2010;68:865–875. doi: 10.1002/ana.22105. doi: http://dx.doi.org/10.1002/ana.22105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringman JM, Diaz-Olavarrieta C, Rodriguez Y, Chavez M, Fairbanks L, Paz F, … Kawas C. Neuropsychological function in nondemented carriers of presenilin-1 mutations. Neurology. 2005;65:552–558. doi: 10.1212/01.wnl.0000172919.50001.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringman JM, Goate A, Masters CL, Cairns NJ, Danek A, Graff-Radford N, … Morris JC. Genetic Heterogeneity in Alzheimer Disease and Implications for Treatment Strategies. Current Neurology and Neuroscience Reports. 2014;14:499. doi: 10.1007/s11910-014-0499-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringman JM, Gylys KH, Medina LD, Fox M, Kepe V, Flores DL, … Leverenz JB. Biochemical, neuropathological, and neuroimaging characteristics of early-onset Alzheimer’s disease due to a novel PSEN1 mutation. Neuroscience Letters. 2011;487:287–292. doi: 10.1016/j.neulet.2010.10.039. doi: http://dx.doi.org/10.1016/j.neulet.2010.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringman JM, Monsell S, Ng DW, Zhou Y, Nguyen A, Coppola G, … Vinters HV. Neuropathology of Autosomal Dominant Alzheimer Disease in the National Alzheimer Coordinating Center Database. Journal of Neuropathology & Experimental Neurology. 2016;75:284–290. doi: 10.1093/jnen/nlv028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringman JM, O’Neill J, Geschwind D, Medina L, Apostolova LG, Rodriguez Y, … Bartzokis G. Diffusion tensor imaging in preclinical and presymptomatic carriers of familial Alzheimer’s disease mutations. Brain: A Journal of Neurology. 2007;130:1767–1776. doi: 10.1093/brain/awm102. doi: http://dx.doi.org/10.1093/brain/awm102. [DOI] [PubMed] [Google Scholar]
- Ringman JM, Younkin SG, Pratico D, Seltzer W, Cole GM, Geschwind DH, … Cummings JL. Biochemical markers in persons with preclinical familial Alzheimer disease. Neurology. 2008;71:85–92. doi: 10.1212/01.wnl.0000303973.71803.81. doi: http://dx.doi.org/10.1212/01.wnl.0000303973.71803.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryman DC, Acosta-Baena N, Aisen PS, Bird T, Danek A, Fox NC, … Bateman RJ. Symptom onset in autosomal dominant Alzheimer disease: A systematic review and meta-analysis. Neurology. 2014;83:253–260. doi: 10.1212/WNL.0000000000000596. doi: http://dx.doi.org/10.1212/WNL.0000000000000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scahill RI, Ridgway GR, Bartlett JW, Barnes J, Ryan NS, Mead S, … Fox NC. Genetic influences on atrophy patterns in familial Alzheimer’s disease: a comparison of APP and PSEN1 mutations. J Alzheimers Dis. 2013;35:199–212. doi: 10.3233/JAD-121255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrington R, Froelich S, Sorbi S, Campion D, Chi H, Rogaeva EA, … St George-Hyslop PH. Alzheimer’s disease associated with mutations in presenilin 2 is rare and variably penetrant. Human Molecular Genetics. 1996;5:985–988. doi: 10.1093/hmg/5.7.985. [DOI] [PubMed] [Google Scholar]
- Shi F, Liu B, Zhou Y, Yu C, Jiang T. Hippocampal volume and asymmetry in mild cognitive impairment and Alzheimer’s disease: Meta-analyses of MRI studies. Hippocampus. 2009;19:1055–1064. doi: 10.1002/hipo.20573. [DOI] [PubMed] [Google Scholar]
- Shohamy D, Myers CE, Kalanithi J, Gluck MA. Basal ganglia and dopamine contributions to probabilistic category learning. Neuroscience and Biobehavioral Reviews. 2008;32:219–236. doi: 10.1016/j.neubiorev.2007.07.008. S0149-7634(07)00080-2 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JR, Gluck MA. Adult age differences in learning and generalization of feedback-based associations. Psychology and Aging. 2013;28:937–947. doi: 10.1037/a0033844. [DOI] [PubMed] [Google Scholar]
- Smith ML, Milner B. The role of the right hippocampus in the recall of spatial location. Neuropsychologia. 1981;19:781–793. doi: 10.1016/0028-3932(81)90090-7. [DOI] [PubMed] [Google Scholar]
- Sperling R. Potential of functional MRI as a biomarker in early Alzheimer’s disease. Neurobiol ogy of Aging. 2011;32(Suppl 1):S37–43. doi: 10.1016/j.neurobiolaging.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng EL, Hasegawa K, Homma A, Imai Y, Larson E, Graves A, … White LR. The Cognitive Abilities Screening Instrument (CASI): A practical test for cross-cultural epidemiological studies of dementia. International Psychogeriatrics. 1994;6:45–58. doi: 10.1017/s1041610294001602. doi: http://dx.doi.org/10.1017/S1041610294001602. [DOI] [PubMed] [Google Scholar]
- Thompson PM, MacDonald D, Mega MS, Holmes CJ, Evans AC, Toga AW. Detection and mapping of abnormal brain structure with a probabilistic atlas of cortical surfaces. Journal of Computer Assisted Tomography. 1997;21:567–581. doi: 10.1097/00004728-199707000-00008. [DOI] [PubMed] [Google Scholar]
- Wenger E, Martensson J, Noack H, Bodammer NC, Kuhn S, Schaefer S, … Lovden M. Comparing manual and automatic segmentation of hippocampal volumes: reliability and validity issues in younger and older brains. Hum Brain Mapp. 2014;35:4236–4248. doi: 10.1002/hbm.22473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Qiu C, Lindberg O, Bronge L, Aspelin P, Backman L, … Wahlund LO. Acceleration of hippocampal atrophy in a non-demented elderly population: the SNAC-K study. International Psychogeriatrics. 2010;22:14–25. doi: 10.1017/S1041610209991396. [DOI] [PubMed] [Google Scholar]