

Abstract
Neuroinflammation plays a major role in brain excitability and may contribute to the development of epilepsy. Prostaglandin E2 (PGE2) is a direct mediator of inflammatory responses and, through EP receptors, plays an important role in neuronal excitability. Pharmacological evidence supports that centrally-administered EP1 and EP3 receptor antagonists reduced acutely evoked seizures in rats. Translation of these findings would benefit from evidence of efficacy with a more clinically relevant route of delivery and validation in another species. In the current study we investigated whether the systemic administration of EP1 and EP3 agonists and antagonists modulate pentylenetetrazole (PTZ)-induced seizures in mice. In addition, it was examined whether these compounds alter Na+, K+-ATPase activity, an enzyme responsible for the homeostatic ionic equilibrium and, consequently, for the resting membrane potential in neurons. While the systemic administration of EP1 and EP3 antagonists (ONO-8713 and ONO-AE3-240, respectively) attenuated, the respective agonists (ONO-DI-004 and ONO-AE-248) potentiated PTZ-induced seizures (all compounds injected at the dose of 10 µg/kg, s.c., 30 min before PTZ challenge). Co-administration of either EP1 or EP3 agonist with the respective antagonists nullified the anticonvulsant effects of EP1/3 receptor blockade. In addition, EP1 and EP3 agonists exacerbated PTZ-induced decrease of Na+, K+-ATPase activity in both cerebral cortex and hippocampus, whereas, EP1 and EP3 antagonists prevented PTZ-induced decrease of Na+, K+-ATPase activity in both structures. Our findings support and extend evidence that EP1 and EP3 receptors may be novel targets for the development of anticonvulsant drugs.
Keywords: Epilepsy, prostaglandin E2, EP1 receptor, EP3 receptor, pentylenetetrazole
Introduction
Epilepsy is a disabling neurological disease that affects about 1% of the general population and is characterized by recurring and unprovoked seizures [1]. One third of epilepsy patients do not respond to currently available treatments. Thus, understanding the causes of seizures and identifying novel anticonvulsant targets is a priority [2-4].
Current evidence strongly supports the involvement of inflammation in the etiopathogenesis of seizures and epilepsy development [5-7]. Major brain injuries known to cause epilepsy are associated with prolonged or excessive neuroinflammation. Proinflammatory cytokines, complement factors [8] and prostaglandins [9] contribute to cell loss and seizures in experimental models. Consequently, inhibiting their production or blocking their receptors decreases seizure activity [6,9].
Prostaglandins are produced from arachidonic acid the via cyclooxygenase (COX) pathway [10]. They are direct mediators of inflammatory responses [11,12]. Prostaglandin E2 (PGE2) is synthesized by the COX-2 pathway and is a crucial mediator of responses to injuries in the brain [12-14]. PGE2 also plays an important role in dynamically maintaining membrane excitability, synaptic transmission, integration, and plasticity in the hippocampus. Notably, depletion of endogenous PGE2 in hippocampal CA1 pyramidal neurons results in a significant reduction of the membrane input resistance and frequency of firing. Such a decrease of membrane excitability is reversed by the exogenous application of PGE2 [13]. In addition, PGE2 facilitates pentylenetetrazole (PTZ)- and methylmalonate-induced seizures [15,16]. PGE2 actions are mediated by G-protein-coupled E-prostanoid (EP) receptors, which are divided into EP1, EP2, EP3, and EP4 [17]. The existence of EP receptor subtypes may account for the multiplicity of biological responses exerted by PGE2, since each EP subtype has a different structure and is coupled to a different signaling pathway [18,19]. It has been shown that the EP1 receptor mediates a G-protein coupled increase of free Ca++ concentration by Ca++ channel gate regulation [18]. This receptor has a splicing isoform that presents a defective signal transduction, suppressing the EP signaling pathway, even though it shows a ligand-binding specificity similar to the EP1 receptor [20]. EP2 and EP4 receptors are coupled to Gs-protein, and their activation increases cAMP production [21,22]. These receptors seem to have different sensitivity to phosphorylation and agonist-dependent desensitization, due to structural differences [23]. The EP3 receptor has three splicing isoforms (α, β and γ) in mouse, with similar ligand-binding specificities [22]. These isoforms are functionally different, as they bind distinct G-proteins (Gi, Gq and Gs) [22]. However, the major EP3 receptor signaling pathway involves Gi activation with consequent adenylatecyclase inhibition [18].
The Na+, K+-ATPase (EC 3.6.3.9) is a heterodimeric integral membrane protein responsible for maintaining the homeostatic ionic equilibrium in almost all tissues and contributing to the resting membrane potential in neurons [24]. Na+, K+-ATPase activity is reduced in the cerebral tissue of patients with epilepsy [25,26], and a mutation in one subunit of the gene encoding this enzyme has been linked with neurological disorders [27,28], including epilepsy in humans [29]. Diminished activity of the Na+, K+-ATPase has also been found in glia within the seizure focus in experimental models of focal epilepsy [26] and in patients [26]. Na+, K+-ATPase activity is also decreased after pentylenetetrazole (PTZ) [30,31], glutaric acid- [30], and methylmalonic acid-induced seizures [32-34]. Consistent with this view, ouabain, an irreversible Na+, K+-ATPase inhibitor, can induce seizures [35,36].
The EP1 and EP3 receptors are key mediators of inflammation, and a seizure protective role has been reported by brain injection of their antagonists in rats [9]. Potentially deleterious effects have been reported for EP2 receptor ligands in seizure models [48] and the EP4 receptor remains less well characterized [49]. Therefore, we focused here on extending earlier findings by investigating whether systemic administration of EP1 or EP3 receptor ligands can modulate PTZ-induced seizures and seizure-induced Na+ K+ATPase changes, in mice.
Material and methods
Animals and reagents
Adult male Swiss mice (25-35 g) were maintained under controlled light and environment (12:12 h light-dark cycle, 24±1°C, 55% relative humidity) with free access to food (SupraTM, Santa Maria, Brazil) and water. All experimental protocols were designed aiming to keep the number of animals used to a minimum, as well as their suffering, in accordance with national and international legislation (guidelines of Brazilian Council of Animal Experimentation, of EU Directive 2010/63/EU for animal experiments, and U.S. National Institute of Health Guide for the Care and Use of Laboratory Animals-NIH Publications No 80-23, revised 1996), and with the approval of the Ethics Committee for Animal Research of the Federal University of Santa Maria (process number 078/2010).
ONO-8713 (an EP1 antagonist), ONO-DI-004 (an EP1 agonist), ONO-AE3-240 (an EP3 antagonist), and ONO-AE- 248 (an EP3 agonist), were generously donated by Ono Pharmaceutical Co. (Osaka, Japan). ONO-8713, ONO-DI-004, ONO-AE3-240, and ONO-AE-248 were dissolved in dimethylsulfoxide (DMSO) and then diluted with sterile saline, in such a way that DMSO concentration did not exceed 1%. PTZ and all other reagents were purchased from Sigma (St. Louis, MO, USA).
Surgical procedures
All animals were anesthetized with ketamine (5 mg/kg) and xylazine (50 mg/kg) and placed in a rodent stereotaxic apparatus. Under stereotaxic guidance, two screw electrodes were placed over the right and left parietal cortices (coordinates in mm: AP -4.5 and L 2.5), along with a ground lead positioned over the nasal sinus [37]. The electrodes were connected to a multipin socket for electroencephalogram (EEG) recordings, and were fixed to the skull with dental acrylic cement. Chloramphenicol (200 mg/kg, i.p.) was administered immediately before the surgical procedure. After surgery, all mice received a single subcutaneous (s.c.) injection of 0.01 mg/kg buprenorphine hydrochloride for amelioration of pain.
Drug administration protocol and EEG recordings
The effect of EP ligands on PTZ-induced seizures was assessed 5-7 days after surgery. Mice were habituated for at least 10 minutes and connected to the lead socket of a swivel, which was connected to the digital encephalographic equipment (Neuromap EQSA260, Neurotec, Brazil) inside a Faraday’s cage. Routinely, a 10 min baseline recording was obtained to establish an adequate control period. After this period, ONO-8713 (10 µg/kg), ONO-DI-004 (10 µg/kg), ONO-AE3-240 (10 µg/kg), ONO-AE-248 (10 µg/kg), or their respective vehicle (1% DMSO in saline) were administered subcutaneously. We selected systemic doses and time elapsed between drug injection based on an extrapolation from our earlier i.c.v. work [9] and pilot tests. Doses of PTZ injections were selected based on pilot experiments. The animals were injected with PTZ (60 mg/kg, i.p.) 30 minutes after antagonist/agonist administration and followed up for 30 min after PTZ administration for the appearance of seizures, by electrographic and behavioral methods. Latency to myoclonic jerks and tonic-clonic seizures were recorded, in seconds. We attributed a cut-off time of 30 minutes for those animals that did not present EEG seizures during the observation period, for statistical purposes. EEG signals were amplified, filtered (0.1 to 70.0 Hz, bandpass), digitalized (sampling rate 256 Hz) and stored in a PC for off-line analysis, as described below. Seizures were defined by the occurrence of ictal episodes characterized by the following alterations in the recording leads: spikes (≥ 2 × baseline) plus slow waves, multispikes (≥ 2 × baseline, ≥ 3 spikes/complex) plus slow waves, multiple sharp waves (≥ 2 × baseline) in long spindle episodes (≥ 5 s) or major seizure (repetitive spikes plus slow waves, ≥ 5 sec) [9]. Rhythmic scratching of the electrode headset rarely caused artifacts, which were easily identified and discarded.A separate set of animals was used to assess whether the EP1 and EP3 agonists (ONO-DI-004 and ONO-AE-248) might pharmacologically prevent the anticonvulsant actions of ONO-8713 and ONO-AE3-240 (EP1 and EP3 antagonists, respectively). This possibility was investigated by injecting a non-effective dose of EP1 (3 µg/kg, s.c.), EP3 (3 µg/kg, s.c.) agonists or their respective vehicles, followed by EP1 (10 µg/kg, s.c.), EP3 (10 µg/kg, s.c.) antagonists or their respective vehicles, 30 and 15 min before PTZ (60 mg/kg, i.p.) seizure induction, respectively.
Na+, K+-ATPase activity measurements
Na+, K+-ATPase has been identified as a target for PGE2-mediated signaling in adult rat hippocampal slices [38]. The effect of EP1 and EP3 ligands on Na+, K+-ATPase activity was measured. Immediately after the EEG recordings, the animals were sacrificed. Cerebral cortices and hippocampi were dissected, weighed and immediately frozen at -80°C. On the experimental day, each sample was gently homogenized (7-10 strokes) in ice-cold 10 mM Tris-HCl (pH 7.4) for Na+, K+-ATPase activity determination [39]. Briefly, the assay medium consisted of 30 mM Tris-HCl (pH 7.4), 0.1 mM EDTA, 50 mM NaCl, 5 mM KCl, 6 mM MgCl2 and 50 µg of protein in the presence or absence of ouabain (1 mM), in a final volume of 350 µL. The reaction was started by the addition of adenosine triphosphate (ATP) to a final concentration of 5 mM. After 30 min, the reaction was stopped by the addition of 70 µL of 50% (w/v) trichloroacetic acid. Appropriate controls were included in the assay for non-enzymatic hydrolysis of ATP. The amount of inorganic phosphate (Pi) released was quantified by the colorimetric method using KH2PO4 as reference standard. Specific Na+, K+-ATPase activity was calculated by subtracting the ouabain-insensitive activity from the overall activity (in the absence of ouabain) and expressed in nmol Pi/mg protein/min. Protein content was measured colorimetrically by the Bradford [40] method using bovine serum albumin (1 mg/ml) as standard.
Statistical analyses
Latencies to myoclonic jerks and to tonic-clonic seizures were analyzed by Kruskal-Wallis, followed by nonparametric Dunn’s multiple comparison test, when indicated. Data are presented as median and interquartile ranges. Total time spent in seizures, mean amplitude of EEG recordings and Na+, K+-ATPase activity were analyzed by one- or two-way ANOVA followed by Bonferroni’s test, depending on the experimental design. Data are expressed as mean + S.E.M. A probability of P < 0.05 was considered significant, and H and F values are shown only if P < 0.05.
Results
Pharmacological modulation of EP1 receptor alters PTZ-induced seizures in mice
Figure 1 shows the effect of the EP1 agonist ONO-DI-004 and the EP1 antagonist ONO-8713 on PTZ-induced seizures, measured as the latency to the first isolated myoclonic jerk, with concomitant spike activity on EEG recordings (A), latency to generalized tonic-clonic seizures (B), and total time spent in generalized seizures (C). Statistical analysis revealed that ONO-8713 increased the latency to myoclonic jerks [H (2) = 14.49, P < 0.05, Figure 1A], as well as to generalized seizures [H (2) = 11.77, P < 0.05, Figure 1B]. ONO-DI-004 did not significantly alter these parameters (Figure 1A and 1B). Figure 1C shows that the EP1 antagonist decreased total time spent in generalized seizures, when compared with vehicle-injected group [F (2, 15) = 22.17, P < 0.05]. However, mice that received the EP1 agonist spent more time in seizures than the respective control group [F (2, 15) = 22.17, P < 0.05]. Quantitative analyses of EEG trace amplitudes before and after PTZ injection are shown in Figure 1D and 1E, for ONO-DI-004 and ONO-8713, respectively. Although ONO-DI-004 did not alter seizure onset, it significantly increased the mean amplitude (in μV) of EEG ictal traces, when compared with the vehicle group [F (2, 18) = 11.86, P < 0.05]. Statistical analysis also revealed a significant decrease in mean amplitude (in μV) of EEG ictal traces of ONO-8713-treated animals, when compared with the vehicle group [F (2, 24) = 20.93, P < 0.05]. Representative EEG patterns are presented in Figure 1F-H (for vehicle-, ONO-DI-004-, and ONO-8713-treated groups, respectively), showing the ictal activity induced by PTZ injection. PTZ injection caused the appearance of multispike plus slow waves and major seizure activity, which coincided with myoclonic jerks. Generalized seizures appeared in the EEG recordings as the major seizure activity, and were characterized by 2-3 Hz high-amplitude activity (Figure 1F-H). After the ictal discharge, postictal EEG suppression and slow waves were observed, correlating with behavioral catalepsy.
Figure 1.

(A-C) Effect of EP1 receptor agonist and antagonist (ONO-DI-004 and ONO-8713, respectively; 10 µg/kg, s.c.) on PTZ-induced seizures (60 mg/kg, i.p.). (A) Latency to myoclonic jerks. (B) Latency to tonic-clonic generalized seizure. (C) Total time spent in generalized seizures. Data expressed as median and interquartile range (A and B), and mean + SEM (C), for n = 6-12 in each experimental group. (D, E) Effect of EP1 receptor agonist (D) and antagonist (E) (ONO-DI-004 and ONO-8713, respectively; 10 µg/kg, s.c.) on the mean amplitude of EEG recordings in the cortex of animal injected with PTZ (60 mg/kg, i.p.). Mean amplitude of EEG recordings was analyzed by two-way ANOVA followed by the Bonferroni’s test and expressed as mean + S.E.M, for n = 6-12 in each experimental group. (F-H) Representative electrocorticographic recordings of animals after PTZ injection are represented as follows: (F) vehicle, (G) ONO-DI-004, and (H) ONO-8713. Black and white arrowheads indicate PTZ injection and seizures latency, respectively, and the y-axis (amplitude) and x-axis (time) calibration bar is the same for all traces. (I) Effect of ONO-DI-004 (3 µg/kg, s.c.) administration, followed or not by ONO-8713 (10 µg/kg, s.c.), on PTZ-induced seizures, measured as latency to tonic-clonic seizure. ONO-DI-004 (3 µg/kg, s.c.) prevented the protective effect of ONO-8713 (10 µg/kg, s.c.) against PTZ-induced seizures. Data expressed as median and interquartile range for n = 3-7 in each group. A probability of P < 0.05 was considered significant. *P < 0.05, and **P < 0.01, when compared with vehicle or vehicle + vehicle group.
Last, we investigated the consequences of co-administration of the EP1 agonist with the EP1 antagonist. ONO-DI-004 (3 µg/kg, s.c.) prevented the anti-seizure effect of ONO-8713 (10 µg/kg, s.c.) on PTZ-induced seizures, measured as the latency to the first generalized tonic-clonic seizure [H (3) = 11.50, P < 0.05, Figure 1I], but not as the latency to myoclonic jerks (P > 0.05, data not shown).
Effect of EP1 receptor modulation on Na+, K+-ATPase activity
Figure 2 shows the effect of ONO-DI-004 (10 µg/kg, s.c., Figure 2A) and ONO-8713 (10 µg/kg, s.c., Figure 2B) on Na+, K+-ATPase activity in homogenates of cerebral cortex and hippocampus. The subcutaneous administration of ONO-DI-004 or ONO-8713 did not alter the Na+, K+-ATPase activity per se. ONO-DI-004 accentuated the PTZ-induced reduction in Na+, K+-ATPase activity in homogenates of both cerebral cortex and hippocampus [F (1, 16) = 5.645 P < 0.05, for cerebral cortex, and F (1, 16) = 11.88, P < 0.05, for hippocampus, Figure 2A]. The EP1 antagonist ONO-8713 prevented the PTZ-induced decrease of Na+, K+-ATPase activity [F (1, 16) = 13.03, P < 0.05, for cerebral cortex, and F (1, 16) = 19.93, P < 0.05, for hippocampus, Figure 2B]. At the non-effective dose of 3 µg/kg, ONO-DI-004 did not alter Na+, K+-ATPase activity per se (P > 0.05, Figure 2C), but also accentuated the PTZ-induced decrease of Na+, K+-ATPase activity. Co-administration of the EP1 agonist ONO-DI-004 prevented the protective effect of the EP1 antagonist ONO-8713 on Na+, K+-ATPase activity in cortical and hippocampal homogenates [F (1, 12) = 10.39, P < 0.05, for cerebral cortex, and F (1, 12) = 7.678, P < 0.05, for hippocampus, Figure 2D].
Figure 2.
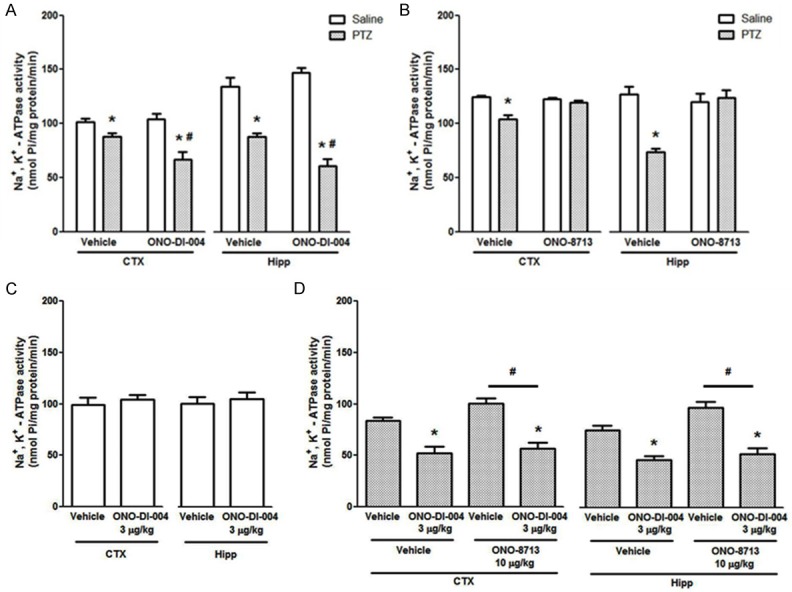
(A-D) EP1 receptors ligand effects on Na+, K+-ATPase activity in the brain. ONO-DI-004 (10 µg/kg, s.c.) increases (A) and ONO-8713 (10 µg/kg, s.c.) prevents (B) PTZ-induced decrease of Na+, K+-ATPase activity in cerebral cortex and hippocampus homogenates in mice. Data are presented in nmol Pi/mg protein/min, as mean + S.E.M., for n = 5 in each group. Data were analyzed by a two-way ANOVA followed by the Bonferroni’s test. (C) ONO-DI-004 (3 µg/kg, s.c.) does not present per se effect on Na+, K+-ATPase (P > 0.05 by Student’s t-test, for n = 5). (D) Two-way ANOVA followed by the Bonferroni’s test revealed that ONO-DI-004 (3 µg/kg, s.c.) accentuated PTZ-induced decrease of Na+, K+-ATPase activity and prevented ONO-8713 (10 µg/kg, s.c.) effect on this enzyme activity in cerebral cortex and hippocampus homogenates in mice. Data are presented in nmol Pi/mg protein/min, as mean + S.E.M., for n = 5 in each experimental group. *P < 0.05, when compared with the respective control group; #P < 0.05, when compared with vehicle group.
Effects of pharmacological modulation of EP3 receptor on PTZ-induced seizures
The effect of the EP3 agonist ONO-AE-248 (10 µg/kg, s.c.) and of the EP3 antagonist ONO-AE3-240 (10 µg/kg, s.c.) on PTZ-induced seizures is shown in Figure 3. Statistical analysis revealed that the EP3 receptor agonist decreased the latency to clonic [H (2) = 14.34, P < 0.05, Figure 3A] and to generalized seizures [H (2) = 18.78, P < 0.05, Figure 3B]. The EP3 receptor antagonist significantly increased both onset parameters [H (2) = 14.34, P < 0.05, Figure 3A, for myoclonic jerks; H (2) = 18.78, P < 0.05, Figure 3B, for generalized seizures]. ONO-AE-248- and ONO-AE3-240-injected animals respectively spent less and more [F (2, 21) = 13.6, P < 0.05] time in generalized seizures compared with control group (Figure 3C). ONO-AE-248 and ONO-AE3-240 respectively increased and decreased the mean amplitude of EEG recordings of PTZ-induced seizures [F (2, 24) = 41.9, P < 0.05, Figure 3D; and F (2, 24) = 184.8, P < 0.05, Figure 3E, respectively]. Representative EEG recordings of animals treated with PTZ and vehicle, ONO-AE-248 or ONO-AE3-240 are shown in Figure 3F-H, respectively.
Figure 3.

(A-C) Effect of EP3 receptor agonist and antagonist (ONO-AE-248 and ONO-AE3-240, respectively; 10 µg/kg, s.c.) on PTZ-induced seizures (60 mg/kg, i.p.). (A) Latency to myoclonic jerk. (B) Latency to tonic-clonic generalized seizure. (C) Total time spent in generalized seizures. Data expressed as median and interquartile range (A and B), and mean + SEM (C), for n = 8 in each experimental group. (D, E) Effect of EP3 receptor agonist (D) and antagonist (E) (ONO-AE-248 and ONO-AE3-240, respectively; 10 µg/kg, s.c.) on the mean amplitude of EEG recordings in the parietal cortex of animal injected with PTZ (60 mg/kg, i.p.). Mean amplitude of EEG recordings was analyzed by two-way ANOVA followed by the Bonferroni’s test and expressed as mean + S.E.M., for n = 8 in each group. (F-H) Representative electrocorticographic recordings of animals after PTZ injection are represented as follows: (F) vehicle, (G) ONO-AE-248, and (H) ONO-AE3-240. Black and white arrowheads indicate PTZ injection and seizures latency, respectively, and the y-axis (amplitude) and x-axis (time) calibration bar is the same for all traces. (I) Effect of ONO-AE-248 (3 µg/kg, s.c.) administration, followed or not by ONO-AE3-240 (10 µg/kg, s.c.), on PTZ-induced seizures, measured as latency to the first tonic-clonic seizure (I). ONO-AE-248 (3 µg/kg, s.c.) prevented the protective effect of ONO-AE3-240 (10 µg/kg, s.c.) against PTZ-induced seizures. Data expressed as median and interquartile range for n = 3-6 in each group. A probability of P < 0.05 was considered significant. *P < 0.05, and **P < 0.01, when compared with vehicle or vehicle + vehicle group.
In co-administration experiments, ONO-AE-248 (3 µg/kg, s.c.) prevented ONO-AE3-240-induced protection against PTZ-induced seizures, measured as the latency to myoclonic jerks [H (3) = 5.745, P < 0.05, data not shown], and to generalized seizure [H (3) = 11.90, P < 0.05, Figure 3I].
Effect of EP3 receptor modulation on Na+, K+-ATPase activity
Similarly to the findings obtained with EP1 ligands, the EP3 receptor agonist decreased Na+, K+-ATPase activity only in those mice which had PTZ-induced seizures [F (1, 16) = 7.082, P < 0.05, for cerebral cortex and F (1, 16) = 6.238, P < 0.05, for hippocampus, Figure 4A]. The EP3 receptor antagonist prevented the expected decrease of Na+, K+-ATPase activity elicited by PTZ [F (1, 16) = 7.024, P < 0.05, for cerebral cortex, and F (1, 16) = 4.615, P < 0.05, for hippocampus, Figure 4B]. Additionally, the EP3 agonist ONO-AE-248 (3 µg/kg, s.c.) accentuated PTZ-induced decrease of Na+, K+-ATPase activity, and prevented the protective effect of the EP3 antagonist (ONO-AE3-240, 10 µg/kg, s.c.) on Na+, K+-ATPase activity in homogenates of cerebral cortex and hippocampus [F (1, 14) = 9.637, and F (1, 14) = 9.244, respectively, P < 0.05, Figure 4D].
Figure 4.
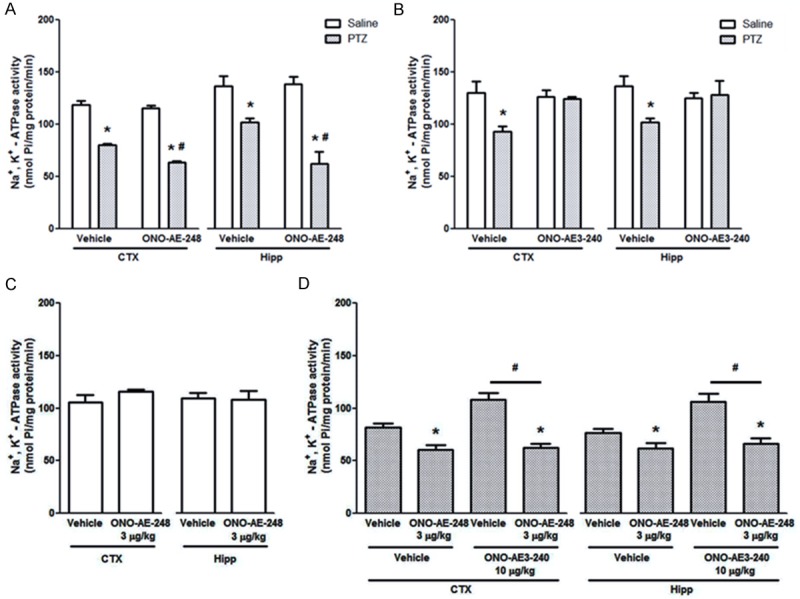
(A-D) Effects of EP3 receptor ligands on Na+, K+-ATPase activity. ONO-AE-248 (10 µg/kg, s.c.) augments (A) and ONO-AE3-240 (10 µg/kg, s.c.) prevents (B) PTZ-induced decrease of Na+, K+-ATPase activity in cerebral cortex and hippocampus homogenates in mice. Data are presented in nmol Pi/mg protein/min, as mean + S.E.M., for n = 5 in each group. The Na+, K+-ATPase activity was analyzed by two-way ANOVA followed by the Bonferroni’s test. (C) ONO-AE-248 (3 µg/kg, s.c.) does not present per SE effect on Na+, K+-ATPase (P > 0.05 by Student’s t-test, for n = 5). (D) Two-way ANOVA followed by the Bonferroni’s test revealed that ONO-AE-248 (3 µg/kg, s.c.) accentuated PTZ-induced decrease of Na+, K+-ATPase activity and prevented ONO-8713 (10 µg/kg, s.c.) effect on this enzyme activity in cerebral cortex and hippocampus homogenates in mice. Data are presented in nmol Pi/mg protein/min, as mean + S.E.M., for n = 5 in each experimental group. *P < 0.05, when compared with the respective control group; #P < 0.05, when compared with vehicle group.
Discussion
There remains a need to identify novel targets for seizure control for the pharmacorefractory epilepsy population. Supporting a role for inflammation in epilepsy, Rumià and colleagues [41] have shown an important increase in PGE2 and TXA2 levels in neocortex from patients with intractable epilepsy, suggesting that selective blockade of prostanoid receptors may constitute a novel strategy for epilepsy treatment. Furthermore, EP1 is an important signaling factor that mediates P-glycoprotein up-regulation at the blood-brain barrier during seizures [45]. Notably, P-glycoprotein overexpression has been proposed as one mechanism contributing to pharmacoresistance in drug-refractory epilepsy [46]. The present study has answered key questions that could facilitate pre-clinical development. Here we tested the effects of systemic injections of ligands acting on the EP1 and EP3 receptors on brief, generalized seizures induced by PTZ. The PTZ model triggers seizures via GABAergic blockade and is widely used for screening compounds with potential anti-convulsant actions. The current study shows that systemic administration of EP1 and EP3 antagonists attenuate PTZ-induced seizures, whereas the systemic administration of EP1 and EP3 agonists facilitated PTZ-induced seizures and, at doses that had no effect on seizures per se, blocked the anti-seizure effects of EP1 and EP3 antagonists. Systemic administration of EP1 and EP3 antagonists also prevented the PTZ-induced decrease of Na+, K+-ATPase activity in the cerebral cortex and hippocampus, whereas EP1 and EP3 agonists potentiated PTZ-induced decrease of Na+, K+-ATPase activity in both cerebral structures. Together, these findings support the targeting of the EP1 and EP3 receptor for the treatment of seizures.
The EP1 and EP3 receptors are key mediators of inflammation. However, they are constitutively expressed in the brain where they may regulate brain excitability. The currently reported reduction of PTZ-induced seizures by EP1 and EP3 antagonists (ONO-DI-004 and ONO-AE3-240, respectively) fully agrees with the previous finding that the i.c.v. administration of an EP1 antagonist (SC-19220) or an EP3 antagonist (L-826266) decreases PTZ-induced seizures in rats [9]. Importantly, it extends these findings to mice and reveals that EP1 and EP3 ligands can be delivered via a clinically-relevant route of administration, and suggests they do not encounter problems crossing the blood-brain barrier. Although the present experiments were not designed to detect side effects of the ligands, and very little is known about their bioactivity [47], we did not observe any obvious behavioral changes in the mice suggestive of toxicity. Additionally, EP1 or EP3 knockout mice were reported to be normal as regards behavior and morphology [44].
From a pharmacological perspective, the current study provides strong evidence for a role of EP1 and EP3 receptors in seizure initiation and/or propagation, because: (i) the anticonvulsant effect initially observed for EP1 and EP3 antagonists [38] was confirmed with chemically diverse compounds, of the same pharmacological class; (ii) EP1 and EP3 agonists facilitated PTZ-induced seizures, the expected effect if these receptors were involved in seizure initiation and/or propagation; (iii) The protective effect of the EP1 and EP3 antagonists were prevented by the respective agonists at doses that had no effect per se on PTZ-induced seizures (non-effective doses), indicating its specificity and further indicating a role for both EP1 and EP3 receptors in this seizure model. In this respect, early evidence that EP1 and EP3 receptors are involved in PTZ-induced seizures came from an experiment that has shown that the protective effect of EP1 and EP3 antagonists against PTZ-induced seizures was prevented by the nonspecific agonist PGE2. It is well known that introducing a nonspecific agonist in a biological system in which a specific antagonist was previously administered, may potentiate the action of co-occurring receptor subtypes. Therefore, one might reasonably argue that previously reported prevention of EP1 and EP3 blockade effects by PGE2 could be due to an agonistic-type interaction with respectively available EP3 and EP1 receptors. The current results largely exclude this possibility, since the EP1 agonist (ONO-DI-004) has a very low affinity for EP3 receptors (Ki > 104 for EP3 receptor). Accordingly, the EP3 agonist ONO-AE-248 has also very low affinity for EP1 receptors (Ki = 590 nM for EP3 receptor), if compared with its affinity to the EP3 receptor (Ki = 0.23 nM) [22].
Another finding of importance in the present study was the comparable effects of EP1 and EP2 receptor antagonists. We did not notice significant differences in seizure suppression between EP1 and EP3 antagonists, although we did not directly compare their effects in a single experiment. Future studies could assess the combination of both antagonists and look for potential synergistic effects on seizure susceptibility. This could represent an important next step before decisions on pre-clinical development.
Seizures are known to influence a multitude of downstream pathways and here we investigated how EP1 and EP3 receptor modulation affected the Na+, K+-ATPase. We found that modulation of EP1 or EP3 altered PTZ-induced decrease of Na+, K+-ATPase activity in the cerebral cortex of mice. This extends previous data by Fighera et al. [30], and Souza and colleagues, who showed a dose-dependent deleterious effect of PTZ on this pump [42]. Moreover, the protective effect of EP1 and EP3 antagonists on PTZ-induced decrease of Na+, K+-ATPase activity in cerebral cortex and hippocampus ex vivo is also, to some degree, in agreement with a previous study that has shown that EP1 and EP3 antagonists prevent PGE2-induced decrease of Na+, K+-ATPase activity in slices of rat hippocampus [38]. While one cannot define whether the currently observed decrease of Na+, K+-ATPase activity is a cause or consequence of the convulsive activity, it is intriguing that manipulation of PGE2 signaling through EP1 and EP3 receptors alter convulsive activity and sodium pump activity with notable congruence. While EP1 and EP3 agonists potentiate PTZ-induced decrease of sodium pump activity and facilitate seizures, the respective antagonists have the opposite effect. In fact, current experimental and clinical evidence suggests that increased neuronal activity increases cyclooxygenase activity by NMDA receptor-mediated activation of nitric oxide synthase and S-nitrosylation of the enzyme [43], with consequent increased production of PGE2. Therefore, it is possible that the currently observed inhibitory effect of PTZ on Na+, K+-ATPase activity may involve primary excitatory-induced production of PGE2, with consequent activation of EP1 and EP3 receptors. In summary, we showed that EP1 and EP3 receptors modulate PTZ-induced seizures and Na+, K+-ATPase activity in mice. Antagonism of either receptor, and perhaps a combination of the two, may represent a novel anticonvulsant strategy for seizure control in epilepsy.
Acknowledgements
The authors would like to thank Ono Pharmaceutical Co. (Osaka, Japan) for the agonists and antagonists donation. This work was supported by grants and fellowships from Coordination for the Improvement of Higher Education Personnel (CAPES), Brazilian National Council for Scientific and Technological Development (CNPq) and Science Foundation Ireland (17/TIDA/5002). We confirm that we have given due consideration to the protection of intellectual property associated with this work and that there are no impediments to publication, including the timing of publication, with respect to intellectual property. In so doing we confirm that we have followed the regulations of our institutions concerning intellectual property.
Disclosure of conflict of interest
None.
References
- 1.Thurman DJ, Beghi E, Begley CE, Berg AT, Buchhalter JR, Ding D, Hesdorffer DC, Hauser WA, Kazis L, Kobau R, Kroner B, Labiner D, Liow K, Logroscino G, Medina MT, Newton CR, Parko K, Paschal A, Preux PM, Sander JW, Selassie A, Theodore W, Tomson T, Wiebe S ILAE Commission on Epidemiology. Standards for epidemiologic studies and surveillance of epilepsy. Epilepsia. 2011;52(Suppl 7):2–26. doi: 10.1111/j.1528-1167.2011.03121.x. [DOI] [PubMed] [Google Scholar]
- 2.Simonato M, Brooks-Kayal AR, Engel J Jr, Galanopoulou AS, Jensen FE, Moshé SL, O’Brien TJ, Pitkanen A, Wilcox KS, French JA. The challenge and promise of anti-epileptic therapy development in animal models. Lancet Neurol. 2014;13:949–60. doi: 10.1016/S1474-4422(14)70076-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perucca E, French J, Bialer M. Development of new antiepileptic drugs: challenges, incentives, and recent advances. Lancet Neurol. 2007;6:793–804. doi: 10.1016/S1474-4422(07)70215-6. [DOI] [PubMed] [Google Scholar]
- 4.Galanopoulou AS, Buckmaster PS, Staley KJ, Moshé SL, Perucca E, Engel J Jr, Löscher W, Noebels JL, Pitkänen A, Stables J, White HS, O’Brien TJ, Simonato M American Epilepsy Society Basic Science Committee and The International League Against Epilepsy Working Group On Recommendations For Preclinical Epilepsy Drug Discovery. Identification of new epilepsy treatments: issues in preclinical methodology. Epilepsia. 2012;53:571–82. doi: 10.1111/j.1528-1167.2011.03391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vezzani A, Granata T. Brain inflammation in epilepsy: experimental and clinical evidence. Epilepsia. 2005;46:1724–43. doi: 10.1111/j.1528-1167.2005.00298.x. [DOI] [PubMed] [Google Scholar]
- 6.Vezzani A, French J, Bartfai T, Baram TZ. The role of inflammation in epilepsy. Nat Rev Neurol. 2011;7:31–40. doi: 10.1038/nrneurol.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vezzani A, Aronica E, Mazarati A, Pittman QJ. Epilepsy and brain inflammation. Exp Neurol. 2013;244:11–21. doi: 10.1016/j.expneurol.2011.09.033. [DOI] [PubMed] [Google Scholar]
- 8.Ravizza T, Noé F, Zardoni D, Vaghi V, Sifringer M, Vezzani A. Interleukin converting enzyme inhibition impairs kindling epileptogenesis in rats by blocking astrocytic IL-1beta production. Neurobiol Dis. 2008;31:327–33. doi: 10.1016/j.nbd.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 9.Oliveira MS, Furian AF, Rambo LM, Ribeiro LR, Royes LF, Ferreira J, Calixto JB, Mello CF. Modulation of pentylenetetrazol-induced seizures by prostaglandin E2 receptors. Neuroscience. 2008;152:1110–8. doi: 10.1016/j.neuroscience.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 10.Simmons DL, Botting RM, Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev. 2004;56:387–437. doi: 10.1124/pr.56.3.3. [DOI] [PubMed] [Google Scholar]
- 11.Phillis JW, Horrocks LA, Farooqui AA. Cyclooxygenases, lipoxygenases, and epoxygenases in CNS: their role and involvement in neurological disorders. Brain Res Rev. 2006;52:201–43. doi: 10.1016/j.brainresrev.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 12.Rojas A, Jiang J, Ganesh T, Yang MS, Lelutiu N, Gueorguieva P, Dingledine R. Cyclooxygenase-2 in epilepsy. Epilepsia. 2014;55:17–25. doi: 10.1111/epi.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen C, Bazan NG. Lipid signaling: sleep, synaptic plasticity, and neuroprotection. Prostaglandins Other Lipid Mediat. 2005;77:65–76. doi: 10.1016/j.prostaglandins.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 14.Milatovic D, Montine TJ, Aschner M. Prostanoid signaling: dual role for prostaglandin E2 in neurotoxicity. Neurotoxicology. 2011;32:312–9. doi: 10.1016/j.neuro.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oliveira MS, Furian AF, Royes LF, Fighera MR, Fiorenza NG, Castelli M, Machado P, Bohrer D, Veiga M, Ferreira J, Cavalheiro EA, Mello CF. Cyclooxygenase-2/PGE2 pathway facilitates pentylenetetrazol-induced seizures. Epilepsy Res. 2008;79:14–21. doi: 10.1016/j.eplepsyres.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 16.Salvadori MG, Banderó CR, Jesse AC, Gomes AT, Rambo LM, Bueno LM, Bortoluzzi VT, Oliveira MS, Mello CF. Prostaglandin E(2) potentiates methylmalonate-induced seizures. Epilepsia. 2012;53:189–98. doi: 10.1111/j.1528-1167.2011.03326.x. [DOI] [PubMed] [Google Scholar]
- 17.Furuyashiki T, Narumiya S. Roles of prostaglandin E receptors in stress responses. Curr Opin Pharmacol. 2009;9:31–8. doi: 10.1016/j.coph.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 18.Tsuboi K, Sugimoto Y, Ichikawa A. Prostanoid receptor subtypes. Prostaglandins Other Lipid Mediat. 2002;68-69:535–56. doi: 10.1016/s0090-6980(02)00054-0. [DOI] [PubMed] [Google Scholar]
- 19.Woodward DF, Jones RL, Narumiya S. International Union of Basic and Clinical Pharmacology. LXXXIII: classification of prostanoid receptors, updating 15 years of progress. Pharmacol Rev. 2011;63:471–538. doi: 10.1124/pr.110.003517. [DOI] [PubMed] [Google Scholar]
- 20.Okuda-Ashitaka E, Sakamoto K, Ezashi T, Miwa K, Ito S, Hayaishi O. Suppression of prostaglandin E receptor signaling by the variant form of EP1 subtype. J Biol Chem. 1996;271:31255–61. doi: 10.1074/jbc.271.49.31255. [DOI] [PubMed] [Google Scholar]
- 21.Katsuyama M, Nishigaki N, Sugimoto Y, Morimoto K, Negishi M, Narumiya S, Ichikawa A. The mouse prostaglandin E receptor EP2 subtype: cloning, expression, and northern blot analysis. FEBS Lett. 1995;372:151–6. doi: 10.1016/0014-5793(95)00966-d. [DOI] [PubMed] [Google Scholar]
- 22.Sugimoto Y, Narumiya S. Prostaglandin E receptors. J Biol Chem. 2007;282:11613–7. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- 23.Nishigaki N, Negishi M, Ichikawa A. Two Gs-coupled prostaglandin E receptor subtypes, EP2 and EP4, differ in desensitization and sensitivity to the metabolic inactivation of the agonist. Mol Pharmacol. 1996;50:1031–7. [PubMed] [Google Scholar]
- 24.Kaplan JH. Biochemistry of Na, K-ATPase. Annu Rev Biochem. 2002;71:511–35. doi: 10.1146/annurev.biochem.71.102201.141218. [DOI] [PubMed] [Google Scholar]
- 25.Rapport RL 2nd, Harris AB, Friel PN, Ojemann GA. Human epileptic brain Na, K ATPase activity and phenytoin concentrations. Arch Neurol. 1975;32:549–54. doi: 10.1001/archneur.1975.00490500069008. [DOI] [PubMed] [Google Scholar]
- 26.Grisar T, Guillaume D, Delgado-Escueta AV. Contribution of Na+, K (+)-ATPase to focal epilepsy: a brief review. Epilepsy Res. 1992;12:141–9. doi: 10.1016/0920-1211(92)90034-q. [DOI] [PubMed] [Google Scholar]
- 27.Heinzen EL, Arzimanoglou A, Brashear A, Clapcote SJ, Gurrieri F, Goldstein DB, Jóhannesson SH, Mikati MA, Neville B, Nicole S, Ozelius LJ, Poulsen H, Schyns T, Sweadner KJ, van den Maagdenberg A, Vilsen B ATP1A3 Working Group. Distinct neurological disorders with ATP1A3 mutations. Lancet Neurol. 2014;13:503–14. doi: 10.1016/S1474-4422(14)70011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benarroch EE. Na+, K+-ATPase: functions in the nervous system and involvement in neurologic disease. Neurology. 2011;76:287–93. doi: 10.1212/WNL.0b013e3182074c2f. [DOI] [PubMed] [Google Scholar]
- 29.Jurkat-Rott K, Freilinger T, Dreier JP, Herzog J, Göbel H, Petzold GC, Montagna P, Gasser T, Lehmann-Horn F, Dichgans M. Variability of familial hemiplegic migraine with novel A1A2 Na+/K+-ATPase variants. Neurology. 2004;62:1857–61. doi: 10.1212/01.wnl.0000127310.11526.fd. [DOI] [PubMed] [Google Scholar]
- 30.Fighera MR, Royes LF, Furian AF, Oliveira MS, Fiorenza NG, Frussa-Filho R, Petry JC, Coelho RC, Mello CF. GM1 ganglioside prevents seizures, Na+, K+-ATPase activity inhibition and oxidative stress induced by glutaric acid and pentylenetetrazole. Neurobiol Dis. 2006;22:611–23. doi: 10.1016/j.nbd.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 31.Silva LF, Hoffmann MS, Rambo LM, Ribeiro LR, Lima FD, Furian AF, Oliveira MS, Fighera MR, Royes LF. The involvement of Na+, K+-ATPase activity and free radical generation in the susceptibility to pentylenetetrazol-induced seizures after experimental traumatic brain injury. J Neurol Sci. 2011;308:35–40. doi: 10.1016/j.jns.2011.06.030. [DOI] [PubMed] [Google Scholar]
- 32.Malfatti CR, Royes LF, Francescato L, Sanabria ER, Rubin MA, Cavalheiro EA, Mello CF. Intrastriatal methylmalonic acid administration induces convulsions and TBARS production, and alters Na+, K+-ATPase activity in the rat striatum and cerebral cortex. Epilepsia. 2003;44:761–7. doi: 10.1046/j.1528-1157.2003.42902.x. [DOI] [PubMed] [Google Scholar]
- 33.Royes LF, Fighera MR, Furian AF, Oliveira MS, Fiorenza NG, Petry JC, Coelho RC, Mello CF. The role of nitric oxide on the convulsive behavior and oxidative stress induced by methylmalonate: an electroencephalographic and neurochemical study. Epilepsy Res. 2007;73:228–37. doi: 10.1016/j.eplepsyres.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 34.Ribeiro LR, Fighera MR, Oliveira MS, Furian AF, Rambo LM, Ferreira AP, Saraiva AL, Souza MA, Lima FD, Magni DV, Dezengrini R, Flores EF, Butterfield DA, Ferreira J, dos Santos AR, Mello CF, Royes LF. Methylmalonate-induced seizures are attenuated in inducible nitric oxide synthase knockout mice. Int J Dev Neurosci. 2009;27:157–63. doi: 10.1016/j.ijdevneu.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 35.Donaldson J, St Pierre T, Minnich J, Barbeau A. Seizures in rats associated with divalent cation inhibition of NA+-K+-ATP’ase. Can J Biochem. 1971;49:1217–24. doi: 10.1139/o71-175. [DOI] [PubMed] [Google Scholar]
- 36.Haas M, Askari A, Xie Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J Biol Chem. 2000;275:27832–7. doi: 10.1074/jbc.M002951200. [DOI] [PubMed] [Google Scholar]
- 37.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Compact 3rd edition. London, UK: Elsevier-Academic Press; 2008. [Google Scholar]
- 38.Oliveira MS, Furian AF, Rambo LM, Ribeiro LR, Royes LF, Ferreira J, Calixto JB, Otalora LF, Garrido-Sanabria ER, Mello CF. Prostaglandin E2 modulates Na+, K+-ATPase activity in rat hippocampus: implications for neurological diseases. J Neurochem. 2009;109:416–26. doi: 10.1111/j.1471-4159.2009.05961.x. [DOI] [PubMed] [Google Scholar]
- 39.Wyse AT, Streck EL, Barros SV, Brusque AM, Zugno AI, Wajner M. Methylmalonate administration decreases Na+, K+-ATPase activity in cerebral cortex of rats. Neuroreport. 2000;11:2331–4. doi: 10.1097/00001756-200007140-00052. [DOI] [PubMed] [Google Scholar]
- 40.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 41.Rumià J, Marmol F, Sanchez J, Carreño M, Bargalló N, Boget T, Pintor L, Setoain X, Bailles E, Donaire A, Ferrer E, Puig-Parellada P. Eicosanoid levels in the neocortex of drug-resistant epileptic patients submitted to epilepsy surgery. Epilepsy Res. 2012;99:127–31. doi: 10.1016/j.eplepsyres.2011.10.034. [DOI] [PubMed] [Google Scholar]
- 42.Souza MA, Oliveira MS, Furian AF, Rambo LM, Ribeiro LR, Lima FD, Dalla Corte LC, Silva LF, Retamoso LT, Dalla Corte CL, Puntel GO, de Avila DS, Soares FA, Fighera MR, de Mello CF, Royes LF. Swimming training prevents pentylenetetrazol-induced inhibition of Na+, K+-ATPase activity, seizures, and oxidative stress. Epilepsia. 2009;50:811–23. doi: 10.1111/j.1528-1167.2008.01908.x. [DOI] [PubMed] [Google Scholar]
- 43.Tian J, Kim SF, Hester L, Snyder SH. S-nitrosylation/activation of COX-2 mediates NMDA neurotoxicity. Proc Natl Acad Sci U S A. 2008;105:10537–40. doi: 10.1073/pnas.0804852105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ushikubi F, Segi E, Sugimoto Y, Murata T, Matsuoka T, Kobayashi T, Hizaki H, Tuboi K, Katsuyama M, Ichikawa A, Tanaka T, Yoshida N, Narumiya S. Impaired febrile response in mice lacking the prostaglandin E receptor subtype EP3. Nature. 1998;395:281–4. doi: 10.1038/26233. [DOI] [PubMed] [Google Scholar]
- 45.Pekcec A, Unkrüer B, Schlichtiger J, Soerensen J, Hartz AM, Bauer B, van Vliet EA, Gorter JA, Potschka H. Targeting prostaglandin E2 EP1 receptors prevents seizure-associated P-glycoprotein up-regulation. J Pharmacol Exp Ther. 2009;330:939–947. doi: 10.1124/jpet.109.152520. [DOI] [PubMed] [Google Scholar]
- 46.Kwan P, Schachter SC, Brodie MJ. Drug-resistant epilepsy. N Engl J Med. 2011;365:919–926. doi: 10.1056/NEJMra1004418. [DOI] [PubMed] [Google Scholar]
- 47.Markovic T, Jakopin Ž, Dolenc MS, Mlinarič-Raščan I. Structural features of subtype-selective EP receptor modulators. Drug Discovery Today. 2017;22:57–71. doi: 10.1016/j.drudis.2016.08.003. [DOI] [PubMed] [Google Scholar]
- 48.Santos A, Temp FR, Marafiga JR, Pillat MM, Hessel AT, Ribeiro LR, Miyazato LG, Oliveira MS, Mello CF. EP2 receptor agonist ONO-AE1-259-01 attenuates pentylenetetrazole- and pilocarpine-induced seizures but causes hippocampal neurotoxicity. Epilepsy Behav. 2017;73:180–188. doi: 10.1016/j.yebeh.2017.03.033. [DOI] [PubMed] [Google Scholar]
- 49.Yokoyama U, Iwatsubo K, Umemura M, Fujita T, Ishikawa Y. The prostanoid EP4 receptor and its signaling pathway. Pharmacol Rev. 2013;65:1010–1052. doi: 10.1124/pr.112.007195. [DOI] [PubMed] [Google Scholar]