

Abstract
Asparagine endopeptidase (AEP), also called legumain, is highly expressed in various solid tumors, promoting cancer cell invasion, migration, and metastasis. It has been proposed to be a prognostic marker and therapeutic target for cancer treatment. However, an effective nonpeptide, smallmolecule inhibitor against this protease has not yet been identified. Here we show that a family of xanthine derivatives selectively inhibit AEP and suppress matrix metalloproteinase (MMP) cleavage, leading to the inhibition of cancer metastasis. Through structure–activity relationship (SAR) analysis, we obtained an optimized lead compound (38u) that represses breast cancer invasion and migration. Chronic treatment of nude mice, which had been inoculated with MDA-MB-231 cells, with inhibitor 38u via oral administration robustly inhibits breast cancer lung metastasis in a dose-dependent manner, associated with blockade of MMP-2 by AEP. Therefore, our study supports that 38u might act as a potent and specific AEP inhibitor useful for cancer treatment.
Graphical abstract
INTRODUCTION
Cancer cell metastasis is a complex process that involves the tumor microenvironment, which is a large contributor to the increased invasiveness and migratory character of neoplastic cells.1 Malignant cells typically interact with a surrounding ecosystem of cells, including myeloid cells, fibroblasts, tumorassociated macrophages, and endothelial cells, to promote angiogenesis, degradation of the extracellular matrix, and cell motility.2 Throughout tumor progression, many extracellular proteases are known to contribute to the changes that occur in the tumor microenvironment and those most commonly associated with aberrant proliferation and metastasis are the matrix metalloproteinases (MMPs).3 The zinc-dependent endopeptidases are involved in a variety of cellular processes, including cell signaling, tissue remodeling, organ development, and inflammatory response.3,4 However, the capability of this enzyme family to degrade the extracellular matrix has implicated it in cancer cell invasion and metastasis.5 For many years, these enzymes were considered to be a promising cancer drug target and metalloproteinase inhibitors were found to be efficacious in animal models, but unfortunately, they proved to be unsuccessful in human clinical trials.6 The clinical failure of metalloproteinase inhibitors is believed to be due to the important roles of the enzyme family in such a wide range of biological processes and their ability to act as tumor suppressors under certain conditions.3
It has been demonstated that MMP-2 contributes migration and invasion in human breast cancer MDA-MB-231 and MDAMB- 435 cells.7-10 Therefore, additional research of MMPs has focused on their regulation in an effort to identify other potential drug target candidates. Asparagine endopeptidase (AEP, legumain) was found to activate MMP-2 by proteolytic removal of an N-terminal propeptide.11-13 AEP is a lysosomal cysteine endoprotease and is the only mammalian enzyme that cleaves C-terminally to asparagine residues.14,15 While only a limited quantity of AEP is detected in normal tissues, the enzyme is overexpressed on the cell surface and in cytoplasmic vesicles of solid tumors.11 The endoprotease activity of AEP has been associated with increased invasive and aggressive behavior of several cancers, including breast, prostate, colorectal, and gastric carcinomas.11,16-18 Thus, it is possible that AEP inhibitors may represent a more promising cancer therapeutic than the aforementioned metalloproteinase inhibitors.
The aberrant expression of AEP in cancer cells has made the enzyme a target for prodrug therapy.19 For instance, an inhibitor of α(v)β(3) integrin was conjugated to an asparaginecontaining peptide to ensure that the prodrug was selectively activated by AEP in acidic tumors and tumor microenvironments. 20 Additionally, the common cancer drug, doxorubicin, has been coupled to peptide-based inhibitors of AEP, and the conjugate was observed to selectively accumulate in murine breast tumor tissue, as opposed to kidney or liver, and reduce the size of the tumor.21 The demonstrated efficacy of AEPtargeted prodrugs validates the high degree of selectivity for tumor tissues that can be achieved by AEP-specific compounds, thereby decreasing potential side effects and toxicity. However, peptide-based compounds tend to be unfavorable drug candidates because of their limited stability and bioavailability. Thus, we have sought to identify small-molecule inhibitors that specifically target AEP and potently abrogate its activity. Herein, we describe the characterization of a lead compound, termed compound 38, from a high throughput screen; we explore its mechanism of inhibition and optimize the positive hit via SAR studies, which led to a second-generation inhibitor with enhanced druggable characteristics. We go on to provide evidence of its ability to preclude invasion and migration in vitro and impede metastasis in vivo, presumably through its inhibition of MMP-2 activation.
RESULTS
Compound 38 Inhibits AEP Activity
To identify smallmolecule inhibitors of AEP, we designed an AEP enzymatic activity assay for high-throughput screen using AEP-rich mouse kidney lysates as the enzyme source.22 Following confirmatory assays with purified AEP, we identified top hits and subsequent triaging produced a lead compound, termed compound 38. A kinetic analysis of the small-molecule inhibitor indicated that compound 38 competitively inhibits AEP activity strongly, KI = 105 ± 37 nM (Figure 1A,B). Substrate reversibility and DTT/l-cysteine reversibility assays further showed the competitive inhibitive feature of compound 38 toward AEP activity (Supporting Information, Figures 1 and 2). Time course inactivation assays produced nonlinear curves, indicating that the compound is a slow-binding inhibitor of AEP; the secondary plot of the rate constants against each inhibitor concentration demonstrates that AEP is inactivated very rapidly and potently, as the second-order rate constant, kinact/KI, for compound 38 is 8.9 × 105 min−1·M−1 (Figure 1C,D)
Figure 1.
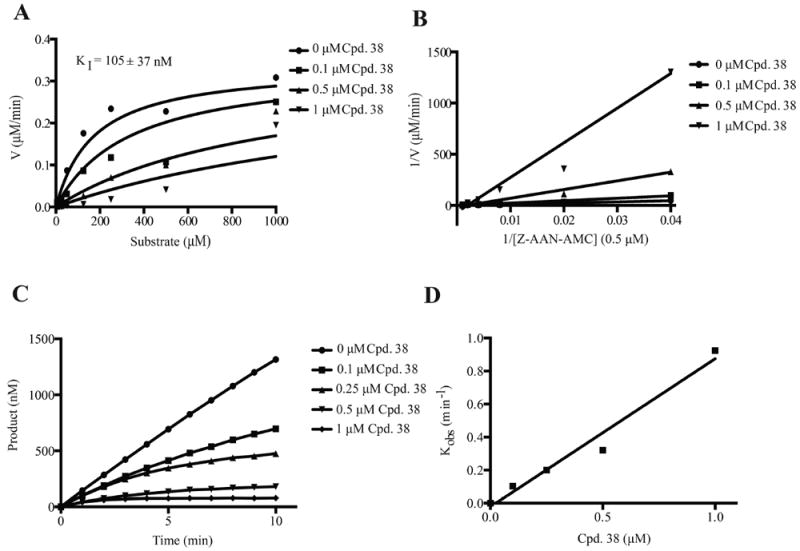
Kinetic analysis of compound 38 inhibition of AEP. (A) Michaelis–Menten plots displaying competitive inhibition of AEP. The inhibition constant, KI, was determined from the globally fit data. (B) Double reciprocal Lineweaver–Burk plots also displaying competitive inhibition. (C) Nonlinear progress curves obtained from the time course inactivation experiments, indicating that compound 38 is a slow-binding inhibitor of AEP. (D) A plot of the corrected kobs values obtained from each curve in (B) versus the concentration of compound 38, which enabled the calculation of the second-order rate constant, kinact/KI, to be determined to measure the potency of the inhibitor.
Figure 2.
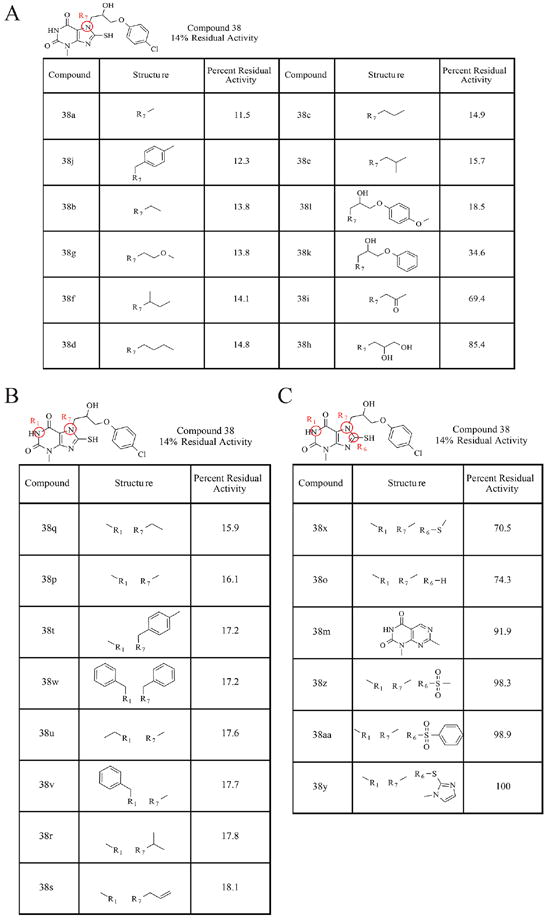
The SAR analysis of compound 38. (A) The structures and percentages of residual AEP activity of the compound 38 derivatives, in which substitutions were made to the nitrogen in the R7 position. (B) The structures and percentages of residual AEP activity of the compound 38 derivatives, in which substitutions were made to the nitrogens at the R7 and R1 positions. (C) The structures and percentages of residual AEP activity of the compound 38 derivatives, in which substitutions were made at the R7, R1, and R6 positions in the xanthine ring.
The xanthine-containing compound also exhibited favorable in vitro ADMET characteristics; it was found to be stable in liver microsomes in the presence or absence of NADPH and did not significantly inhibit any of the major cytochrome P450 isozymes.22 However, its Caco-2 cell permeability was minimal, suggesting that the compound may not be readily absorbed upon oral administration23 (Table 1).
Table 1.
Caco-2 Permeability
| Compound | A ->B Papp (10-6 cm•s-1) | B ->A Papp (10-6 cm•s-1) | RE |
|---|---|---|---|
| Ranitidine | 0.2 | 1.3 | 8.1 |
| Warfarin | 35.5 | 8.6 | 0.2 |
| Talinolol | 0.3 | 5.4 | 16.8 |
| 38 | 1.1 | 20.3 | 18.2 |
| 38D-16 | 0.9 | 3.0 | 3.3 |
| 38D-21 | 2.7 | 7.0 | 2.6 |
| 38D-22 | 10.0 | 30.5 | 3.0 |
|
| |||
| |||
Structure–Activity Relationship (SAR) Analysis of Compound 38
In an effort to improve the druggability of the compound, structure–activity relationship studies were performed with various compound 38 derivatives. One μM of each compound was incubated with 50 nM purified, active AEP and 5 μM substrate peptide, and the percentage of residual enzyme activity was calculated compared to a control reaction. Figure 2 displays the results of this analysis. Substitution at the R7 position alone with smaller hydrophobic alkane chains did not seem to affect the activity of the parent molecule. Interestingly, compound 38u, which contains an electron-donating methoxy group in place of the chloride of the parent compound, has similar reactivity as compound 38, suggesting that the electron-withdrawing −Cl is not critical for the effective inhibition of AEP. However, there is greater than a 2- fold loss in activity when nothing is placed in the para- position of the benzene ring, as in the case of compound 38k. There was also a 5–6-fold loss of activity of the parent compound, when a ketone or diol group was substituted at the R7 position, demonstrating that although some manipulations at this position can be tolerated while others cannot (Figure 2A). Nonetheless, the simultaneous addition of a methyl or benzyl group at the nitrogen at R1 and substitution of a small alkyl group or a bulky benzene-containing group at R7 did not seem to have any deleterious effects on the compound’s inhibitory activity (Figure 2B). Conversely, all of the observed alterations made to the thiol group of compound 38 had profoundly adverse effects on its inhibitory activity (Figure 2C), indicating that the thiol group is critical for its inhibitory activity. In addition, we also carried out the rapid elimination of swill (REOS) and pan assay interference compounds (PAINS) filter analysis. None of the compound has PAINS structure, and those compounds were identified as REOS due to the presence of the thiol group. Although xanthine and its derivatives has been reported for other activities, such as A2BAdoR (G-protein coupled receptors) antagonists24 and DPP (dipeptidyl peptidase)-4 inhibitor,25 no bioactivity of the compound has been reported except for 38o (caffeine). Altogether, these results demonstrate that the R1 and R7 positions can be manipulated to generate second-generation inhibitors of compound 38 with more favorable qualities and potentially enhanced potency.
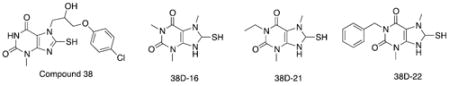
Compound 38u displays Potent Caco-2 Permeability and Inhibition Specificity
On the basis of the above SAR analysis and chemical availability, three of the compound 38 derivatives, 38p, 38u, and 38v, were chosen for further characterization. Addition of alkyl groups on the R1 position in the xanthine ring was believed to increase the hydrophobic character, leading to higher Caco-2 permeability. Moreover, these derivatives possess the inhibitory activities similar to that of the parent compound. As expected, Caco-2 permeability assays revealed that the derivatives displayed much higher permeability than the parent compound 38 (Table 1).26 Compound 38u exhibited the most favorable efflux:influx ratio of all of the derivatives tested; thus it might be the most bioavailable derivative.23 The inhibitory activity of the derivatives was also assessed in more depth to ensure that their inhibitory potential was indeed comparable to the parent compound; inhibition assays revealed that they possessed submicromolar IC50 values and that they inhibited AEP with about 2-fold increased potency compared to compound 38 (Figure 3). The derivatives also had similar, and in some cases improved, selectivity for AEP over other major cysteine proteases. Just like compound 38, compounds 38p, 38u, and 38v had IC50 values that were greater than 200 μM against cathepsin-S (Figure 3). Although compounds 38p and 38u were able to inhibit cathepsin-L, they inhibited AEP 500-fold and 1000-fold, respectively, more selectively. All of the compound 38 derivatives were less potent inhibitors of caspase-3 and caspase-8 and were therefore more specific for AEP as compared to the parent compound 38.
Figure 3.
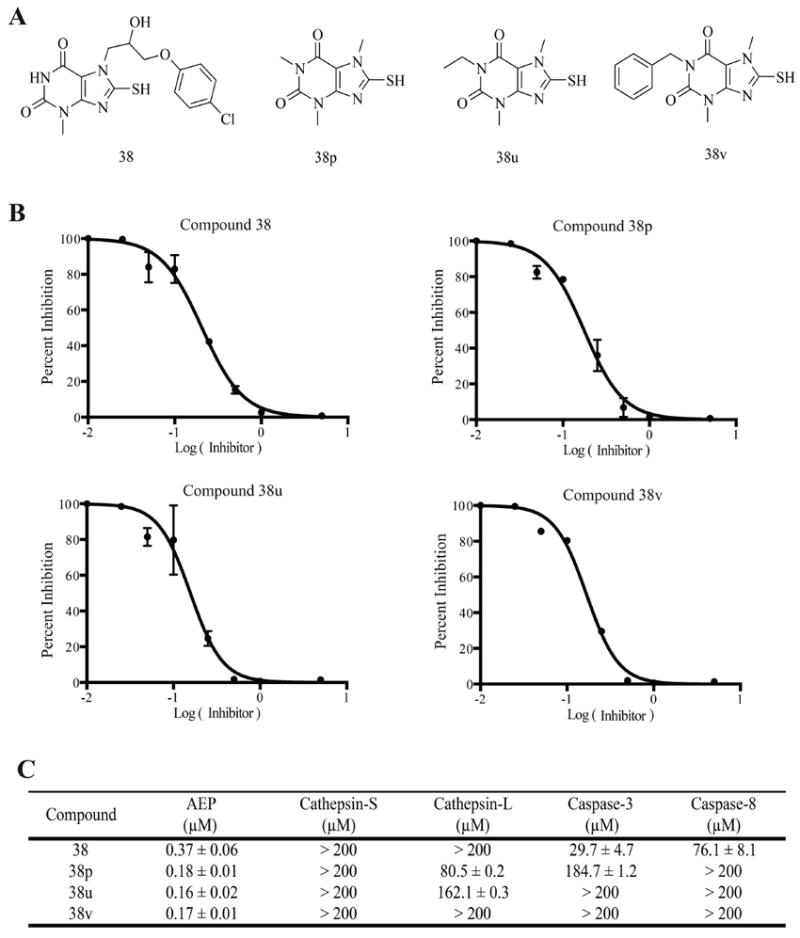
Compound 38u specifically inhibits AEP. (A) Structures of compound 38 and its derivatives with increased hydrophobicity. (B) IC50 curves for compound 38 and its derivatives against pure AEP. (C) Inhibition specificity assay. IC50 values of compound 38 and its derivatives against AEP and other related major cysteine proteases.
Compound 38u Inhibits Breast Cancer Cell Invasion in Vitro
Becaused compound 38u displayed favorable, druggable characteristics and was able to strongly inhibit AEP, its potential as an anticancer drug was assessed, initially in a cellular model. The moderately and highly metastatic human breast cancer cell lines, MDA-MB-231 and MDA-MB-435, respectively, were used to determine the efficacy of the compound 38 derivative toward the inhibition of cancer cell invasion and migration. Cell proliferation assays showed that compound 38u was not cytotoxic to either cell line (Figure 4A,B). For the invasion assays, breast cancer cells were seeded in matrigel-coated inserts, which were placed above wells containing fetal bovine serum as the chemoattractant, and various concentrations of the inhibitor was added to each insert. After the number of invading cells was counted, it was apparent that compound 38u was able to inhibit the invasion in a dosedependent manner. At 2 μM, 38u inhibited approximately 75% of cell invasion of both breast cancer cell types (Figure 4A,B). Similarly, 38u was able to inhibit the migration of about 25% of the less metastatic breast cancer cells, MDA-MB-231, whereas it inhibited about 40% of the more detrimental MDA-MB-435 cells from migrating in vitro (Figure 4A,B). AEP activities from cells treated by 38u were also suppressed in a dose-dependent manner (Figure 4C). To confirm the role of AEP in 38u induced inhibition of cell invasion, we carried out invasion assay with cells treated by AEP siRNA. As shown in Figure 4D, AEP knockdown decreased cell invasion significantly and abolished the inhibitive actions of 38u. Hence, AEP inhibitors block cancer cell invasion without impinging on the cell viability.
Figure 4.
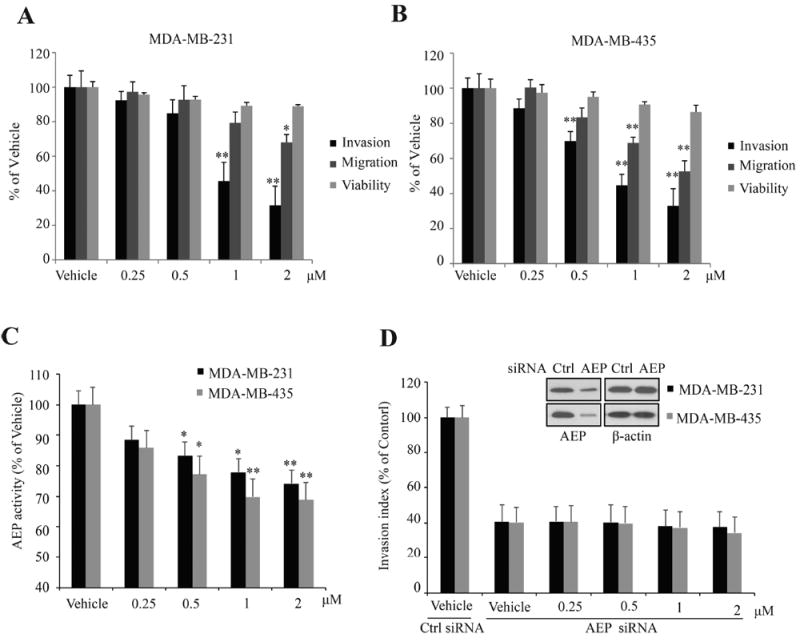
Compound 38u inhibits invasion in vitro. (A) Effect of compound 38u on cell proliferation, invasion, and migration of MDA-MB-231 cells. Cell proliferation was determined by MTT assay, following incubation with the compound. (B) Effect of compound 38u on cell proliferation, invasion, and migration of MDA-MB-435 cells. Cell proliferation was determined by MTT assay, following incubation with the compound. (C) Effect of compound 38u on AEP activity in MDA-MB-231 and MDA-MB-435 cells. AEP activity were examined with lysate of cells treated by 38u (D) Effect of compound 38u on invasion of MDA-MB-231 and MDA-MB-435 cells treated by control and AEP siRNA.
Compound 38u Inhibits Breast Cancer Metastasis in Vivo
Because compound 38u successfully attenuated the invasion and migration of MDA-MB-231 and MDA-MB-435 cells in vitro, the compound’s ability to inhibit the metastasis of breast tumor cells in vivo was subsequently examined. A murine model of breast cancer metastasis was developed by subcutaneously injecting MDA-MB-231 cells into the mammary fat pad of nude mice.27,28 Compound 38u was administered to the mice via oral gavage at doses of 3 or 10 mg/kg, or vehicle alone was given to the control group. After 42 days of treatment, the lungs were assessed for the presence of metastatic nodules; 3 mg/kg of compound 38u significantly decreased the number of nodules present on the mice, while the 10 mg/kg dosage almost completely prevented lung metastasis (Figure 5). There was no significant difference between the mammary tumor weight or volume between the control and drug-treated mice (data not shown). The percentage of mice with metastatic nodules on their lungs was 100% for the control mice and decreased to 33% for the mice that received the 3 mg/kg and 17% for the mice that received 10 mg/kg (Figure 5). Although treatment with compound 38u impeded breast cancer metastasis, no significant toxicity was observed, as demonstrated by the similarity in body weight between the control group and drug-treated mice (Figure 6B). Pathological examination of various major organs, such as kidney, liver, and bone marrow, did not exhibit any significant amount of toxicity (Figure 6A). Moreover, complete blood count (CBC) analysis revealed no significant difference between the drug- and vehicle-treated mice (Supporting Information, Table 1). Nonetheless, there was a slight increase in the size of the spleen in the drug-treated mice (Figure 6C), fitting with previous observations that knockout of AEP leads to the splenomegaly.29
Figure 5.
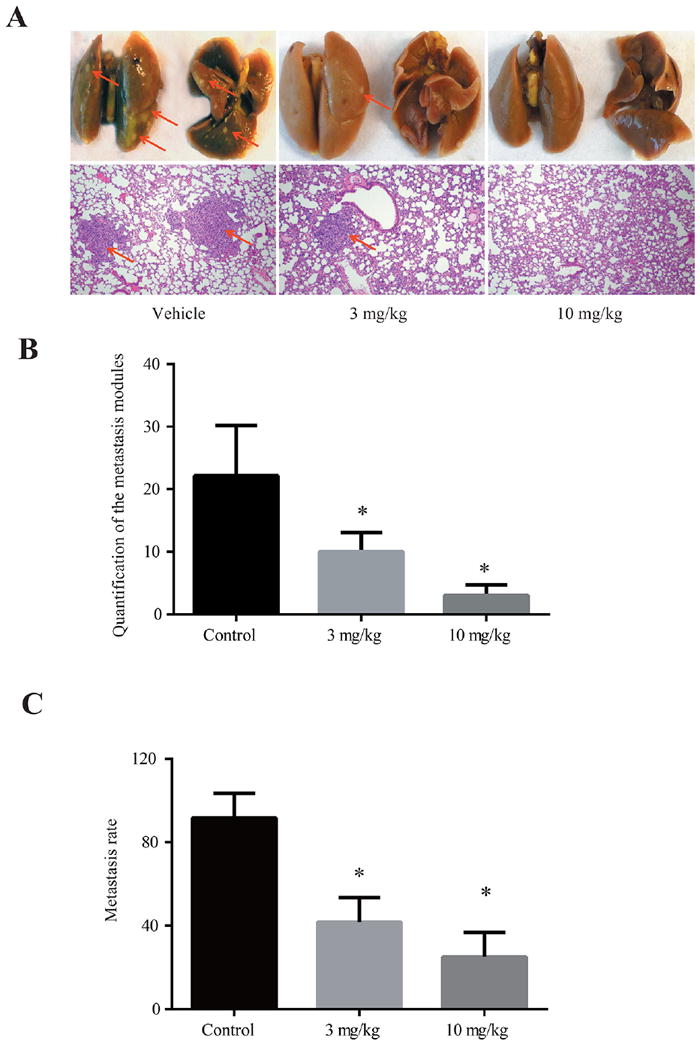
Compound 38u prevents breast-to-lung metastasis in vivo. (A) Representative mouse lungs of animals treated with vehicle control or 3 or 10 mg/kg compound 38u. Bottom panel depicts IHC staining. (B) Quantification of metastasized cancer nodules found on the treated and untreated mouse lungs demonstrates that significantly less nodules are present in the lungs of the drug-treated mice. (C) The proportion of mice exhibiting metastatic nodules in lungs decreased as the drug treatment dosage increased.
Figure 6.
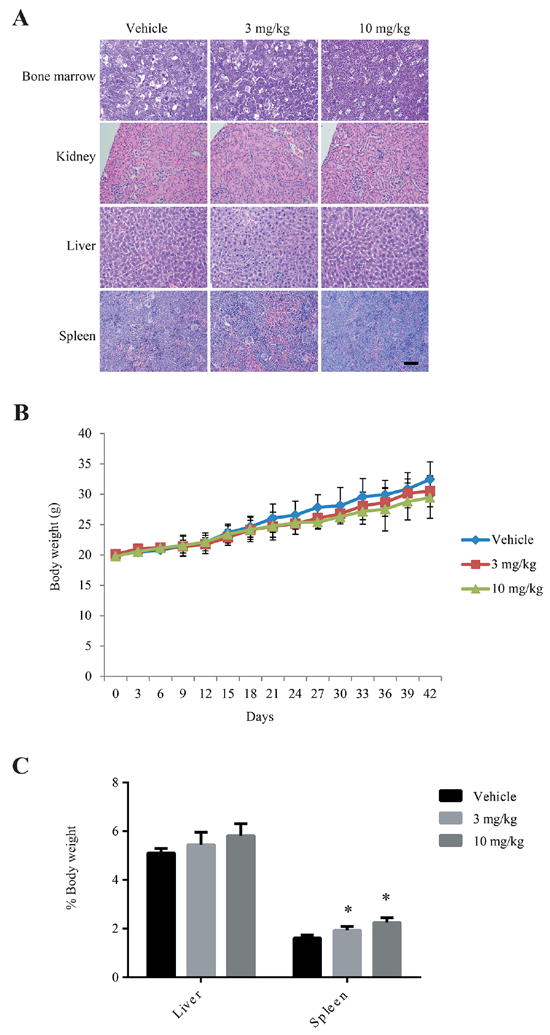
Compound 38u does not exhibit toxic effects in mice. (A) IHC sections of bone marrow, kidney, liver, and spleen from animals treated with vehicle control or 3 or 10 mg/kg of compound 38u show similar results in vehicle and drug-treated tissues. (B) There was no significant difference in body weight observed in mice treated with vehicle control or compound 38u. (C) The size of the liver remained similar in vehicle and drug-treated animals, however, the spleen was slightly enlarged in drug-treated animals as compared to control.
Compound 38u Inhibits Cleavage of MMP-2 in Vitro and in Vivo
Because the AEP inhibitor was able to prevent mammary tumor metastasis to the lungs of mice, we wondered whether this observation was due to the inhibition of AEPmediated cleavage of matrix metalloproteinase, MMP-2.12,13 The linkage between the overexpression of AEP and the increased invasive and metastatic potential of cancer cells, along with the observation that MMP-2 is a major substrate of AEP, suggests that AEP may exacerbate the migratory potential of tumor tissues through its cleavage of MMP-2. Therefore, we used gelatin zymography to assess whether the inhibition of AEP was concomitant with the inhibition of MMP-2 cleavage and mammary tumor metastasis. Initially, compound 38u was found to inhibit MMP-2 cleavage in the presence of purified active AEP in vitro. As the AEP inhibitor concentration gradually increased, MMP-2 cleavage progressively decreased (Figure 7A). Similarly, endogenous MMP-2 cleavage was inhibited in a dose-dependent manner in MDA-MD-231 breast cancer cells, however, the cell viability was not affected by the drug (Figure 7B,C). AEP activities from the cells treated by 38u were inhibited in a dose-dependent manner (Figure 7D). To rule out the off-target effect of 38u, MMP-2 cleavages in AEP knockdown cells were also examined. As shown in Figure 7E, following AEP knockdown, inhibition of MMP-2 conversion were abolished. Interestingly, the ratio of cleaved MMP-2 to full-length MMP-2 (pro-MMP-2) was observed to decrease in the mammary tumor tissue of mice treated with 3 or 10 mg/kg compound 38u (Figure 7F). Collectively, our data support the conclusion that compound 38u inhibits AEP, subsequently inhibiting MMP-2 from being cleaved by AEP, resulting in suppression of the metastasis of the mammary tumor cells.
Figure 7.
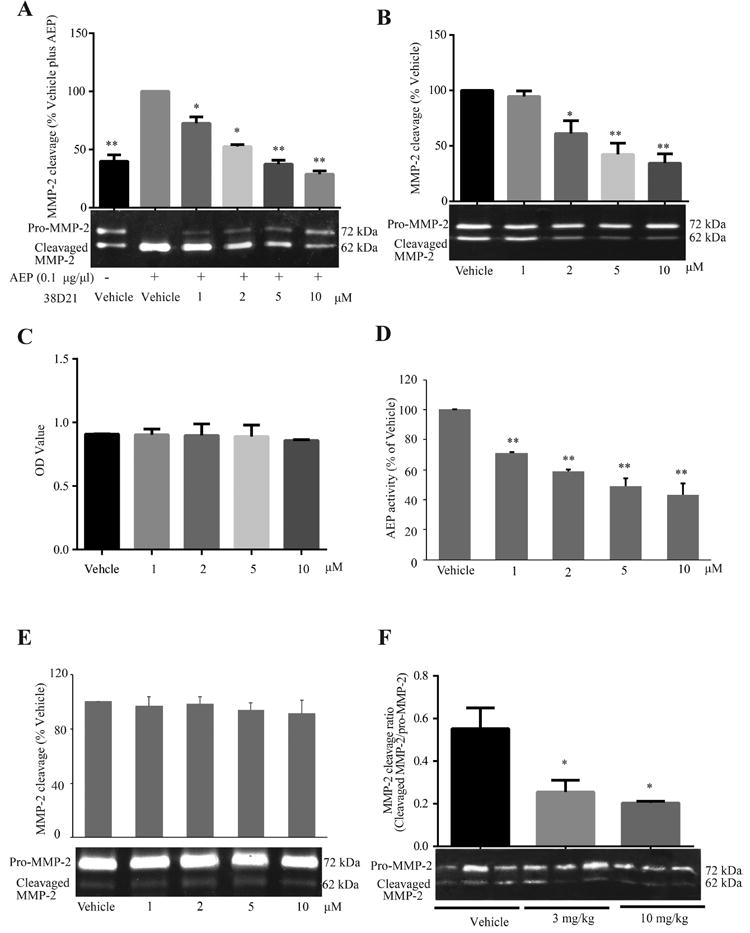
Compound 38u inhibits AEP cleavage of MMP-2 in vitro and in vivo. (A) Gelatin zymography was used to demonstrate that in the presence of AEP, MMP-2 is cleaved when only vehicle control is added to the reaction. In the presence of only 1 μM compound 38u, there is a significant decrease in the observed cleavage of MMP-2 and at the highest drug dose, 10 μM, MMP-2 cleavage returns to basal levels, as observed inthe absence of AEP. (B) Compound 38u was administered to MDA-MB-231 cells, and the inhibition of endogenous MMP-2 cleavage was observed in the presence of 1, 2, 5, and 10 μM compound. (C) The cell viability was not affected by the different doses of drug used in the experiment. (D) Effect of compound 38u on AEP activity in MDA-MB-231 cells. Cells were treated with indicated concentrations of 38u. AEP activity were examined with lysate of cells treated by 38u. (E) Effect of compound 38u on MMP-2 cleavage in MDA-MB-231 cells treated by AEP siRNA. Depletion of AEP abolished Pro-MMP-2 cleavage, and 38u lost its inhibitory effect on MMP-2 cleavage. (F) The ratio of cleaved MMP-2 to full-length MMP-2 (pro- MMP-2) significantly decreased in drug-treated mammary tumor tissue.
DISCUSSION AND CONCLUSION
The aberrant expression of AEP in cancer cells and on the surface of tumor-associated macrophages has been linked to the enzyme’s involvement in tumor development and metastasis.30 There is a plethora of evidence suggesting that AEP is a viable drug target and a biomarker for the diagnosis and progression of various cancers.16,21,31-33 Recent studies suggest that legumain expression could be a prognostic factor in patients with colorectal cancer, breast cancer, and ovarian cancer as well as a potential target for tumor therapy.34-36 Although peptidebased AEP-targeted prodrugs have been generated to specifically target common cancer drugs to cancer cells,37,38 no small molecular AEP inhibitors have been reported. Here, we have described the identification and optimization of an AEP-specific, small-molecule inhibitor, compound 38u, which has favorable ADME and toxicity characteristics. The inhibitor was also found to inhibit the migration and invasion of MDAMB- 231 and MDA-MB-435 breast cancer cells in vitro; however, it did not impede the proliferation of the cells. This is consistent with a previous report which showed that AEP does not affect the proliferation of SGC7901 human gastric cancer cells.17 Our observations suggest that the inhibition of AEP alone may not be sufficient to eradicate primary tumors. Nonetheless, the efficacious inhibition of mammary tumor metastasis exhibited by 38u suggests that AEP inhibitors would be successful in containing a primary tumor in its original environment and preventing the spread of tumorous tissues to more vital tissues. Thus, the AEP inhibitor may be utilized in conjunction with other known cancer therapeutics to eradicate the tumor at the primary site and simultaneously prevent the migration of the tumor cells to a secondary site. There have been reports of peptide-based AEP inactivators that, when conjugated to a nanoparticle and the anticancer compound, doxorubicin, are able to specifically target doxorubicin to cancer cells, mitigating any systemic toxicity.21 In this case, the AEP inhibitor is used as a targeting molecule to direct the cancer drug specifically to cancer cells by exploiting the fact that AEP is extracellularly expressed on tumors and in tumor microenvironments; interestingly, the inhibitor conjugated nanoparticle alone is not sufficient to significantly decrease the size of the primary tumor.20,21 However, the small size of compound 38u and its oral bioavailability would be advantageous in conjunction with other orally bioavailable anticancer agents, such as the breast cancer drug, lapatinib, which would decrease the discomfort of drug administration to cancer patients and concomitantly protect them from the formation of metastatic lesions.39
The in vivo mouse studies showed that the orally active AEP inhibitor does not cause any undesirable toxicity other than a slight enlargement of the spleen (Figure 6). Nonetheless, pathological examination of the spleen sections and CBC (complete blood chemistry) analysis did not reveal any demonstrable abnormality. Conceivably, the limited animal numbers (6 mice/group) in each dose of drug treatment group may somehow overstate the variation. However, we still cannot definitively exclude the possibility that 38u may incur some unknown minor side effects in the spleen. Finally, we also determined that the small molecular AEP inhibitor is likely imparting its effects by inhibiting the cleavage and activation of MMP-2 (Figure 7). The matrix metalloproteinase is a known substrate of AEP, which cleaves a propeptide from the Nterminus of MMP-2, thus enabling the enzyme to degrade the extracellular matrix and promote more aggressive and invasive tumor growth.40,41 It is reported that promoting the processing of pro-MMP-2 to MMP-2 by a artificial receptor for pro-MMP- 2 enhances metastatic ability of U87 cells.42 Chang et. al showed that epigallocatechingallate suppressed metastasis uveal melanoma cells and showed that secreted MMP-2 activity was dose-dependently inhibited by epigallocatechingallate, whereas the MMP-2 expression at protein and mRNA levels was not affected.43
Studies have shown that, similarly to AEP, there is an overexpression of MMPs in the majority of human cancers, which is associated with an increase in invasive and metastatic behavior and an overall poor prognosis because patients overexpressing these enzymes tend to have shorter survival rates.17 Additionally, in gastric cancer, the enhanced expression of MMP-2 has been most strongly correlated with a poor prognosis in comparison to any of the other MMPs.44 The inhibition of MMPs has not proven to be a successful strategy in cancer treatment because of the existence of multiple isozymes and their importance in various cellular processes.6 MMP-2 cleavage in MDA-MB-231 cells and mammary tissue was inhibited by compound 38u in a dose-dependent manner (Figure 7), suggesting that the AEP inhibitor successfully regulates the activity of MMP-2. It has been shown before that legumain could degrade fibronection, the main component of extracellular matrix.45 Conceivably, inhibition of AEP by its small inhibitors may potently block the breast-to-lung metastasis in mice. Therefore, we have described a novel approach to prevent breast tumor metastasis through the attenuation of MMP-2 activity by precluding its activation through the inhibition of AEP. These results suggest that it may be plausible to prevent cancer metastasis or even proliferation in different types of cancer, in which other AEP substrates are overexpressed.
EXPERIMENTAL SECTION
Reagents, Compounds, and Cells
Asparagine endopeptidase (AEP, legumain) was obtained from Sino Biological. Biocoat Matrigel Invasion Inserts were purchased from BD Biosciences. SiRNAs against AEP (sc-60930 and control scrambled siRNA (SC-37007) were from Santa Cruze ln. All the compounds were purchased and the purities (≥95%) were confirmed by the commercial vendors. HPLC and LC/MS were usually employed to determine the purity of the commercial compounds. Detailed information (company name, catalogue number) is listed below: Compounds 38a (BAS 00162656), 38b (BAS 00162658), 38d (BAS 00162664), 38e (BAS 00286887), 38j (BAS 00964091), and 38l (BAS 00458008) were from Asinex. Compounds 38 (R279900), 38c (S990175), 38f (S315095), 38h (L216518), and 38o (C0750) were purchased from Sigma. Compounds 38g (9007265), 38k (5647063), 38p (5216008), 38r (5994286), 38y (5839553), and 38z (7646613) were from ChemBridge. Compounds 38i (D090–0126), and 38m (C066–5678) were from ChemDiv. Compounds 38q (STK869195), 38s (STK868784), 38t (STK834902), 38w (STK834600), 38x (STK760770), and 38aa (STK357374) were from Vitas M Laboratories. Compounds 38u (F3260–0907) and 38v (F3260–0165) were from Life Chemicals. MDA-MB-231 and MDA-MB-435 cells were a gift from Dr. Lily Yang, Winship Cancer Institute, Emory University School of Medicine; the cells were cultured in DMEM (supplemented with 10% FBS, 2 mM L-glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin) and incubated at 37 °C with 5% CO2 in a humidified incubator.
IC50 Assays
Various concentrations of the appropriate compound were incubated with AEP reaction buffer (50 mM sodium citrate pH 5.5, 0.1% CHAPS, 60 mM Na2HPO4, 1 mM EDTA, final pH 6.0) and peptide substrate, 10 μM Cbz-Ala-Ala-Asn-AMC (Bachem). The reaction was initiated upon addition of 50 nM AEP, and fluorescent product formation was monitored over 15 min. The IC50 values were calculated from the following equation: Fractional Enzymatic Activity = 1/(1 + ([I]/IC50)), in which [I] = inhibitor concentration and IC50 = inhibitor concentration that yields half-maximal activity. Data were analyzed with GraFit version 5.0.11 software package.
Inhibition Kinetics Assays
To determine the inhibition constants and the mechanism by which compound 38 inhibits AEP, the steadystate kinetic parameters for the hydrolysis of the peptide substrate, ZAAN- AMC, were determined in the absence or presence of increasing concentrations of inhibitor. In these assays, specified concentrations of the inhibitor were preincubated with substrate for 10 min at 37 °C, then 50 nM AEP was added to initiate the reaction, which was quenched after 10 min. The RFU values of the reaction product were converted to micromolar values with an AMC standard curve, and the final reaction rates were plotted against substrate concentration and globally fit to equations representative of competitive inhibition (eq 1), noncompetitive inhibition (eq 2), mixed inhibition (eq 3), and uncompetitive inhibition (eq 4) using a nonlinear least fit squares approach by GraFit version 5.0.11.
| (1) |
| (2) |
| (3) |
| (4) |
In the equations, Kii is the intercept Ki, and Kis is the slope Ki. The mode of the inhibition induced by the compounds on AEP was determined by the best fit of the data to eqs 1-4. Visual inspection of the fits, and a comparison of the standard errors, was used to confirm these assignments.
Time Course Inactivation Assays
Progress curves were generated by incubating 5 μM Z-AAN-AMC and the specified concentration of inhibitor in assay buffer at 37 °C for 10 min. The reaction was initiated by the addition of 50 nM AEP and quenched after 10 min. The concentration of the product was determined from an AMC standard curve, and the data was fit by nonlinear regression. Because the curves were nonlinear, they were fit to eq 5, using the GraFit version 5.0.11 software package,
| (5) |
where vi is the initial velocity, kobs·app is the apparent pseudo-first-order rate constant for inactivation, and t is time. Equation 6,
| (6) |
was used to correct the apparent pseudo-first-order inactivation rate constants, obtained from this analysis, for substrate concentration and the pseudo-first-order inactivation rate constants, i.e., kobs, thus obtained, were plotted against the tested inhibitor concentrations. As the data are consistent with a two-step mechanism of inactivation, they were fit to eq 7,
| (7) |
using the GraFit version 5.0.11 software, where KI is the concentration of inactivator that yields half-maximal inactivation, kinact is the maximal rate of inactivation, and [I] is the concentration of inactivator.
Caco-2 Permeability Assay
CaCo-2 cells grown in tissue culture flasks are trypsinized, suspended in medium, and the suspensions applied to wells of a Millipore 96-well Caco-2 plate. The cells were allowed to grow and differentiate for 3 weeks, feeding at 2-day intervals. For apical to basolateral (A → B) permeability, the test agent was added to the apical (A) side and amount of permeation was determined on the basolateral (B) side; for basolateral to apical (B > A) permeability, the test agent was added to the B side and the amount of permeation was determine on the A side. The A-side buffer contains 100 μM Lucifer yellow dye, in transport buffer (1.98 g/L glucose in 10 mM HEPES, 1× Hank’s Balanced Salt Solution), pH 6.5, and the B-side buffer is transport buffer, pH 7.4. CaCo-2 cells were incubated with these buffers for 2 h, and the receiver side buffer was removed for analysis by LC/MS/MS. Cells were incubated with 10 μM of each compound in buffer for 2 h, and the receiver side buffer was removed for analysis by LC/MS/MS. Efflux ratio RE > 2 indicates a significant efflux activity and indication of potential substrate for PGP or other active transporters.
Cell Viability Assay
Cells were seeded and cultured in 96-well plates (4000 cells/well). The next day, the medium was replaced with fresh medium containing different concentrations of the drugs or vehicle controls. The cells were then incubated at 37 °C for the indicated times. After treatment, the cells were incubated for another 4 h with 0.5 mg/mL MTT solution at 37 °C. The culture medium was discarded, and 0.1 mL DMSO was used to dissolve precipitate. The absorbance was measured at 570 nm using an automated microplated reader (Synergy 2, BioTek, VT, USA).
Chamber Invasion and Migration Assay
Invasion of cells through Matrigel was determined using a Transwell system (10 mm diameter, 8 μm pore size with polycarbonate membrane; Corning Costar). Briefly, cells (3 × 104) were suspended in serum free medium with different concentrations of drug and seeded onto Matrigel-coated transwell chamber. Medium with 5% serum was used as a chemoattractant in the lower chambers. After desired times of incubation at 37 °C under 5% CO2/95% air atmosphere, medium was aspirated, and cells on the upper side of the membrane were removed with a cotton swab. The invading cells on the bottom of the filter were stained with 0.5% crystal violet in 25% methanol and quantified under invert microscope. In the transwell chamber migration assay, the BD Falcon Cell Culture Insert System containing membranes with 8 μm pore size was utilized in the assay; l.5 × 104 cells were suspended in serum free medium with different concentrations of drug and seeded onto 10 mm upper chamber of transwell system. Medium with 5% serum and same concentrations of drugs in corresponding upper chambers was added to the lower chamber. After a fixed time of incubation, the migration cells were stained and quantified as in the invasion assay.
In Vivo Spontaneous Metastasis Assay
MDA-MB-231 cells were trypsinized and resuspended in serum free media at a density of 1 × 107/mL and implanted into the mammary fat pad of the mice (200 μL/mouse). When the volumes of xenografts reached to 100 mm3, mice were randomized to receive vehicle control (12 mice per group) or tested compounds (six mice per group). Test compound or vehicle was then administered orally for 42 consecutive days. Tumor volume in mm3 was determined using the formula (length × width2)/2, where length was the longest axis and width being the measurement at right angles to the length. Then 24 h after the last drug administration, the animals were sacrificed and tumors/orgns were collected for various experiments. Animals were maintained in a pathogen-freee environment, and procedures were operated in accordace with Emory Institutional Animal Care and Use Committee guidelines. For lung metastasis, after the mice were killed, the lungs were removed, washed, and fixed with Bouin’s solution for 24 h and the number of the tumor nodules on the whole surface of the lungs was counted under a dissecting microscope. Sections of the lungs were stained with hematoxylin and eosin (HE) to confirm the formation of metastases.
Gelatin Zymography Assay
Gelatinase-containing samples were dissolved in Laemmli sample buffer in the absence of reducing agents and electrophoresed in 8% polyacrylamide SDS gels copolymerized with gelatin (1 mg/mL). Following electrophoresis, the gels were washed twice (30 min each time) and once in calcium assay buffer (40 mM Tris, 0.2 M NaCl, 10 mM CaCl2, pH 7.5) and then incubated in the calcium assay buffer at 37 °C for certain times with gently shaking. Gels were then fixed in 45% methanol/10% glacial acidic acid containing 0.5% Commassie Blue G-250 for 1 h, followed by destaining with 10% acetic acid, 10% methanol. Enzyme-digested regions were observed as white bands against a blue background. Zones of enzymatic activity were seen as negatively stained bands.
Statistical Analysis
For statistical analysis, data shown in the study were obtained in at least two independent experiments performed in a parallel manner unless otherwise indicated, which are presented as mean ± SD. Statistical evaluation was carried out by Student’s t test or one-way ANOVA. Significance of difference was indicated as *P < 0.05 and **P < 0.01. The statistical analysis was performed by program Prism (GraphPad Software, La Jolla, CA).
Supplementary Material
Acknowledgments
This work is supported by a grant from National Institute of Health (RO1, CA186918) to K.Y. H.F. is a GRA/Georgia Cancer Coalition Distinguished Cancer Scholar. The high throughput screening was carried out at the Emory Chemical Biology Discovery Center.
ABBREVIATIONS USED
- AEP
asparagine endopeptidase
- MMP
matrix metalloproteinases
- SAR
structure–activity relationship
- REOS
rapid elimination of swill
- PAINS
pan assay interference compounds
- DPP
dipeptidyl peptidase
- ADMET
absorption, distribution, metabolism, excretion, and toxicity
- CBC
complete blood chemistry
Footnotes
Supporting Information
The Supporting Information, is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem. 7b00228.
Complete blood count (CBC) of vehicle and compound 38u treated nude mice; substrate reversibility of compound 38 toward AEP; DTT and l-cysteine reversiblitiy of compound 38 toward AEP (PDF) (Molecular formula stringsCSV)
Notes
The authors declare no competing financial interest.
References
- 1.Coghlin C, Murray GI. Current and emerging concepts in tumour metastasis. J Pathol. 2010;222:1–15. doi: 10.1002/path.2727. [DOI] [PubMed] [Google Scholar]
- 2.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Husemann Y, Geigl JB, Schubert F, Musiani P, Meyer M, Burghart E, Forni G, Eils R, Fehm T, Riethmuller G, Klein CA. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13:58–68. doi: 10.1016/j.ccr.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 5.Liotta LA, Tryggvason K, Garbisa S, Hart I, Foltz CM, Shafie S. Metastatic potential correlates with enzymatic degradation of basement membrane collagen. Nature. 1980;284:67–68. doi: 10.1038/284067a0. [DOI] [PubMed] [Google Scholar]
- 6.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 7.Ukaji T, Lin Y, Okada S, Umezawa K. Inhibition of MMP-2- mediated cellular invasion by NF-kappaB inhibitor DHMEQ in 3D culture of breast carcinoma MDA-MB-231 cells: A model for early phase of metastasis. Biochem Biophys Res Commun. 2017;485:76–81. doi: 10.1016/j.bbrc.2017.02.022. [DOI] [PubMed] [Google Scholar]
- 8.Kim D, Rhee S. Matrix metalloproteinase 2 regulates MDAMB231 breast cancer cell invasion induced by active mammalian diaphanous-related formin 1. Mol Med Rep. 2016;14:277–282. doi: 10.3892/mmr.2016.5282. [DOI] [PubMed] [Google Scholar]
- 9.Qi Q, Gu H, Yang Y, Lu N, Zhao J, Liu W, Ling H, You QD, Wang X, Guo Q. Involvement of matrix metalloproteinase 2 and 9 in gambogic acid induced suppression of MDA-MB-435 human breast carcinoma cell lung metastasis. J Mol Med (Heidelberg, Ger) 2008;86:1367–1377. doi: 10.1007/s00109-008-0398-z. [DOI] [PubMed] [Google Scholar]
- 10.Munoz-Najar UM, Neurath KM, Vumbaca F, Claffey KP. Hypoxia stimulates breast carcinoma cell invasion through MT1- MMP and MMP-2 activation. Oncogene. 2006;25:2379–2392. doi: 10.1038/sj.onc.1209273. [DOI] [PubMed] [Google Scholar]
- 11.Gawenda J, Traub F, Luck HJ, Kreipe H, von Wasielewski R. Legumain expression as a prognostic factor in breast cancer patients. Breast Cancer Res Treat. 2007;102:1–6. doi: 10.1007/s10549-006-9311-z. [DOI] [PubMed] [Google Scholar]
- 12.Chen JM, Fortunato M, Stevens RA, Barrett AJ. Activation of progelatinase A by mammalian legumain, a recently discovered cysteine proteinase. Biol Chem. 2001;382:777–783. doi: 10.1515/BC.2001.093. [DOI] [PubMed] [Google Scholar]
- 13.Liu C, Sun C, Huang H, Janda K, Edgington T. Overexpression of legumain in tumors is significant for invasion/metastasis and a candidate enzymatic target for prodrug therapy. Cancer Res. 2003;63:2957–2964. [PubMed] [Google Scholar]
- 14.Chen JM, Dando PM, Rawlings ND, Brown MA, Young NE, Stevens RA, Hewitt E, Watts C, Barrett AJ. Cloning, isolation, and characterization of mammalian legumain, an asparaginyl endopeptidase. J Biol Chem. 1997;272:8090–8098. doi: 10.1074/jbc.272.12.8090. [DOI] [PubMed] [Google Scholar]
- 15.Chen JM, Rawlings ND, Stevens RA, Barrett AJ. Identification of the active site of legumain links it to caspases, clostripain and gingipains in a new clan of cysteine endopeptidases. FEBS Lett. 1998;441:361–365. doi: 10.1016/s0014-5793(98)01574-9. [DOI] [PubMed] [Google Scholar]
- 16.Ohno Y, Nakashima J, Izumi M, Ohori M, Hashimoto T, Tachibana M. Association of legumain expression pattern with prostate cancer invasiveness and aggressiveness. World J Urol. 2013;31:359–364. doi: 10.1007/s00345-012-0977-z. [DOI] [PubMed] [Google Scholar]
- 17.Li N, Liu Q, Su Q, Wei C, Lan B, Wang J, Bao G, Yan F, Yu Y, Peng B, Qiu J, Yan X, Zhang S, Guo F. Effects of legumain as a potential prognostic factor on gastric cancers. Med Oncol. 2013;30:621. doi: 10.1007/s12032-013-0621-9. [DOI] [PubMed] [Google Scholar]
- 18.Haugen MH, Johansen HT, Pettersen SJ, Solberg R, Brix K, Flatmark K, Maelandsmo GM. Nuclear legumain activity in colorectal cancer. PLoS One. 2013;8:e52980. doi: 10.1371/journal.pone.0052980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stern L, Perry R, Ofek P, Many A, Shabat D, Satchi-Fainaro R. A novel antitumor prodrug platform designed to be cleaved by the endoprotease legumain. Bioconjugate Chem. 2009;20:500–510. doi: 10.1021/bc800448u. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, Bajjuri KM, Liu C, Sinha SC. Targeting cell surface alpha(v)beta(3) integrin increases therapeutic efficacies of a legumain protease-activated auristatin prodrug. Mol Pharmaceutics. 2012;9:168–175. doi: 10.1021/mp200434n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liao D, Liu Z, Wrasidlo W, Chen T, Luo Y, Xiang R, Reisfeld RA. Synthetic enzyme inhibitor: a novel targeting ligand for nanotherapeutic drug delivery inhibiting tumor growth without systemic toxicity. Nanomedicine. 2011;7:665–673. doi: 10.1016/j.nano.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Z, Obianyo O, Dall E, Du Y, Fu H, Liu X, Kang SS, Song M, Yu SP, Cabrele C, Schubert M, Li X, Wang JZ, Brandstetter H, Ye K. Inhibition of delta-secretase improves cognitive functions in mouse models of Alzheimer’s disease. Nat Commun. 2017;8:14740. doi: 10.1038/ncomms14740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas VH, Bhattachar S, Hitchingham L, Zocharski P, Naath M, Surendran N, Stoner CL, El-Kattan A. The road map to oral bioavailability: an industrial perspective. Expert Opin Drug Metab Toxicol. 2006;2:591–608. doi: 10.1517/17425255.2.4.591. [DOI] [PubMed] [Google Scholar]
- 24.Basu S, Barawkar DA, Ramdas V, Patel M, Waman Y, Panmand A, Kumar S, Thorat S, Naykodi M, Goswami A, Reddy BS, Prasad V, Chaturvedi S, Quraishi A, Menon S, Paliwal S, Kulkarni A, Karande V, Ghosh I, Mustafa S, De S, Jain V, Banerjee ER, Rouduri SR, Palle VP, Chugh A, Mookhtiar KA. Design and synthesis of novel xanthine derivatives as potent and selective A2B adenosine receptor antagonists for the treatment of chronic inflammatory airway diseases. Eur J Med Chem. 2017;134:218–229. doi: 10.1016/j.ejmech.2017.04.014. [DOI] [PubMed] [Google Scholar]
- 25.Huang Y, He X, Wu T, Zhang F. Synthesis and characterization of process-related impurities of antidiabetic drug linagliptin. Molecules. 2016;21:1041. doi: 10.3390/molecules21081041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 27.Huang Y, He Y, Ye S, Li X, Zhong Q, Chen Z, Jin X. Combined use of cyclophosphamide and Chalone 19-peptide in experimental breast cancer. OncoTargets Ther. 2013;6:861–867. doi: 10.2147/OTT.S44564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang Y, Chen Y, Yu L, Zheng C, Qi Y, Li Z, Yang Z, Zhang Y, Shi T, Luo J, Liu M. Inhibition of breast cancer metastases by a novel inhibitor of TGFbeta receptor 1. J Natl Cancer Inst. 2013;105:47–58. doi: 10.1093/jnci/djs485. [DOI] [PubMed] [Google Scholar]
- 29.Chan CB, Abe M, Hashimoto N, Hao C, Williams IR, Liu X, Nakao S, Yamamoto A, Zheng C, Henter JI, Meeths M, Nordenskjold M, Li SY, Hara-Nishimura I, Asano M, Ye K. Mice lacking asparaginyl endopeptidase develop disorders resembling hemophagocytic syndrome. Proc Natl Acad Sci U S A. 2009;106:468–473. doi: 10.1073/pnas.0809824105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin Y, Wei C, Liu Y, Qiu Y, Liu C, Guo F. Selective ablation of tumor-associated macrophages suppresses metastasis and angiogenesis. Cancer Sci. 2013;104:1217–1225. doi: 10.1111/cas.12202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reisfeld RA. The tumor microenvironment: a target for combination therapy of breast cancer. Crit Rev Oncog. 2013;18:115–133. doi: 10.1615/critrevoncog.v18.i1-2.70. [DOI] [PubMed] [Google Scholar]
- 32.Wang L, Chen S, Zhang M, Li N, Chen Y, Su W, Liu Y, Lu D, Li S, Yang Y, Li Z, Stupack D, Qu P, Hu H, Xiang R. Legumain: a biomarker for diagnosis and prognosis of human ovarian cancer. J Cell Biochem. 2012;113:2679–2686. doi: 10.1002/jcb.24143. [DOI] [PubMed] [Google Scholar]
- 33.Edgington LE, Verdoes M, Ortega A, Withana NP, Lee J, Syed S, Bachmann MH, Blum G, Bogyo M. Functional imaging of legumain in cancer using a new quenched activity-based probe. J Am Chem Soc. 2013;135:174–182. doi: 10.1021/ja307083b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murthy RV, Arbman G, Gao J, Roodman GD, Sun XF. Legumain expression in relation to clinicopathologic and biological variables in colorectal cancer. Clin Cancer Res. 2005;11:2293–2299. doi: 10.1158/1078-0432.CCR-04-1642. [DOI] [PubMed] [Google Scholar]
- 35.Wu W, Luo Y, Sun C, Liu Y, Kuo P, Varga J, Xiang R, Reisfeld R, Janda KD, Edgington TS, Liu C. Targeting cellimpermeable prodrug activation to tumor microenvironment eradicates multiple drug-resistant neoplasms. Cancer Res. 2006;66:970–980. doi: 10.1158/0008-5472.CAN-05-2591. [DOI] [PubMed] [Google Scholar]
- 36.Luo Y, Zhou H, Krueger J, Kaplan C, Lee SH, Dolman C, Markowitz D, Wu W, Liu C, Reisfeld RA, Xiang R. Targeting tumor-associated macrophages as a novel strategy against breast cancer. J Clin Invest. 2006;116:2132–2141. doi: 10.1172/JCI27648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ovat A, Muindi F, Fagan C, Brouner M, Hansell E, Dvorak J, Sojka D, Kopacek P, McKerrow JH, Caffrey CR, Powers JC. Aza-peptidyl Michael acceptor and epoxide inhibitors–potent and selective inhibitors of Schistosoma mansoni and Ixodes ricinus legumains (asparaginyl endopeptidases) J Med Chem. 2009;52:7192–7210. doi: 10.1021/jm900849h. [DOI] [PubMed] [Google Scholar]
- 38.Loak K, Li DN, Manoury B, Billson J, Morton F, Hewitt E, Watts C. Novel cell-permeable acyloxymethylketone inhibitors of asparaginyl endopeptidase. Biol Chem. 2003;384:1239–1246. doi: 10.1515/BC.2003.136. [DOI] [PubMed] [Google Scholar]
- 39.Guan Z, Xu B, DeSilvio ML, Shen Z, Arpornwirat W, Tong Z, Lorvidhaya V, Jiang Z, Yang J, Makhson A, Leung WL, Russo MW, Newstat B, Wang L, Chen G, Oliva C, Gomez H. Randomized trial of lapatinib versus placebo added to paclitaxel in the treatment of human epidermal growth factor receptor 2- overexpressing metastatic breast cancer. J Clin Oncol. 2013;31:1947–1953. doi: 10.1200/JCO.2011.40.5241. [DOI] [PubMed] [Google Scholar]
- 40.Chen WT. Proteases associated with invadopodia, and their role in degradation of extracellular matrix. Enzyme Protein. 1996;49:59–71. doi: 10.1159/000468616. [DOI] [PubMed] [Google Scholar]
- 41.Corcoran ML, Hewitt RE, Kleiner DE, Jr, Stetler-Stevenson WG. MMP-2: expression, activation and inhibition. Enzyme Protein. 1996;49:7–19. doi: 10.1159/000468613. [DOI] [PubMed] [Google Scholar]
- 42.Nishida Y, Miyamori H, Thompson EW, Takino T, Endo Y, Sato H. Activation of matrix metalloproteinase-2 (MMP-2) by membrane type 1 matrix metalloproteinase through an artificial receptor for proMMP-2 generates active MMP-2. Cancer Res. 2008;68:9096–9104. doi: 10.1158/0008-5472.CAN-08-2522. [DOI] [PubMed] [Google Scholar]
- 43.Chang CW, Hsieh YH, Yang WE, Yang SF, Chen Y, Hu DN. Epigallocatechingallate inhibits migration of human uveal melanoma cells via downregulation of matrix metalloproteinase-2 activity and ERK1/2 pathway. Biomed Res Int. 2014;2014:141582. doi: 10.1155/2014/141582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kubben FJ, Sier CF, van Duijn W, Griffioen G, Hanemaaijer R, van de Velde CJ, van Krieken JH, Lamers CB, Verspaget HW. Matrix metalloproteinase-2 is a consistent prognostic factor in gastric cancer. Br J Cancer. 2006;94:1035–1040. doi: 10.1038/sj.bjc.6603041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morita Y, Araki H, Sugimoto T, Takeuchi K, Yamane T, Maeda T, Yamamoto Y, Nishi K, Asano M, Shirahama-Noda K, Nishimura M, Uzu T, Hara-Nishimura I, Koya D, Kashiwagi A, Ohkubo I. Legumain/asparaginyl endopeptidase controls extracellular matrix remodeling through the degradation of fibronectin in mouse renal proximal tubular cells. FEBS Lett. 2007;581:1417–1424. doi: 10.1016/j.febslet.2007.02.064. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.