

Abstract
AIM
To explore hepatitis C virus (HCV) adaptive mutations or combinations thereof responsible for enhanced viral production and investigate the underlying mechanisms.
METHODS
A series of plasmids with adaptive mutations were constructed. After the plasmids were transfected into Huh7.5 cells, we determined the infectious HCV particle titers by NS5A immunofluorescence assays, and detected HCV RNA replication by real-time PCR and protein expression by Western blot. Then we carried out immunoblotting of supernatants and cell lysates with anti-NS3 to analyze the virus release level. In addition, co-localization of lipid droplets (LDs) with NS5A was measured using confocal laser scanning microscopy. The ratio between the p56 and p58 phosphoforms of NS5A was analyzed further.
RESULTS
The plasmids named JFH1-mE2, JFH1-mp7, JFH1-mNS4B, JFH1-mNS5A, JFH1-mE2/NS5A, JFH1-mp7/NS5A, JFH1-mNS4B/NS5A, JFH1-mE2/p7/NS5A, and mJFH1 were constructed successfully. This study generated infectious HCV particles with a robust titer of 1.61 × 106 focus-forming units (FFUs)/mL. All of the six adaptive mutations increased the HCV particle production at varying levels. The NS5A (C2274R, I2340T, and V2440L) and p7 (H781Y) were critical adaptive mutations. The effect of NS5A (C2274R, I2340T, and V2440L), p7 (H781Y), and NS4B (N1931S) on infectious HCV titers was investigated by measuring the HCV RNA replication, protein expression, and virion release. However, the six adaptive mutations were not required for the LD localization of NS5A proteins or the phosphorylation of NS5A.
CONCLUSION
In this study, we generated infectious HCV particles with a robust titer of 1.61 × 106 FFUs/mL, and found that the viral replication and release levels could be enhanced by some of the adaptive mutations.
Keywords: Hepatitis C virus, JFH1, Adaptive mutation, RNA replication, Virion release, Lipid droplet localization
Core tip: In this study, we explored hepatitis C virus (HCV) adaptive mutations or combinations thereof responsible for enhanced viral production and investigated the underlying mechanisms. We generated infectious HCV particles with a robust titer of 1.61 × 106 focus-forming units (FFUs)/mL, and confirmed that the adaptive mutations could enhance viral replication and release. The results were established at the levels of infectious particle titers, HCV RNA, protein expression, virus release, lipid droplet, and NS5A co-localization, and further the ratio between p56 and p58 phosphoforms of NS5A.
INTRODUCTION
Hepatitis C virus (HCV) is a member of the flaviviridae family. HCV infection is a major public health challenge, with an estimated number of 130 to 170 million individuals infected worldwide[1,2]. HCV causes acute and chronic hepatitis, and also leads to permanent liver damage and hepatocellular carcinoma in a significant number of patients, via oxidative stress, insulin resistance, fibrosis, liver cirrhosis, and HCV-induced steatosis[3]. Interferon-α-based therapy, in combination with ribavirin, has limited efficacy in approximately 50% of patients and is associated with severe side effects[4]. Direct-acting antivirals (DAAs) targeting NS3/4A, NS5A, and NS5B proteins can lead to higher sustained virological responses than interferon-based regimens, have shorter treatment duration, are orally administered, and have fewer side effects[5].
HCV is an enveloped RNA virus whose replication occurs in the cytoplasm. It consists of a single-stranded 9.6-kb RNA genome of positive polarity with a 5’ internal ribosome entry site (IRES). IRES-driven HCV RNA produces a polyprotein of approximately 3000 amino acids localized to the rough endoplasmic reticulum (ER), where it is cleaved into at least four structural proteins (C, E1, E2, and p7) and six nonstructural proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) that play a key role in viral replication, assembly, and pathogenesis[6].
Elucidation of the viral structure and virus-host interaction is an important goal of anti-HCV drug discovery and vaccine development[7]. HCV replicon system has contributed to the study of HCV in the human hepatoma cell line Huh-7[8,9]. The infectious HCV JFH1 cell culture system represents a major advance in anti-HCV drug discovery research[7,10-12]. This model generates infectious viral particles in cell culture (HCVcc) and facilitates the study of HCV life cycle[7,11]. However, HCV JFH1 variant genome (genotype 2a) results in relatively low viral titers[7,13,14].
Several studies suggested that cell culture-adaptive mutations in HCV genomic RNA might potentially increase the production of infectious HCV particles[13,15-18]. Recently, an adaptive HCV JFH1 reporter isolate designated as JFH1-∆V3-EGFP was identified[19], which produced higher titers (106 focus-forming units [FFUs]/mL) of HCV-EGFP reporter virus. Whole genome sequencing analysis showed that JFH1-ΔV3-EGFP included six mutations located in the E2, p7, NS4B, and NS5A regions as follows: D657G in E2; H781Y in p7; N1931S in NS4B; and C2274R, I2340T, and V2440L in NS5A. V2440L and H781Y improved the infectious HCV titers[20,21], while data pertaining to the other mutations are not available. In this study, we explored these mutations or combinations thereof responsible for enhanced viral production and investigated the underlying mechanisms.
MATERIALS AND METHODS
Cell culture
The human hepatoma cell line Huh7.5 was generously provided by Dr. Charles M. Rice[22] (Rockefeller University) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen) supplemented with 100 U/mL of penicillin, 100 μg/mL of streptomycin, non-essential amino acids, and 10% fetal bovine serum (Invitrogen) at 37 °C in 5% CO2. All the experiments described in this study were performed using these cells.
Antibodies
The monoclonal antibody to NS5A protein (Abcam), the goat anti-mouse IgG conjugated with horseradish peroxidase (Sigma), and goat anti-mouse IgG conjugated with Alexa Fluor 594 (Invitrogen) were all obtained commercially.
Plasmid construction
Plasmid constructs were based on the consensus sequence of HCV pJFH1, which was kindly provided by Dr. Wakita[10]. JFH1-∆V3-EGFP and JFH1-AM120 plasmids were kindly provided by Dr. C.H. Hagedorn and Shuang-Hu Liu[19]. The mutations located in HCV genomic RNA are shown in Figure 1. A series of primers for construction of adaptive variants of wild-type HCV JFH1 listed in Table 1 were designed using the pJFH1 sequence and mutations. The pJFH1 plasmid was used as a template for subsequent PCR with Phushion High-Fidelity PCR Master Mix with GC buffer (New England Biolabs) according to the manufacturer’s instructions. The preliminary PCR products (mE2-1, mE2-2, mp7-1, mp7-2, mNS4B-1, mNS4B-2, mNS5A-1, mNS5A-2, mNS5A-3, and mNS5A-4) were analyzed by 1% agarose gel electrophoresis, and used for overlap PCR following the combination showed in Tables 2 and 3 to obtain adaptive mutation fragments. The above fragments (mE2, mp7, mNS4B, mNS5A, mE2/NS5A, mp7/NS5A, mNS4B/NS5A, and mE2/p7/NS5A) were sub-cloned into pJFH1 using the appropriate unique restriction enzyme sites such as Bsiw I, Kpn I, Nsi I, Rsr II, or BsrG I, to produce JFH1-mE2, JFH1-mp7, JFH1-mNS4B, JFH1-mNS5A, JFH1-mE2/ NS5A, JFH1-mp7/NS5A, JFH1-mNS4B/NS5A, JFH1-mE2/p7/ NS5A, and also mJFH1, which contained all the six mutations. All new clones were sequenced using an ABI 3700-XL (Shanghai Sangon Biotech).
Figure 1.
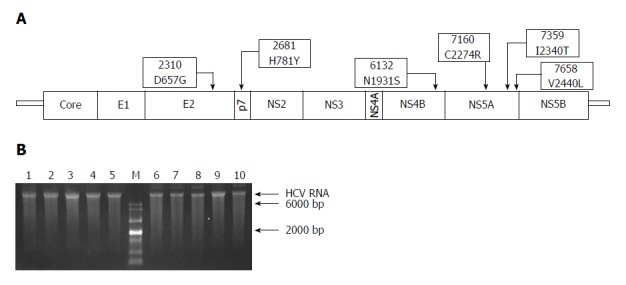
Schematic representation of adaptive mutations used in this study (A) and the electrophoresis results of each mutant virus RNA (B). A: Both nucleotide substitutions (2310, 2681, 6132, 7160, 7359, and 7658) and amino acid substitutions (D657G, H781Y, N1931S, C2274R, I2340T, and V2440L) are shown; B: HCV RNA (500 ng) was analyzed using formaldehyde agarose gel electrophoresis. Lane 1: JFH1; Lane 2: JFH1-mE2; Lane 3: JFH1-mP7; Lane 4: JFH1-mNS4B; Lane 5: JFH1-mNS5A; Lane 6: JFH1-mE2/NS5A; Lane 7: JFH1-mp7/NS5A; Lane 8: JFH1-mNS4B/NS5A; Lane 9: mJFH1; Lane 10: JFH1-mE2/p7/NS5A; M: RNA marker. HCV: hepatitis C virus.
Table 1.
Sequence of primers used for adaptive mutation plasmid construction
| Primer | Sequence (5’-3’) |
| 1340-F | CTGGCGTACGTGATGCG |
| m2310-R | TGTCCCTGTCCTCCAAGCCGCAGCGAT |
| m2310-F | ATCGCTGCGGCTTGGAGGACAGGGACA |
| 3500-R | GCCCCGTCATACTCACCAC |
| m2681-R | TGATGTACCAAGCTGCCACGAAGAAG |
| m2681-F | CTTCTTCGTGGCAGCTTGGTACATCA |
| 5249-F | AATGAGGTCACCCTCACACA |
| m6132-R | ACGTGGCTTCCTCTGGAAGCAAAGGCA |
| m6132-F | TGCCTTTGCTTCCAGAGGAAGCCACGT |
| 7791-R | GATGTTGTACAGTACACCTTG |
| 5249-F | AATGAGGTCACCCTCACACA |
| m7160-R | AGCATGCGCTCCGATGGTATTGAG |
| m7160-F | CTCAATACCATCGGAGCGCATGCT |
| m7359-R | TTCTGATGTGGTGCTCTCGCTCAG |
| m7359-F | CTGAGCGAGAGCACCACATCAGAA |
| m7658-R | GCACAGGGTGGTATCGTCCTCCT |
| m7658-F | AGGAGGACGATACCACCCTGTGC |
| 7966-R | CTTGGATCTTGCAGAAT |
Table 2.
Primer combinations used in adaptive mutation plasmid construction
| Fragment | Template |
Primers |
|
| Sense | Anti-sense | ||
| mE2-1 | JFH1 | 1340-F | m2310-R |
| mE2-2 | m2310-F | 3500-R | |
| mp7-1 | 1340-F | m2681-R | |
| mp7-2 | m2681-F | 3500-R | |
| mNS4B-1 | 5249-F | m6132-R | |
| mNS4B-2 | m6132-F | 7791-R | |
| mNS5A-1 | 5249-F | m7160-R | |
| mNS5A-2 | m7160-F | m7359-R | |
| mNS5A-3 | m7359-F | m7658-R | |
| mNS5A-4 | m7658-F | 7966-R | |
| mNS4B/NS5A-1 | JFH1-mNS5A | 5249-F | m6132-R |
| mNS4B/NS5A-2 | m6132-F | 7791-R | |
| mE2/p7 -1 | JFH1-mp7/NS5A | 1350-F | m2310-R |
| mE2/p7 -2 | m2310-F | 3500-R | |
Table 3.
Primers and templates for overlap PCR
| Fragment | Template |
Primer |
|
| Upstream | Downstream | ||
| mE2 | mE2-1 + mE2-2 | 1340-F | 3500-R |
| mp7 | mp7-1 + mp7-2 | 1340-F | 3500-R |
| mNS4B | mNS4B-1 + mNS4B-2 | 5249 F | 7791-R |
| mNS5A-3/4 | mNS5A-3 + mNS5A-4 | m7359-F | 7966-R |
| mNS5A-2/3/4 | mNS5A-2 + mNS5A-3/4 | m7160-F | 7966-R |
| mNS5A | mNS5A-1 + mNS5A-2/3/4 | 5249-F | 7966-R |
| mNS4B/NS5A | mNS4B/NS5A-1 + mNS4B/NS5A-2 | 5249 F | 7791-R |
| mE2/p7 | mE2/p7 -1 + mE2/p7-2 | 1340-F | 3500-R |
Transfection with HCV RNA
To generate the full-length genomic RNA, pJFH-1 and all plasmids were linearized with Xba I. The linearized plasmid DNA was purified and then used as a template for T7 in vitro transcription (MEGAscript; Ambion). The RNA genomes were detected by formaldehyde agarose gel electrophoresis as described previously[23], and transfected into cells by electroporation[13].
Immunofluorescence assay
Cells seeded on glass coverslips were infected with HCV. After 48 h, the slips were washed with PBS. Then, the cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and blocked with 1% BSA and 1% normal goat serum. The NS5A in cells was detected with a monoclonal antibody and a secondary goat anti-mouse Alexa Fluor 594 antibody (Invitrogen) and visualized by fluorescence microscopy.
Virus titration
The titer of infectious HCV was determined by immunofluorescence assay[7]. Virus titers from supernatants and cell lysates as well were determined using FFUs.
Cell lysates were prepared as described previously[24]. Briefly, cell pellets harvested after trypsinization were washed with PBS, re-suspended in completed culture medium, and lysed in four freeze/thaw cycles at -80 °C and 37 °C. The cell lysates were centrifuged at 4000 rpm for 5 min prior to inoculation into naïve Huh7.5 cells.
Virus titration analysis was conducted by serially diluting the cell supernatants or cell lysates 10-fold in DMEM. The supernatants were used to infect 1 × 104 naïve Huh7.5 cells in 96-well plates. The cells were incubated with virus for 2 h at 37 °C, washed, and incubated with complete DMEM. The level of HCV infection was analyzed 3 and 9 d post-infection by immune- fluorescence staining for NS5A.
Western blot analysis
The Huh7.5 cells infected with HCV RNA were lysed in 50 mmol/L Tris-HCl (pH 7.5) containing 150 mmol/L sodium chloride, 1% Nonidet P40, 0.5% sodium deoxycholate, 0.1% SDS, and proteinase inhibitors (Complete Mini, Roche). Samples were separated by 10% SDS-PAGE and then transferred onto nitrocellulose membranes. The HCV NS5A (p56/p58) was analyzed as described previously[13].
Quantification of HCV RNA by qPCR
Total RNA was extracted with TRIzol (Invitrogen). HCV RNA was measured by qPCR analysis as described previously[25]. β-actin was used as the internal control. The relative quantity of HCV RNA in control and HCV samples was calculated by the comparative Ct (cycling threshold) method using LightCycler480.
Confocal laser scanning microscopy
Cells transfected with HCV RNA with adaptive mutations were seeded onto 24-well plates with cover slips. The cells were treated as previously described[13]. After 48 h, the cells were washed with PBS, fixed with 4% paraformaldehyde, and then permeabilized with 0.2% Triton X-100. Fixed cells were blocked with 1% bovine serum albumin and 1% normal goat serum in PBS. Next, HCV NS5A was analyzed in cells using a NS5A monoclonal antibody and a secondary goat anti-mouse IgG conjugated with Alexa 488 (Invitrogen, dilution of 1:1000). LipidTOX Deep Red (Invitrogen) was used to detect neutral lipids present in lipid droplets (LDs). The slides were counterstained using DAPI (Invitrogen), and examined using an Zeiss LSM 510 Meta confocal laser scanning microscope.
RESULTS
Effect of individual mutations or combinations of adaptive mutations on the production of infectious HCV
A previous study demonstrated that JFH-∆V3-EGFP variant produces a higher titer of reporter virus[19]. The six adaptive mutations in this variant were located in the E2 (D657G), p7 (H781Y), NS4B (N1931S), and NS5A (C2274R, I2340T, and V2440L) (Figure 1A), respectively. To determine the individual or synergistic combination of these mutations responsible for the increased viral production, recombinant JFH1 genomes containing only one of the selected mutations or four different combinations as shown in Figure 1A were constructed.
Next, ten in vitro-transcribed mutant JFH1 RNAs (Figure 1B) were electroporated into Huh7.5 cells to produce recombinants of adapted virus. The transfected cells were sub-cultured every three days. The infectivity titers of supernatants at day 3 (P1) and day 9 (P3) were measured (Figure 2). Viral titers are expressed as FFUs/mL and were assayed in duplicate, which was repeated three times. The data are presented as mean ± SD (n = 3). The HCV titers of wild type JFH1 (JFH1) were extremely low, with a typical titer of 102 FFUs/mL. The adaptive mutations in E2, p7, NS4B, and NS5A individually increased the production of infectious HCV titers 2- to 4-fold compared with the levels of JFH1. The NS5A mutations exhibited the greatest effect on the production of infectious HCV particles, with a titer of 1.30 × 106 FFUs/mL at P3. The p7 mutation followed closely, generating a titer of 2.10 × 105 FFUs/mL. Briefly, except for E2, the HCV titers of other variants at P3 were partially higher than those at P1.
Figure 2.
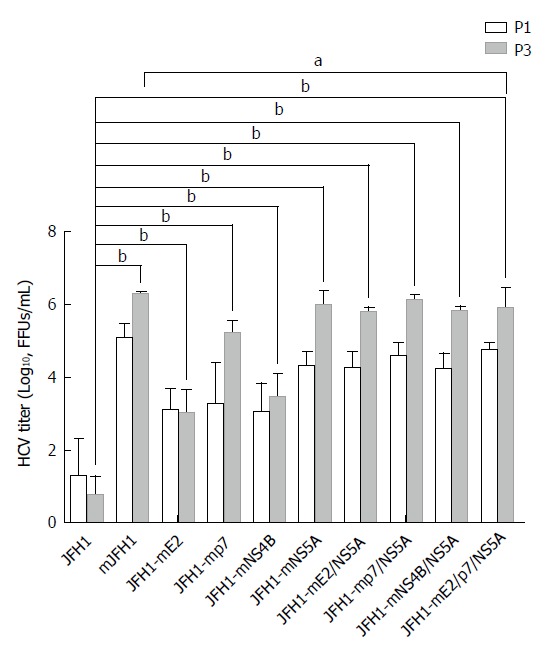
Generation of high titer cell culture-adaptive JFH1 virus. Hepatitis C virus RNA was electroporated into Huh7.5 cells to produce the recombinants of adapted virus in cell culture. The transfected cells were passaged every three days. The infectivity titers of the culture supernatants at day 3 (P1) and day 9 (P3) were measured. Viral titers are expressed as focus-forming units per milliliter (FFUs/mL). The data are presented as mean ± SD (n = 3). HCV: Hepatitis C virus. aP < 0.05; bP < 0.01.
To further determine any synergistic effect of the six adaptive mutations on HCV production, we focused on the recombinant viruses with adaptive mutations in different combinations. As shown in Figure 2, JFH1-mE2/NS5A, JFH1-mp7/NS5A, JFH1-mNS4B/NS5A, and JFH1-mE2/p7/NS5A remarkably enhanced the production of infectious HCV, and the mJFH1 produced infectious HCV particles with a robust titer of 1.61 × 106 FFUs/mL 9 d post-transfection.
These results suggest that all the six adaptive mutations increase the HCV particle production. NS5A (C2274R, I2340T, and V2440L) and p7 (H781Y) are the critical adaptive mutations.
HCV RNA replication and protein expression are up-regulated by adaptive mutations
HCV RNA genome replication and structural or nonstructural protein expression are early steps in the HCV life cycle. To further confirm our speculation that the robust HCV titers and enhanced virion release were both related to up-regulated RNA replication and protein expression, we determined the relative HCV RNA, NS5A immunofluorescence, and NS3 protein levels in the RNA-transfected Huh7.5 cells on day 3 (P1) and day 9 (P3).
As shown in Figure 3, the expression of NS5A (Figure 3A) and NS3 (Figure 3B) in mutants was up-regulated at different levels during serial passages. The trend was extraordinary obvious in NS5A or p7 mutants. Anti-NS3 Western blot analysis, which was the most widely used for quantitative experiment, yielded consistent results.
Figure 3.
Effects of the adaptive mutations on the hepatitis C virus RNA replication. A: Hepatitis C virus (HCV) RNA was electroporated into Huh7.5 cells to produce the recombinants of adapted virus. The transfected cells were passaged every 3 d. Cells were fixed 48 h after passage and infected cells were identified by fluorescence immunostaining and microscopy. Nuclear DNA was stained with DAPI (blue); B: HCV RNA was electroporated into Huh7.5 cells to produce the recombinants of adapted virus in cell culture. The transfected cells were passaged every 3 d. Cells were lysed at 72 h after passage. The HCV NS3 protein levels were analysis by Western blot. bP < 0.01; C: HCV RNA levels in cells 3 d after transfection. Intracellular HCV RNA levels were analyzed by quantitative RT-PCR. The mean ± SD for three independent experiments are presented (qPCR assays, n = 3). aP < 0.05; bP < 0.01; cP < 0.001.
As shown in Figure 3C, the RNA levels of all the mutants were increased compared with JFH1, and mJFH1 was increased 18.7-fold. Interestingly, the results indicated that JFH1-mNS4B expression increased 6.1-fold, and the combination of mutants showed a 7.4-16.8-fold increase.
Taken together, we confirmed that the effect of NS5A (C2274R, I2340T and V2440L), p7 (H781Y), and NS4B (N1931S) on infectious HCV titers was robust, and started with HCV RNA replication and protein expression, followed by virion release.
Adapted variants enhance the efficiency of virus release
Virion release is the last step of the HCV life cycle. To further explore the mechanism underlying the enhanced virus production, the role of adaptive mutations was examined. Ten HCV RNAs (JFH1, JFH1-mE2, JFH1-mP7, JFH1-mNS4B, JFH1-mNS5A, JFH1-mE2/NS5A, JFH1-mp7/NS5A, JFH1-mNS4B/NS5A, mJFH1, and JFH1-mE2/p7/NS5A) were electroporated into Huh7.5 cells. After 3 d, we collected the supernatants and cell lysates, and measured the HCV titers using NS5A immunofluorescence assays. Furthermore, to confirm the infectivity of virions, we carried out immunoblotting of supernatants and cell lysates with anti-NS3, which was extraordinarily consistent with the infectious HCV titers (Figure 4A). As shown in Figure 4B, we also calculated the proportion of extracellular (supernatant) or intracellular (cell lysate) HCV titers using total titers as 100%. Typically, the supernatant and intracellular HCV titers of JFH1 were 76.31% and 23.69%, respectively, and those of mJFH1 were 94.00% and 6.00%, respectively. Taken together, these findings provide evidence suggesting that the ten variant viruses enhanced the virion release, and the high viral production was linked to the effective virion release. NS5A (C2274R, I2340T, and V2440L) and p7 (H781Y) showed the highest levels compared with the others.
Figure 4.
Effect of the adaptive mutations on the virion release. A: Hepatitis C virus (HCV) RNA was electroporated into Huh7.5 cells to produce the recombinants of adapted virus. At 72 h after transfection, the infectivity titers of the culture supernatants and cell lysates were measured. Viral titers are expressed as FFUs/mL. The data are presented as mean ± SD (n = 3); B: HCV RNA was electroporated into Huh7.5 cells to produce the recombinants of adapted virus. At 72 h after transfection, the infectivity titers of the culture media and cell lysates were measured. The extracellular and intracellular viral titers were measured. The relative ratios of infectious virions are shown. The results were from three independent experiments; C: The naive Huh7.5 cells were infected with the culture media and cell lysates. At 72 h after infection, cells were lysed with RIPA buffer, and analyzed by Western blot.
Adaptive mutations are not essential for intracellular LD localization of the NS5A protein
LDs have been reported to play an important role in the HCV virion assembly process[26]. To determine if the six adaptive mutations increased the assembly of HCV at this step, LDs and NS5A were stained in JFH1 and mJFH1 transfected cells and the co-localization of LDs with NS5A was measured. As shown in Figure 5, the LDs were totally covered with NS5A in all cases. However, no significant difference was observed between JFH1 and mJFH1 groups using Image J software and Pearson’s correlation coefficient analysis. These results indicated that the six adaptive mutations were not required for LD localization of the NS5A proteins.
Figure 5.
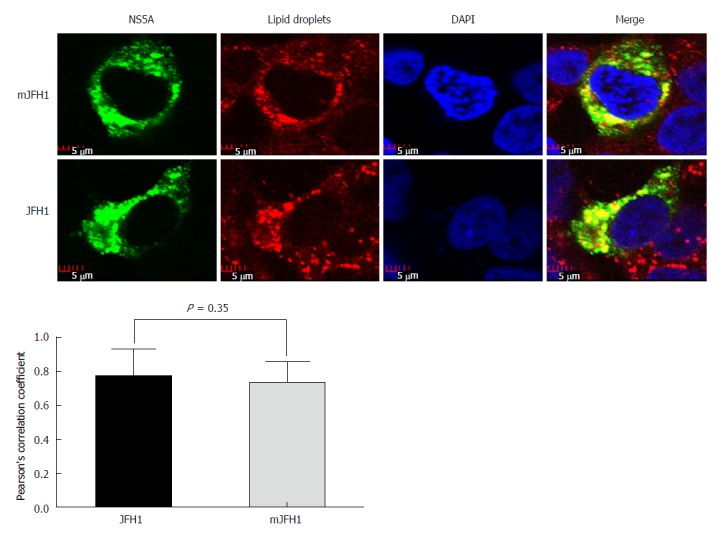
Colocalization analysis of lipid droplets and hepatitis C virus NS5A. JFH1 and mJFH1 RNA was electroporated into Huh7.5 cells to produce the recombinants of adapted virus. At 48 h after transfection, the cells were fixed. Lipid droplets were stained with LipidTOXRed (Red). The HCV NS5A was stained with anti-NS5A antibody (Green). The nucleus was stained with DAPI (Blue). Each triplicate sample of 25 cells was analyzed using Image J software. The degree of co-localization was quantified and compared using Pearson’s correlation coefficients.
Adapted mutations do not affect hyper-phosphorylation of NS5A
Previous studies showed that a ratio between the p56 and p58 phosphoforms of NS5A is required for optimal HCV RNA replication[13,27-29]. JFH1-AM120 is a robust adaptive mutant selected by Liu et al[13]. which displays significant switch of p56/p58. In this study, JFH1, JFH1-AM120, and mJFH1 RNA were transfected into Huh7.5 cells, and the total protein was used for Western blot analysis after 3 d (Figure 6). However, we observed no difference in p56 and p58 between the two groups. These results demonstrated that the phosphorylation level of NS5A was not affected by adaptive mutations.
Figure 6.
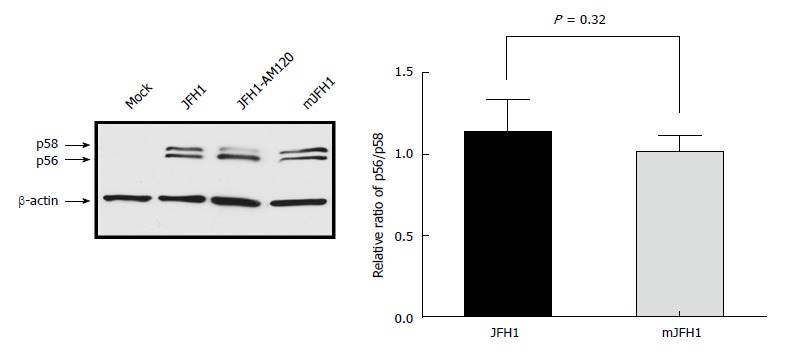
Phosphorylation of NS5A during JFH1 and mJFH1 replication. Huh7.5 cells were transfected with JFH1 or mJFH1 RNA. After three days of culture, cells were lysed for western blot using anti-NS5A and anti-β-actin antibodies. The quantity of p56 and p58 was determined using Image J software and the ratios of p56/p58 are shown. Data are presented as mean ± SD (n = 3). JFH1-AM120 was used as the positive control.
DISCUSSION
Previous studies suggested that in vitro adaptive mutations enhance the production of infectious virus[13-17,20,21,30-34]. A high mutation rate in HCV RNA genome is a challenge for successful HCV treatment and vaccine research, although a method to obtain a robust clonal culture of HCV has been unavailable[13]. Liu et al[19] demonstrated that a JFH1-∆V3-EGFP variant produced higher titer of reporter virus. The six adaptive mutations in this variant were located in the E2 (D657G), p7 (H781Y), NS4B (N1931S), and NS5A (C2274R, I2340T, and V2440L) (Figure 1A). However, the mutations responsible for enhanced viral production were not clear. The six mutations in this study were simultaneously located in JFH1-∆V3-EGFP, which was a reporter EGFP gene chimera virus[19]. The mJFH1 refers to JFH1-ΔV3-EGFP that yielded a robust titer up to 1.61 × 106 FFUs/mL in this study, suggesting that the six mutations are effective adaptive mutations.
HCV is a single, positive-strand RNA virus. We focused on key life cycle events in the virus such as replication, expression, assembly, and release. Infectious virion release is the last step and the final objective of the JFH1 system. In our study, we detected variant virus titers initially. Consistent with previous reports[7,14], JFH1 only exhibited a decreased titer of 102 FFUs/mL. The other mutants showed increased titers with several orders of magnitude compared with JFH1. Synergistic enhancement of HCV titer was demonstrated obviously. Jiang et al[35] suggested that adaptive mutations enhance specific protein-protein interactions among viral proteins and promote the assembly of infectious HCV particles. We speculated that the six mutations involved refer to unknown life cycle phases and mechanism as well. Therefore, we analyzed the effect of viral mutations on the distribution of virions in the supernatant and the cell lysate, co-localization of LDs and NS5A, HCV RNA level, NS3 expression, and p56/p58. We found that the adaptive mutations were associated with diverse effects on the life cycle events. The virion release and RNA genome replication were specifically associated with NS5A and p7 mutations.
The transmembrane domains of chimeric E1 and/or E2 HCV glycoproteins were modified to allow transport to and assembly at the cell surface[36]. E2 consisted of three critical domains: a receptor-binding domain (RBD; residues 384-661), the membrane proximal stem-like region of E2 (residues 675-699), and a hydrophobic heptad repeat linking the two domains[37]. Within the RBD, the E2 bound the cellular receptor CD81, leading to receptor-mediated endocytosis of virions[38,39]. Serial studies showed that the mutations in E2 play a role in the HCV life cycle via different mechanisms. Tao et al[30] demonstrated that the E2 (I414T) mutation had no significant effect on HCV RNA replication and viral entry. However, it enhanced the production of infectious viral particles and decreased the receptor-mediated viral entry. E2 (G451R) altered the relationship between particle density and infectivity, disrupted the co-receptor dependence, and increased virion sensitivity to receptor mimics[40]. The T563I mutation in the E2 protein increased virion viability at 37 °C. Unfortunately, D657G in E2 improved the HCV titer via an unknown mechanism, without any effect on HCV RNA replication or virion release.
As a small membrane polypeptide, the HCV p7 channel plays multiple roles in virus life cycle and mediates several biological functions in HCV infection[41]. The p7 consists of six equivalent hydrophobic pockets between the peripheral and pore-forming helices[42]. Generally, p7 is not essential for HCV RNA replication, but required for virion assembly and release[43]. The adaptive mutation N765D in p7 influenced early stages of the HCV life cycle, and increased the infectious HCV titer[15]. Y781H enhances the level of HCV core in the supernatant three- to five-fold, and moderately increases virion assembly and release[21]. In our study, we found similar results, and Y781H enhanced HCV RNA replication 3.1-fold, suggesting its role as a critical initiating agent and a novel mechanism during the HCV life cycle.
HCV NS4B plays an important role in RNA genome replication and virion assembly[44]. NS4B triggers the formation of a viral replication complex[45] similar to the “sponge-like inclusions” observed in the liver of HCV-infected chimpanzee[46]. NS4B (K1846T) increased HCV RNA replication nearly 30-fold[47]. N1931S is located between helices 1 and 2 of the NS4B C-terminus, and was first determined by Li et al[48] during HCV RNA replication and virion assembly. Our data suggested that the N1931S increased HCV titer to 103 FFUs/mL, which was 103-fold compared with JFH1. It significantly enhanced HCV genome replication, and slightly improved virion release. N1931S is a novel mutation in the JFH1 system, and comprehensive studies investigating its role in HCV infection are needed.
HCV NS5A is a phosphoprotein existing in two different forms: a basic phosphorylated NS5A, p56, and a hyperphosphorylatedNS5A, p58. It appears to play an important role in viral replication, since most of the adaptive mutations determined so far are located within the region of NS5A[47]. The three domains in NS5A include: domain I (aa 28-213) coordinating a single zinc atom, and domains II (aa 250-342) and III (aa 356-447), which are less well characterized but are important in RNA replication and/or virion assembly[28]. A previous report suggested that V2440L was located at the NS5A-B cleavage site and decreased the cleavage kinetics[20]. Thus, the mutation C2274R is located in domain II, and the other mutations (I2340T and V2440L) occur in domain III. We analyzed the HCV RNA replication and protein expression. The results showed that the three mutations enhanced HCV RNA replication, which is consistent with the structure.
A previous study demonstrated that HCV p7 promotes a late step of assembly and release of infectious virions[49] and NS5A plays a major role in regulating the release of infectious virus particles in cell culture[32]. In this study, there were three mutants (C2274R, I2340T, and V2440L) located in NS5A and one located (H781Y) in p7. Our results showed that these mutants obviously promoted the HCV viral particles release (Figure 4). HCV core is located on the cytosolic side of the ER membrane, assembly probably initiates in the cytosol before further maturation, and release occurs by transfer of nascent particles across the ER membrane to enable access to the secretory pathways in hepatocytes[50]. The amino acid changes induced by mutants in NS5A and P7 may be involved in these steps. The specific mechanism needs to be further studied in future.
Previous studies suggested that the up-regulation of p56/p58 ratio might be a critical factor for HCV titer[13] and increased HCV replication since specific mutations reduced NS5A hyper-phosphorylation activating RNA replication[27]. NS5A-p58 levels increased following overexpression of CKI-alpha, CKI-delta, and CKI-epsilon, whereas RNA interference of CKI-alpha alone reduced NS5A hyper-phosphorylation[51]. Here, we detected the status of p56/p58. However, there was no switch between the JFH1 and mJFH1 groups. The two viral proteins including the core and NS5A were localized to LDs, which play an important role in the intracellular assembly of HCV[24,26]. The recruitment of NS5A to LDs was a prerequisite for virion assembly in Huh7.5 cells[26]. Our analysis of the co-localization of NS5A and LDs showed no significant difference between mJFH1 and JFH1, suggesting that these adaptive mutations did not alter virion formation.
The life cycle of HCV is extremely complex, and several details remain unknown. Regulation of host gene expression[52,53], altered association between viral proteins and/or host-cell proteins, and changes in virus per se[54] represent obvious mechanisms. In our study, we confirmed that the adaptive mutations led to a robust infectious titer via enhanced viral replication and release. It is recommended that DAA regimens can be used for treatment of patients with hepatitis C rather than pegylated interferon/rabivirin[5]. Meanwhile, our study was limited by the reaction of DAAs to above adaptive mutations. Further studies investigating the underlying mechanisms of viral morphogenesis are needed.
In conclusion, we generated infectious HCV particles with a robust titer of 1.61 × 106 FFUs/mL in this study. All of the six adaptive mutations increased the HCV particle production at varying levels. The NS5A (C2274R, I2340T, and V2440L) and p7 (H781Y) were critical adaptive mutations. This study confirmed that the JFH1 is still a promising system to study the HCV life cycle. To use adaptive mutations is an effective means to establish a new system with higher infectious HCV virions titer. And the research on molecular mechanism of interaction between viral proteins and/or host-cell proteins should be carried out in depth.
ARTICLE HIGHLIGHTS
Research background
Hepatitis C virus (HCV) causes acute and chronic hepatitis, and leads to permanent liver damage and hepatocellular carcinoma. The infectious HCV JFH1 cell culture system represents a major advance in anti-HCV drug discovery research and facilitates the study of HCV life cycle. However, HCV JFH1 (genotype 2a) merely generates relatively low viral titers. JFH1-∆V3-EGFP, which includes six mutations located in the E2, p7, NS4B, and NS5A regions, could produce higher titers of HCV-EGFP reporter virus. However, there were no data about which mutations or combinations thereof are responsible for enhanced viral production and the underlying mechanisms.
Research motivation
This JFH1 model generated infectious viral particles in cell culture and facilitated the study of the HCV life cycle, but the low infectious virion titer limits its application range. Some previous studies have confirmed that adaptive mutations could enhance the virion titer, but the mechanism has not yet been fully elucidated. In this study, we focused on the positive effect of six adaptive mutations located in the E2, p7, NS4B, and NS5A regions, and found that the mechanism was different among them during the procession. These results gave us some new insights into the infectious HCV cell culture system and adaptive mutations.
Research objectives
The main objective of this study was to establish an infectious HCV cell culture system with a robust titer, and to discuss the underlying mechanisms of the adaptive mutations found in previous studies. The results of this study have supplied the researchers with a useful tool. We hope it will be used for the study of viral structure, virus-host interaction, anti-HCV drug discovery, and vaccine development.
Research methods
We investigated JFH1-mE2, JFH1-mp7, JFH1-mNS4B, JFH1-mNS5A, JFH1-mE2/NS5A, JFH1-mp7/NS5A, JFH1-mNS4B/NS5A, JFH1-mE2/p7/ NS5A, and mJFH1, carrying all the six mutations. We analyzed the infectious HCV titer, HCV RNA and NS3 protein levels, viral release capacity, assembly and hyper-phosphorylation of NS5A to determine the role of these mutations in the HCV life cycle. These methods were the routine ways adopted widely in virological and molecular biological research.
Research results
The main findings in this study were as follows: (1) we generated infectious HCV particles with a robust titer of 1.61 × 106 FFUs/mL; (2) The six adaptive mutations increased the HCV particle production at varying levels. The NS5A (C2274R, I2340T, and V2440L) and p7 (H781Y) are critical adaptive mutations. The effect of NS5A (C2274R, I2340T, and V2440L), p7 (H781Y), and NS4B (N1931S) on infectious HCV titers was investigated by measuring the HCV RNA replication, protein expression, and virion release; and (3) the six adaptive mutations were all not required for the lipid droplet localization of NS5A proteins or the phosphorylation of NS5A. To our knowledge, this is a new robust titer related to adaptive mutations from JFH1. The problems that remain to be solved in the future include: (1) how could the adaptive mutations be translated to clinical conditions? (2) are these mutation patterns observed in vivo? and (3) would these results be relevant to the resistance to direct-acting antivirals (DAAs)?
Research conclusions
First, this study generated infectious HCV particles with a robust titer of 1.61 × 106 FFUs/mL. Second, all of the six adaptive mutations increased the HCV particle production at varying levels. Third, the NS5A (C2274R, I2340T, and V2440L) and p7 (H781Y) were critical adaptive mutations, but they were not required for the LD localization of NS5A proteins or the phosphorylation of NS5A. Based on the new findings of this study, we proposed that more important adaptive mutations would be addressed in the future, and unknown mechanism of the HCV life cycle would be explained.
Research perspectives
This study re-confirmed that the JFH1 was still a promising system to study the HCV life cycle. To use adaptive mutations was an effective way to establish a new system with higher infectious HCV virion titer. In addition, we also re-confirmed that the molecular mechanism of interaction between viral proteins and/or host-cell proteins is more complex and important.
ACKNOWLEDGMENTS
We thank Dr. Wakita T for providing the plasmid containing the HCV JFH1 plasmid. We also thank Dr. Xu HT for providing Huh7.5 cells, and Dr. Hagedorn CH and Dr. Liu SH for providing JFH1-∆V3-EGFP and JFH1-AM120 plasmids.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): D
Grade E (Poor): 0
Supported by Beijing Natural Science Foundation, No. 7161006; and Beijing Municipal Administration of Hospitals’ Youth Program, No. QML20161801 and No. QML20171801.
Institutional review board statement: This study did not involve any animal experiments or human specimens, and thus was exempted from ethical review according to the Human Research Management Stipulation of the Beijing Ditan Hospital Affiliated to the Capital University of Medical Sciences.
Conflict-of-interest statement: The authors declare that there are no conflicts of interest in this study.
Data sharing statement: Technical appendix, statistical code, and data set available from the corresponding author at chengj0817@ccmu.edu.cn. No additional data are available.
Peer-review started: December 1, 2017
First decision: December 20, 2017
Article in press: January 17, 2018
P- Reviewer: Bock CT, Shi ZJ, Sipos F S- Editor: Wang JL L- Editor: Wang TQ E- Editor: Huang Y
Contributor Information
Qi Wang, Center of Liver Diseases, Beijing Ditan Hospital, Capital Medical University, Beijing 100015, China; Beijing Key Laboratory of Emerging Infectious Diseases, Beijing 100015, China.
Yue Li, Department of Pathology, Beijing Ditan Hospital, Capital Medical University, Beijing 100015, China.
Shun-Ai Liu, Institute of Infectious Diseases, Beijing Ditan Hospital, Capital Medical University, Beijing 100015, China; Beijing Key Laboratory of Emerging Infectious Diseases, Beijing 100015, China.
Wen Xie, Center of Liver Diseases, Beijing Ditan Hospital, Capital Medical University, Beijing 100015, China.
Jun Cheng, Center of Liver Diseases, Beijing Ditan Hospital, Capital Medical University, Beijing 100015, China; Institute of Infectious Diseases, Beijing Ditan Hospital, Capital Medical University, Beijing 100015, China; Beijing Key Laboratory of Emerging Infectious Diseases, Beijing 100015, China. chengj0817@ccmu.edu.cn.
References
- 1.Li YP, Ramirez S, Jensen SB, Purcell RH, Gottwein JM, Bukh J. Highly efficient full-length hepatitis C virus genotype 1 (strain TN) infectious culture system. Proc Natl Acad Sci USA. 2012;109:19757–19762. doi: 10.1073/pnas.1218260109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the United States. Clin Infect Dis. 2012;55 Suppl 1:S10–S15. doi: 10.1093/cid/cis361. [DOI] [PubMed] [Google Scholar]
- 3.Jahan S, Ashfaq UA, Qasim M, Khaliq S, Saleem MJ, Afzal N. Hepatitis C virus to hepatocellular carcinoma. Infect Agent Cancer. 2012;7:2. doi: 10.1186/1750-9378-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoofnagle JH, di Bisceglie AM. The treatment of chronic viral hepatitis. N Engl J Med. 1997;336:347–356. doi: 10.1056/NEJM199701303360507. [DOI] [PubMed] [Google Scholar]
- 5.World Health Organization. Guidelines for the Screening Care and Treatment of Persons with Chronic Hepatitis C Infection: Updated Version. Geneva: World Health Organization;; 2016. [PubMed] [Google Scholar]
- 6.Moradpour D, Penin F. Hepatitis C virus proteins: from structure to function. Curr Top Microbiol Immunol. 2013;369:113–142. doi: 10.1007/978-3-642-27340-7_5. [DOI] [PubMed] [Google Scholar]
- 7.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci USA. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lohmann V, Körner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 9.Blight KJ, Kolykhalov AA, Rice CM. Efficient initiation of HCV RNA replication in cell culture. Science. 2000;290:1972–1974. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- 10.Kato T, Furusaka A, Miyamoto M, Date T, Yasui K, Hiramoto J, Nagayama K, Tanaka T, Wakita T. Sequence analysis of hepatitis C virus isolated from a fulminant hepatitis patient. J Med Virol. 2001;64:334–339. doi: 10.1002/jmv.1055. [DOI] [PubMed] [Google Scholar]
- 11.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Kräusslich HG, Mizokami M, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindenbach BD, Evans MJ, Syder AJ, Wölk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 13.Liu S, Xiao L, Nelson C, Hagedorn CH. A cell culture adapted HCV JFH1 variant that increases viral titers and permits the production of high titer infectious chimeric reporter viruses. PLoS One. 2012;7:e44965. doi: 10.1371/journal.pone.0044965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delgrange D, Pillez A, Castelain S, Cocquerel L, Rouillé Y, Dubuisson J, Wakita T, Duverlie G, Wychowski C. Robust production of infectious viral particles in Huh-7 cells by introducing mutations in hepatitis C virus structural proteins. J Gen Virol. 2007;88:2495–2503. doi: 10.1099/vir.0.82872-0. [DOI] [PubMed] [Google Scholar]
- 15.Kim CS, Keum SJ, Jang SK. Generation of a cell culture-adapted hepatitis C virus with longer half life at physiological temperature. PLoS One. 2011;6:e22808. doi: 10.1371/journal.pone.0022808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gorzin AA, Ramsland PA, Tachedjian G, Gowans EJ. Identification of residues involved in NS2 homodimerization and elucidation of their impact on the HCV life cycle. J Viral Hepat. 2012;19:189–198. doi: 10.1111/j.1365-2893.2011.01504.x. [DOI] [PubMed] [Google Scholar]
- 17.Chan K, Robinson M, Yang H, Cornew S, Delaney Iv WE. Development of a robust luciferase reporter 1b/2a hepatitis C virus (HCV) for characterization of early stage HCV life cycle inhibitors. Antiviral Res. 2013;98:85–92. doi: 10.1016/j.antiviral.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 18.Liu S, Nelson CA, Xiao L, Lu L, Seth PP, Davis DR, Hagedorn CH. Measuring antiviral activity of benzimidazole molecules that alter IRES RNA structure with an infectious hepatitis C virus chimera expressing Renilla luciferase. Antiviral Res. 2011;89:54–63. doi: 10.1016/j.antiviral.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu S, Chen R, Hagedorn CH. Direct visualization of hepatitis C virus-infected Huh7.5 cells with a high titre of infectious chimeric JFH1-EGFP reporter virus in three-dimensional Matrigel cell cultures. J Gen Virol. 2014;95:423–433. doi: 10.1099/vir.0.055772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaul A, Woerz I, Meuleman P, Leroux-Roels G, Bartenschlager R. Cell culture adaptation of hepatitis C virus and in vivo viability of an adapted variant. J Virol. 2007;81:13168–13179. doi: 10.1128/JVI.01362-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brohm C, Steinmann E, Friesland M, Lorenz IC, Patel A, Penin F, Bartenschlager R, Pietschmann T. Characterization of determinants important for hepatitis C virus p7 function in morphogenesis by using trans-complementation. J Virol. 2009;83:11682–11693. doi: 10.1128/JVI.00691-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol. 2002;76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bryant S, Manning DL. Formaldehyde gel electrophoresis of total RNA. Methods Mol Biol. 1998;86:69–72. doi: 10.1385/0-89603-494-1:69. [DOI] [PubMed] [Google Scholar]
- 24.Gastaminza P, Kapadia SB, Chisari FV. Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J Virol. 2006;80:11074–11081. doi: 10.1128/JVI.01150-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papic N, Maxwell CI, Delker DA, Liu S, Heale BS, Hagedorn CH. RNA-sequencing analysis of 5’ capped RNAs identifies many new differentially expressed genes in acute hepatitis C virus infection. Viruses. 2012;4:581–612. doi: 10.3390/v4040581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T, Zayas M, Bartenschlager R, Wakita T, Hijikata M, Shimotohno K. The lipid droplet is an important organelle for hepatitis C virus production. Nat Cell Biol. 2007;9:1089–1097. doi: 10.1038/ncb1631. [DOI] [PubMed] [Google Scholar]
- 27.Tellinghuisen TL, Foss KL, Treadaway J. Regulation of hepatitis C virion production via phosphorylation of the NS5A protein. PLoS Pathog. 2008;4:e1000032. doi: 10.1371/journal.ppat.1000032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiu D, Lemm JA, O’Boyle DR 2nd, Sun JH, Nower PT, Nguyen V, Hamann LG, Snyder LB, Deon DH, Ruediger E, Meanwell NA, Belema M, Gao M, Fridell RA. The effects of NS5A inhibitors on NS5A phosphorylation, polyprotein processing and localization. J Gen Virol. 2011;92:2502–2511. doi: 10.1099/vir.0.034801-0. [DOI] [PubMed] [Google Scholar]
- 29.Huang Y, Staschke K, De Francesco R, Tan SL. Phosphorylation of hepatitis C virus NS5A nonstructural protein: a new paradigm for phosphorylation-dependent viral RNA replication? Virology. 2007;364:1–9. doi: 10.1016/j.virol.2007.01.042. [DOI] [PubMed] [Google Scholar]
- 30.Tao W, Xu C, Ding Q, Li R, Xiang Y, Chung J, Zhong J. A single point mutation in E2 enhances hepatitis C virus infectivity and alters lipoprotein association of viral particles. Virology. 2009;395:67–76. doi: 10.1016/j.virol.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 31.Kaul A, Stauffer S, Berger C, Pertel T, Schmitt J, Kallis S, Zayas M, Lohmann V, Luban J, Bartenschlager R. Essential role of cyclophilin A for hepatitis C virus replication and virus production and possible link to polyprotein cleavage kinetics. PLoS Pathog. 2009;5:e1000546. doi: 10.1371/journal.ppat.1000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han Q, Xu C, Wu C, Zhu W, Yang R, Chen X. Compensatory mutations in NS3 and NS5A proteins enhance the virus production capability of hepatitis C reporter virus. Virus Res. 2009;145:63–73. doi: 10.1016/j.virusres.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 33.Murayama A, Sugiyama N, Suzuki R, Moriyama M, Nakamura N, Mochizuki H, Wakita T, Kato T. Amino Acid Mutations in the NS4A Region of Hepatitis C Virus Contribute to Viral Replication and Infectious Virus Production. J Virol. 2017;91:pii: e02124–16. doi: 10.1128/JVI.02124-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan Y, He Y, Boson B, Wang X, Cosset FL, Zhong J. A Point Mutation in the N-Terminal Amphipathic Helix α0 in NS3 Promotes Hepatitis C Virus Assembly by Altering Core Localization to the Endoplasmic Reticulum and Facilitating Virus Budding. J Virol. 2017;91:pii: e02399–16. doi: 10.1128/JVI.02399-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang J, Luo G. Cell culture-adaptive mutations promote viral protein-protein interactions and morphogenesis of infectious hepatitis C virus. J Virol. 2012;86:8987–8997. doi: 10.1128/JVI.00004-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bartosch B, Vitelli A, Granier C, Goujon C, Dubuisson J, Pascale S, Scarselli E, Cortese R, Nicosia A, Cosset FL. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J Biol Chem. 2003;278:41624–41630. doi: 10.1074/jbc.M305289200. [DOI] [PubMed] [Google Scholar]
- 37.Drummer HE, Boo I, Maerz AL, Poumbourios P. A conserved Gly436-Trp-Leu-Ala-Gly-Leu-Phe-Tyr motif in hepatitis C virus glycoprotein E2 is a determinant of CD81 binding and viral entry. J Virol. 2006;80:7844–7853. doi: 10.1128/JVI.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, et al. Binding of hepatitis C virus to CD81. Science. 1998;282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 39.Codran A, Royer C, Jaeck D, Bastien-Valle M, Baumert TF, Kieny MP, Pereira CA, Martin JP. Entry of hepatitis C virus pseudotypes into primary human hepatocytes by clathrin-dependent endocytosis. J Gen Virol. 2006;87:2583–2593. doi: 10.1099/vir.0.81710-0. [DOI] [PubMed] [Google Scholar]
- 40.Grove J, Nielsen S, Zhong J, Bassendine MF, Drummer HE, Balfe P, McKeating JA. Identification of a residue in hepatitis C virus E2 glycoprotein that determines scavenger receptor BI and CD81 receptor dependency and sensitivity to neutralizing antibodies. J Virol. 2008;82:12020–12029. doi: 10.1128/JVI.01569-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Du QS, Wang SQ, Chen D, Meng JZ, Huang RB. In depth analysis on the binding sites of adamantane derivatives in HCV (hepatitis C virus) p7 channel based on the NMR structure. PLoS One. 2014;9:e93613. doi: 10.1371/journal.pone.0093613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.OuYang B, Xie S, Berardi MJ, Zhao X, Dev J, Yu W, Sun B, Chou JJ. Unusual architecture of the p7 channel from hepatitis C virus. Nature. 2013;498:521–525. doi: 10.1038/nature12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lohmann V. Hepatitis C virus RNA replication. Curr Top Microbiol Immunol. 2013;369:167–198. doi: 10.1007/978-3-642-27340-7_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jones DM, Patel AH, Targett-Adams P, McLauchlan J. The hepatitis C virus NS4B protein can trans-complement viral RNA replication and modulates production of infectious virus. J Virol. 2009;83:2163–2177. doi: 10.1128/JVI.01885-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gawlik K, Baugh J, Chatterji U, Lim PJ, Bobardt MD, Gallay PA. HCV core residues critical for infectivity are also involved in core-NS5A complex formation. PLoS One. 2014;9:e88866. doi: 10.1371/journal.pone.0088866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pfeifer U, Thomssen R, Legler K, Böttcher U, Gerlich W, Weinmann E, Klinge O. Experimental non-A, non-B hepatitis: four types of cytoplasmic alteration in hepatocytes of infected chimpanzees. Virchows Arch B Cell Pathol Incl Mol Pathol. 1980;33:233–243. doi: 10.1007/BF02899184. [DOI] [PubMed] [Google Scholar]
- 47.Lohmann V, Hoffmann S, Herian U, Penin F, Bartenschlager R. Viral and cellular determinants of hepatitis C virus RNA replication in cell culture. J Virol. 2003;77:3007–3019. doi: 10.1128/JVI.77.5.3007-3019.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li YP, Ramirez S, Gottwein JM, Scheel TK, Mikkelsen L, Purcell RH, Bukh J. Robust full-length hepatitis C virus genotype 2a and 2b infectious cultures using mutations identified by a systematic approach applicable to patient strains. Proc Natl Acad Sci U S A. 2012;109:E1101–E1110. doi: 10.1073/pnas.1203829109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steinmann E, Penin F, Kallis S, Patel AH, Bartenschlager R, Pietschmann T. Hepatitis C virus p7 protein is crucial for assembly and release of infectious virions. PLoS Pathog. 2007;3:e103. doi: 10.1371/journal.ppat.0030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jones DM, McLauchlan J. Hepatitis C virus: assembly and release of virus particles. J Biol Chem. 2010;285:22733–22739. doi: 10.1074/jbc.R110.133017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quintavalle M, Sambucini S, Di Pietro C, De Francesco R, Neddermann P. The alpha isoform of protein kinase CKI is responsible for hepatitis C virus NS5A hyperphosphorylation. J Virol. 2006;80:11305–11312. doi: 10.1128/JVI.01465-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ding Q, Huang B, Lu J, Liu YJ, Zhong J. Hepatitis C virus NS3/4A protease blocks IL-28 production. Eur J Immunol. 2012;42:2374–2382. doi: 10.1002/eji.201242388. [DOI] [PubMed] [Google Scholar]
- 53.Li X, Jiang H, Qu L, Yao W, Cai H, Chen L, Peng T. Hepatocyte nuclear factor 4α and downstream secreted phospholipase A2 GXIIB regulate production of infectious hepatitis C virus. J Virol. 2014;88:612–627. doi: 10.1128/JVI.02068-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alisi A, Arciello M, Petrini S, Conti B, Missale G, Balsano C. Focal adhesion kinase (FAK) mediates the induction of pro-oncogenic and fibrogenic phenotypes in hepatitis C virus (HCV)-infected cells. PLoS One. 2012;7:e44147. doi: 10.1371/journal.pone.0044147. [DOI] [PMC free article] [PubMed] [Google Scholar]