

Abstract
The elucidation of Δ9-tetrahydrocannabinol as the active principal of Cannabis sativa in 1963 initiated a fruitful half-century of scientific discovery, culminating in the identification of the endocannabinoid signaling system, a previously unknown neuromodulatory system. A primary function of the endocannabinoid signaling system is to maintain or recover homeostasis following psychological and physiological threats. We provide a brief introduction to the endocannabinoid signaling system and its role in synaptic plasticity. The majority of the article is devoted to a summary of current knowledge regarding the role of endocannabinoid signaling as both a regulator of endocrine responses to stress and as an effector of glucocorticoid and corticotrophin-releasing hormone signaling in the brain. We summarize data demonstrating that cannabinoid receptor 1 (CB1R) signaling can both inhibit and potentiate the activation of the hypothalamic-pituitary-adrenal axis by stress. We present a hypothesis that the inhibitory arm has high endocannabinoid tone and also serves to enhance recovery to baseline following stress, while the potentiating arm is not tonically active but can be activated by exogenous agonists. We discuss recent findings that corticotropin-releasing hormone in the amygdala enables hypothalamic-pituitary-adrenal axis activation via an increase in the catabolism of the endocannabinoid N-arachidonylethanolamine. We review data supporting the hypotheses that CB1R activation is required for many glucocorticoid effects, particularly feedback inhibition of hypothalamic-pituitary-adrenal axis activation, and that glucocorticoids mobilize the endocannabinoid 2-arachidonoylglycerol. These features of endocannabinoid signaling make it a tantalizing therapeutic target for treatment of stress-related disorders but to date, this promise is largely unrealized.
Introduction to the Components and Paradigms of CNS Endocannabinoid Signaling
Cannabis sativa has been cultivated and used for thousands of years for medicinal and, more recently, recreational purposes (159). Among other things, preparations of C. sativa are often used to reduce the physiological and psychological consequences of stress and fear (13, 36, 67). Cannabis users increase consumption during times of increased stress (76) and social anxiety triggers craving for cannabis in some users (12).
There are several biologically active chemicals in C. sativa; the most well studied is Δ9-tetrahydrocannabinol (THC). There is considerable evidence that THC is the constituent of C. sativa primarily responsible for its characteristic psychoactive and physiological effects (117). THC affects the body and brain because it is a high affinity, partial agonist of two G-protein coupled receptors (GPCRs), the 1 and 2 subtypes of cannabinoid receptors (CB1R and CB2R, respectively). CB1Rs are expressed by neurons throughout the body, including the brain and spinal cord, and autonomic and enteric nervous systems (91). CB2Rs are primarily expressed by circulating immune cells, such as B cells, macrophages, and T cells as well as by tissue-resident immune cells, including brain microglia (95). There is some evidence that CB2Rs are expressed by neurons (146), and CB1Rs are expressed by circulating immune cells (7); however, in both cases, the densities are far lower that seen in the cells of primary expression.
The endocannabinoids are lipids “made on demand” and regulated by catabolism as well as synthesis
Two endogenous lipids bind to the cannabinoid receptors: 2-arachidonoylglycerol (2-AG) and N-arachidonylethanolamine (AEA) (Fig. 1) (60). AEA and 2-AG have been named endocannabinoids (eCBs); both can function as endogenous agonists at CB1Rs, with 2-AG exhibiting greater efficacy, but lower potency than AEA (59). 2-AG is a full agonist at CB2Rs; AEA also binds with reasonable affinity to these receptors, but has very low efficacy (61). AEA is an agonist of the vanilloid subtype of the transient potential receptor family (140) and, together with other N-acylethanolamines (NAEs), activates several subtypes of the peroxisome proliferator activated receptor family (118).
Figure 1.
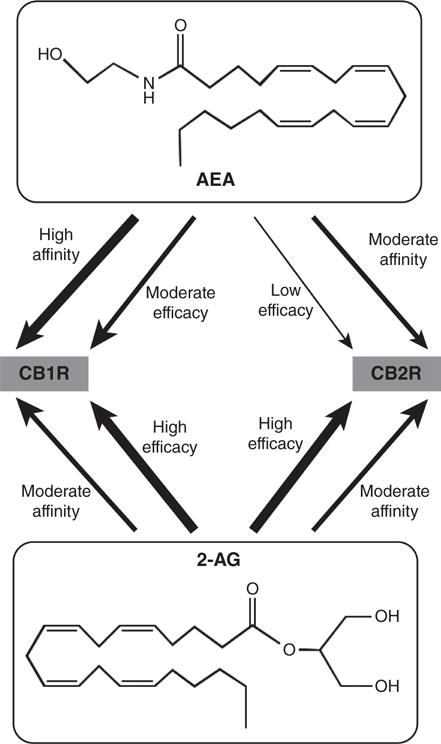
Two eCBs act at CB1 and CB2 receptors with differing affinities and efficacies. AEA, N-arachidonylethanolamine; CB1R, subtype 1 of the cannabinoid receptor; CB2R, subtype 2 of the cannabinoid receptor; 2-AG, 2-arachidonoylglycerol. Affinity refers to the ability of the eCB and receptor to form a bound complex and efficacy refers to the ability of the eCB to induce signaling via the receptor.
Both AEA and 2-AG are synthesized from phospholipid precursors through the actions of phospholipases (60). The precursor for AEA is a low abundance phospholipid, N-arachidonyl-phosphatidylethanolamine (N-arach-PE; Fig. 2A). Several enzymatic pathways have been proposed that can convert N-arach-PE to AEA, including N-acyl-PE-specific phospholipase D (NAPE-PLD) (60). 2-AG is synthesized from diacylglycerol (DAG) by DAG lipase (DAGL); DAG is produced from phosphatidylinositol bisphosphate (PIP2) by phospholipase C (PLC; Fig. 2B). Thus, receptors that couple to the activation of PLC, including many GPCRs and growth factor receptors, are capable of initiating 2-AG synthesis, provided that DAGL is available to convert DAG to 2-AG.
Figure 2.
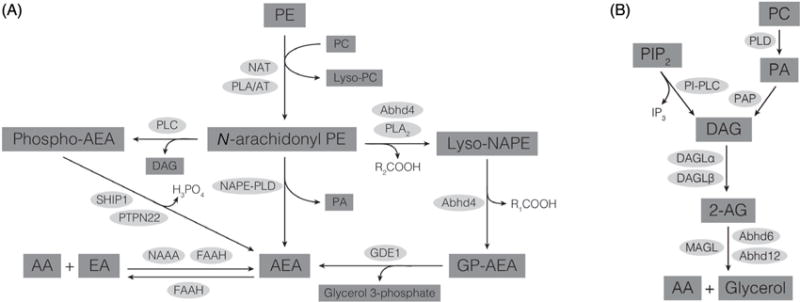
(A) Biosynthetic and catabolic pathways for AEA. (B) Biosynthetic and catabolic pathways for 2-AG. AA, arachidonic acid; Abhd, alpha-beta-hydrolase domain protein; DAG, diacylglycerol; DAGL, diacylglycerol lipase; EA, ethanolamine; FAAH, fatty acid amide hydrolase; GDE1, glycerophosphodiesterase 1; GP-AEA, glycerophospho-AEA; NAAA, N-acylethanolamine-hydrolyzing acid amidase; NAPE-PLD, N-acyl phosphatidylethanolamine-specific phospholipase D; NAT, N-acyltransferase; PA, phosphatidic acid; PAP, phosphatidic acid phosphatase; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PIP2, phosphatidylinositol bis phosphate; PI-PLC, phosphatidylinositol-specific phospholipase C; PLA/AT, phospholipase A/acyltransferase; PLA2, phospholipase A2; PLC, phospholipase C; PLD, phospholipase D; PTPN22, protein tyrosine phosphatase, nonreceptor type 22; SHIP1, Src homology 2-containing inositol phosphatase-1.
Both AEA and 2-AG are inactivated with respect to cannabinoid receptor agonism by enzymatic hydrolysis to free arachidonic acid (Fig. 2A and 2B). AEA and other NAEs are substrates for the serine hydrolase, fatty acid amide hydrolase (FAAH) (23). FAAH is present in the brain (143) and is an important regulator of brain concentrations of AEA as well as other NAEs (6, 112). A second amidohydrolase, N-acylethanolamine-hydrolyzing acid amidase, hydrolyzes AEA in the periphery (144). 2-AG is hydrolyzed by multiple enzymes, including monoacylglycerol lipase (MAGL) (30) and alpha-beta hydrolase 6 (97). Genetic deletion studies in mice (22) and studies using pharmacologic inhibitors of FAAH (40) and MAGL (111) consistently demonstrate that the activities of these hydrolytic enzymes play a very important role in the regulation of CB1R activation. In particular, AEA and 2-AG concentrations rise when FAAH and MAGL are inhibited, respectively, suggesting that there is ongoing synthesis and degradation of both eCBs. MAGL is also important in the regulation of free arachidonic acid concentrations in brain and thereby plays an important role in neuroinflammation (108). Like free arachidonic acid, AEA and 2-AG are substrates for cyclooxygenase 2, which converts them to ethanolamide- and glycerol-substituted prostaglandins, respectively (51).
The CB1R is presynaptic and subserves retrograde suppression of neurotransmission
CB1R activate Gαi/o family G proteins which couple receptor activation with inhibition of adenylyl cyclase (64) and inhibition of the opening of voltage-dependent calcium channels (16, 92). Early autoradiographic studies of the distribution of CB1R revealed the predominant expression of the receptor on axon terminals in the cerebellum (45) and this distribution pattern has been confirmed throughout the brain (79, 80, 119). Like other Gαi/o-linked GPCRs on axon terminals, activation of CB1R signaling results in inhibition of neurotransmitter release through its ability to block the opening of voltage-gated calcium channels (135). MAGL is also found on axonal terminals (43), while DAGLα, which is required for 2-AG synthesis, (157), is in dendritic compartments (Fig. 3A).
Figure 3.
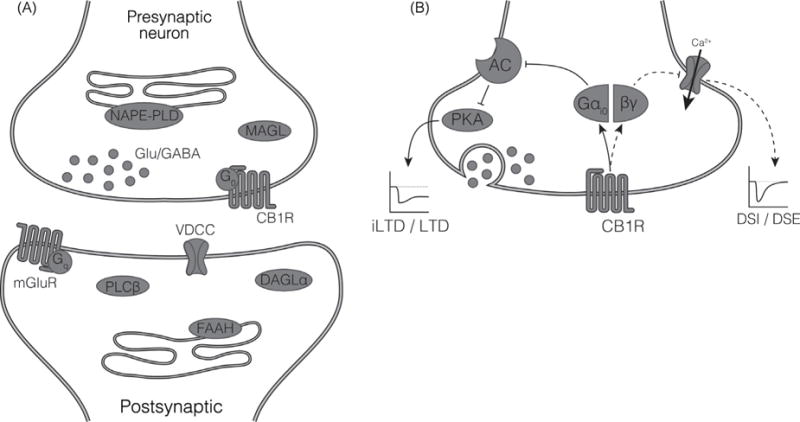
(A) Components of eCB signaling are expressed at the synapse. Shown is the common pattern of distribution of the CB1R and the proteins involved in the synthesis and degradation of the eCBs AEA and 2-AG. See the legend to Figure 2 for abbreviations. (B) CB1R, via Gαi/0, inhibit neurotransmitter release via multiple mechanisms. Inhibition of the opening of voltagegated calcium channels results in the short-term changes in synaptic plasticity of depolarization-induced suppression of inhibition (DSI) or excitation (DSE). Inhibition of cAMP generation and the subsequent reduction in activation of protein kinase A results in long term depression of inhibition (iLTD) and long term depression of excitation (LTD).
This distribution of proteins is consistent with a role for 2-AG/CB1R as a retrograde signaling system (37). 2-AG is synthesized and released from the postsynaptic neuron and targets CB1R on the presynaptic terminal and is degraded by MAGL. The synthesis of 2-AG in the postsynaptic neuron is coupled to excitatory activity. For example, 2-AG is synthesized in postsynaptic dendritic spines following the activation of type I metabotropic glutamate receptors (mGluRs), a subtype of glutamate receptors that couple via Gαq to PLC activation; in response to an increase in intracellular calcium; or by a combination of subthreshold Gαq activation and an increase in calcium (75). The newly synthesized 2-AG diffuses from the dendritic spine into the extracellular space and activates CB1Rs present on axon terminals in the vicinity. As a result of Gαo-mediated inhibition of voltage-operated calcium channel opening, activation of CB1R results in rapid and short-lived inhibition of neurotransmitter release (Fig. 3B). CB1R activation can also lead to longer-lasting suppression of neurotransmitter release, likely through changes in protein kinase A activity mediated by Gai activation (Fig. 3B) (75).
This basic paradigm of eCBs/CB1R signaling links post-synaptic activity to presynaptic neurotransmitter release in many brain regions. In some brain regions, CB1R are present on glutamatergic terminals, in which case 2-AG/CB1R signaling functions as a feedback mechanism (Fig. 4A). For example, eCB synthesis is driven by glutamate and inhibits glutamate release in a negative feedback manner at granule neuron-Purkinje cell synapses in the cerebellum (87). Conversely, in the hippocampus (80) and medial prefrontal cortex (mPFC) (57), the majority of CB1R are on GABAergic interneurons such that mobilization of 2-AG results in enhanced excitatory transmission because gamma aminobutyric acid (GABA) release is reduced (Fig. 4B). CB1R are also present on serotonergic (50) and noradrenergic (109) axon terminals in the brain and can play a role in the regulation of their release as well.
Figure 4.
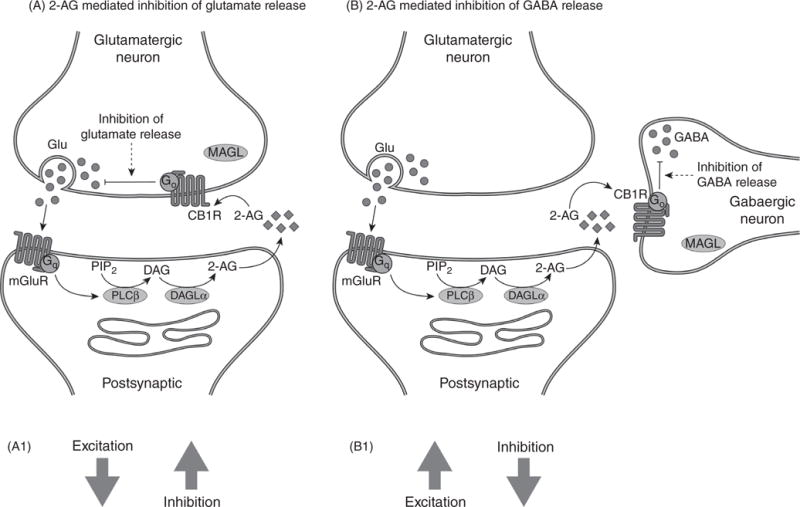
mGluR and 2-AG regulation of synaptic activity. (A) In this scenario, the CB1R are present on glutamatergic terminals so increased 2-AG synthesis in response to mGluR receptor activation results in inhibition of glutamate release. As a result, the synaptic balance is shifted to reduced excitation and increased inhibition (A1). (B) In this scenario, the CB1R are present on GABA terminals and increased 2-AG synthesis results in inhibition of GABA release. As a result, the synaptic balance is shifted to greater excitation and reduced inhibition. 2-AG, 2-arachidonoylglycerol; CB1R, subtype 1 of the cannabinoid receptor; Glu, glutamate; mGluR, metabotropic glutamate receptor. Other abbreviations are in the legend to Figure 2.
NAPE-PLD and FAAH are also present in neuronal elements that are close to the synapse (Fig. 3A); NAPE-PLD has a prominent presynaptic distribution (119), while FAAH in present in large outflow neurons throughout the brain (143). The role of AEA in the regulation of synaptic activity is not well understood. Inhibitors of MAGL, but not FAAH potentiate eCB-mediated signaling in slices from hippocampus and cerebellum (111), suggesting that 2-AG and not AEA is the primary eCB mediator. On the other hand, estradiol affects synaptic plasticity in the hippocampus via a CB1R-AEA mediated mechanism (65) and AEA mediates a form of long-term depression in the amygdala (2), indicating that there are some circumstances under which AEA is the primary endogenous activator of synaptic CB1R.
CB1R Regulation of the Hypothalamic-Pituitary-Adrenal Axis
Threats to well being and physical injuries activate neuronal and endocrine responses that allow for an immediate response to the threat; restoration of homeostasis after the danger has passed; and enhanced memory formation to allow for avoidance in the future. When appropriately mobilized, these responses increase the probability that the individual will survive the threat. The neuronal arm of the stress response is the sympathetic nervous system, which results in rapid and short-lived increases in heart rate, blood pressure, and blood flow to muscles.
The endocrine responses to physiological and psychological threats are mediated by primarily by the glucocorticoids [cortisol in humans and corticosterone (CORT) in rodents], which are released from the adrenal cortex as a result of hypothalamic-pituitary-adrenal (HPA) axis activation. Glucocorticoid signaling is responsible for many of the physiological and psychological changes that follow stress, including increased food consumption, suppression of the immune system, and changes in memory and mood through effects on glucocorticoid receptors (GRs) (71, 99). In addition to being stress responsive, the HPA axis also exerts effects at lower concentrations than occur during stress that are mediated by mineralocorticoid receptor (MR) activation. There is a profound circadian rhythm in circulating glucocorticoid concentrations; concentrations of cortisol and CORT reach their peak early in the active period of the day in both diurnal and nocturnal species.
Stress-induced activation of the HPA axis is mediated by corticotrophin-releasing hormone (CRH)-expressing neurons in the PVN of the hypothalamus (Fig. 5). These neurons respond to and integrate inputs from the amygdaloid complex, mPFC, hippocampus, and brain stem (47). Release of CRH via the median eminence stimulates pituitary adrenocorticotropic hormone (ACTH) release into the circulation. ACTH acts via MC2 receptors in the zona fasiculata of the adrenal cortex to induce glucocorticoid synthesis and release into the circulation. CRH is also expressed in nonhypothalamic brain regions, including the amygdala, bed nucleus of the stria terminalis and dorsal hippocampus (154). GR receptors are found in the hypothalamus as well as limbic regions, including the hippocampus, amygdala, septum, and prefrontal cortex (49).
Figure 5.
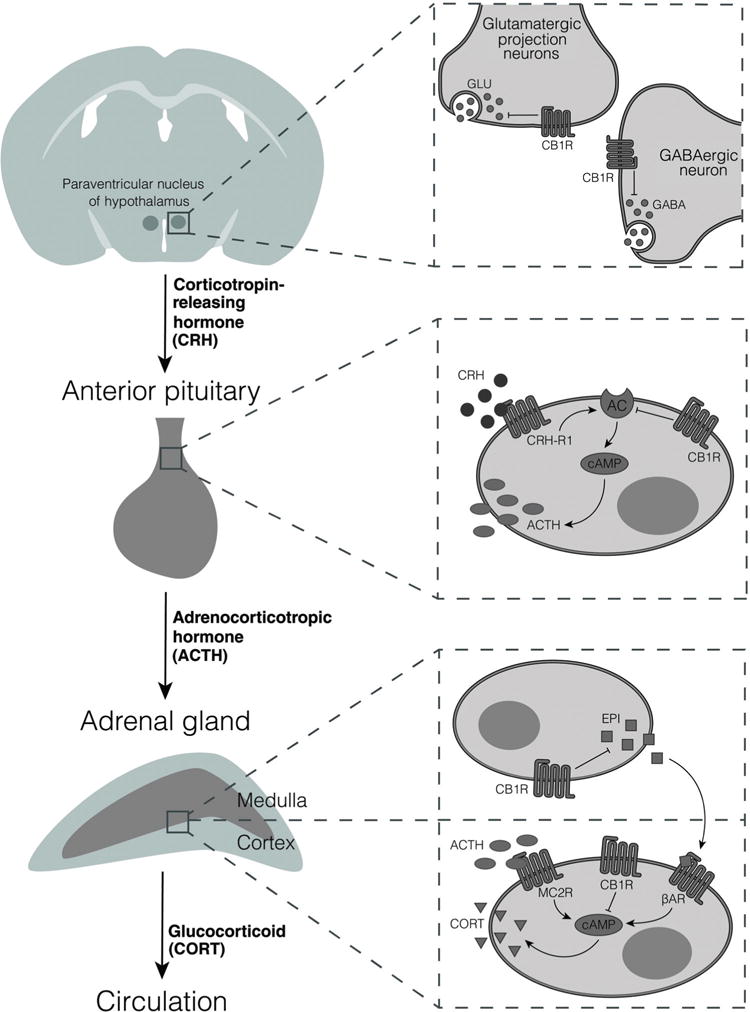
Hypothesized expression and function of CB1R along the HPA axis. Data described in the text support the expression and hypothesized inhibitory roles of the CB1R at all three levels of the HPA axis. In the paraventricular nucleus, CB1R have been identified on both glutamatergic and GABAergic terminals. Corticotropes of the anterior pituitary express CB1R, which we hypothesize, could inhibit CRH-induced activation of adenylyl cyclase (AC), thereby reducing cAMP concentrations. In the adrenal gland, CB1R have been identified on cortical cells, where we hypothesize they could oppose the elevation of cAMP induced by ACTH acting through melanocortin 2 receptors (MC2R). CB1R have also been proposed to be present on medullary cells of the adrenal gland and to inhibit the release of epinephrine (EPI) from these cells. This would result in reduced activation of ß-adrenergic receptors (ßAR) on the cortical cells.
CB1R are present throughout the HPA axis
CB1R density in the hypothalamus is low compared to other brain regions (46) in spite of the fact that the cannabinoids have many effects on hypothalamically mediated processes (34). CB1R are heterogeneously expressed within subregions of the hypothalamus; in particular, CB1R are present at greater density within the PVN relative to other hypothalamic regions (155). Subcellularly, CB1Rs are present on both symmetrical and nonsymmetrical synapses in the hypothalamus and most immunoreactivity is found in axon terminals. Hypothalamic CB1R density differs between male and female rodents (129) which could contribute to sex-related differences in cannabinoid effects between males and females (21).
Low but measureable amounts of CB1R protein are detected in the intermediate and anterior lobes of the pituitary gland (110) and CB1R mRNA is present in the external zone of the median eminence (46, 155). CB1R mRNA is expressed in the adrenal gland of rodents (11) and humans (160). Thus, there is evidence for CB1R expression at all levels of the HPA axis.
CB1R activation can both inhibit and potentiate HPA axis activation by stress
High efficacy CB1R agonists, when systemically administered, exert biphasic effects on activation of the HPA axis by stress; low doses reduce, while high doses increase circulating concentrations of CORT in rodents (113, 138). Studies using CB1R antagonists and CB1R null (CB1R−/−) mice demonstrate that loss of CB1R results in increased CORT release in response to an acute stress (19, 57, 113, 149) that exhibits sex differences (127). Similarly, pharmacological antagonism of central CB1Rs increases circulating CORT and ACTH concentrations in rat (96). These findings lead to the hypothesis that the functional pool of CNS CB1Rs that inhibit HPA axis activation is endogenously activated (Fig. 6). In support of this hypothesis, FAAH (113) and MAGL (127) inhibitors reduce stress-induced CORT release, although FAAH inhibition is not consistently observed to do so (127). The increase in HPA axis activity that occurs in response to high doses of CB1R agonists could be secondary to activation of monoaminergic hindbrain nuclei as both noradrenergic and serotonergic blockade reduce these stimulatory effects (101).
Figure 6.
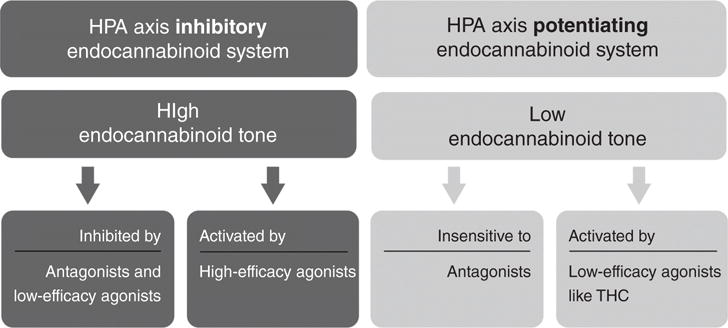
High-efficacy cannabinoid agonists have biphasic effects on HPA axis activation by stress, while the partial agonist, THC, and antagonists increase HPA axis activation. Our hypothesis for these findings is displayed in this figure. The CB1R-mediated inhibition of HPA axis activation by stress has high eCB tone (as is shown in Fig. 7). As a result, only high efficacy agonists can increase this tone further. Low-efficacy agonists and antagonists will inhibit this tone and “uncover” the HPA axis potentiating CB1R function. We hypothesize based on the data available that the HPA axis potentiating CB1R has low eCB tone.
Interestingly, THC does not produce the inhibitory effects of the synthetic agonists, instead exerts a monophasic, dose-related increase in basal (5, 116) and stress-induced CORT release (113). Given that THC has low efficacy at the CB1R [i.e., it is a weak partial agonist (81)] and in light of the data that these receptors are endogenously activated, it is possible that THC acts as an antagonist in this circumstance, blocking the effects of eCBs to reduce stress-induced CORT release (Fig. 6). An older study suggests that the effects of THC on “basal” CORT secretion may actually be stress related, with the stressor being THC-induced hypothermia (5).
There are multiple mechanisms by which increased CB1R signaling, through either increased eCBs or exogenous agonist administration, can reduce HPA axis activation. First, agonist activation of presynaptic CB1R on glutamatergic inputs onto CRH neurons in the PVN inhibits release of glutamate through the mechanisms described above (28,29). Recent studies indicate that CB1R signaling is not recruited by mGluR1 activation (32), however, it is possible that the primary mechanism for activation of eCB signaling at this synapse is CORT activation of membrane GR (104) and a primary role for this process is glucocorticoid-mediated negative feedback. Studies utilizing intrahypothalamic injections in rats indicate that mobilization of 2-AG and inhibition of HPA axis activity occurs within 15 min of the elevation of CORT, findings that further support the participation of a nongenomic GR and a mechanism that allows for fairly rapid accumulation of 2-AG (33).
Second, intra-BLA injections of a CB1R agonist and antagonist decrease and increase, respectively, CORT responses to acute stress in male rats (39,56). These data indicate that a pool of CB1R in the BLA is also important for HPA axis activation. Recent studies by Hill and colleagues indicate that the BLA AEA/CB1R couple “gates” HPA axis activation (Fig. 7) (41). When BLA AEA concentrations are high, CB1R signaling inhibits HPA axis activation. However, acute stress reduces AEA concentrations in the BLA through CRH-mediated activation of FAAH, which allows for HPA axis activation (41). Thus, CB1R signaling in the BLA likely plays an important role in the initiation of HPA axis activation by stress.
Figure 7.
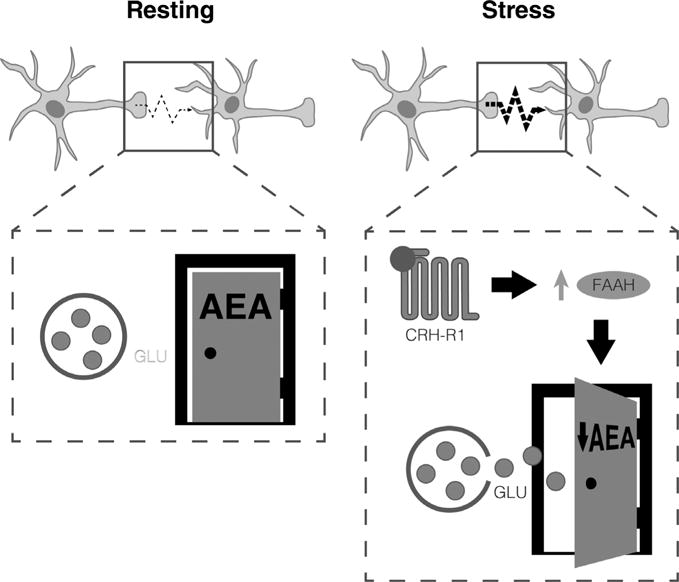
Depiction of the amygdalar AEA gate hypothesis. According to this hypothesis, AEA concentrations are high at rest, resulting in tonic activation of CB1R on glutamatergic terminals in the basolateral amygdala. This prevents glutamate release. Increased CRH as occurs during stress results in activation of FAAH by an as-yet unidentified mechanism, resulting in reduced AEA and relief of AEA/CB1R-mediated inhibition of glutamate release.
Third, activation of CB1R within the PFC (57) and hippocampus (152) both result in reduced excitatory drive into the PVN through multisynaptic circuits (Fig. 8). As is discussed further later, there is evidence that 2-AG is mobilized in these brain regions by CORT-mediated activation of GR receptors. These data support the hypothesis that 2-AG/CB1R in these brain regions are involved in long-loop negative feedback regulation of HPA axis activation by CORT. It is interesting that the timing of glucocorticoid-mediated elevation of 2-AG contents is slower in hippocampus and mPFC compared to PVN (33). This matches the role of these structures in recovery of the HPA axis to baseline over hours rather than the rapid, negative feedback that glucocorticoids induce through effects in the PVN (48, 94).
Figure 8.
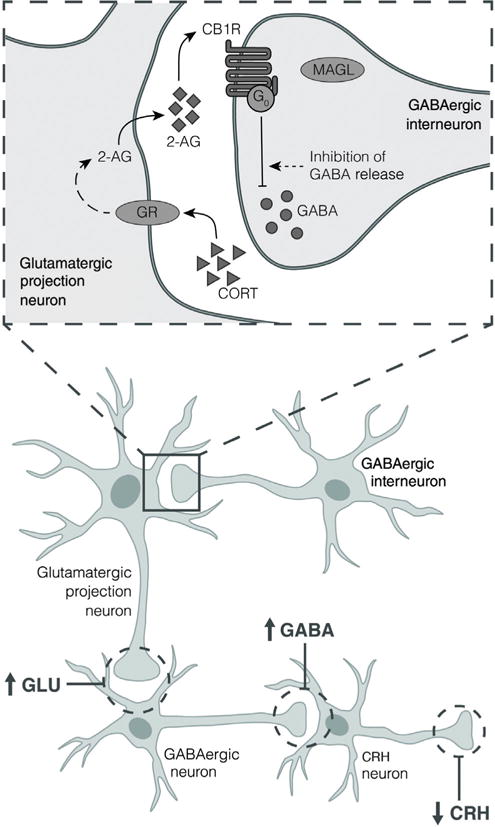
In both the prefrontal cortex and hippocampus, studies demonstrate that 2-AG synthesis is enhanced by glucocorticoid (CORT) through activation of GRs. CB1R on GABAergic terminals are subsequently activated, resulted in reduced GABA release and disinhibition of glutamatergic projection neurons. These neurons project onto GABAergic neurons in other areas of the brain which ultimately inhibit CRH release.
Pituitary cells isolated from CB1R−/− mice exhibited greater secretion of ACTH in response to both CRH and forskolin stimulation than those isolated from wild-type mice (19). These findings suggest that activation of CB1R in the pituitary exerts an inhibitory effect on ACTH release (Fig. 5). Finally, CB1R agonist treatment decreases basal and stimulated adrenocortical steroidogenesis (160) and decreases epinephrine release from adrenal medullary cells (106), both changes that would be expected to reduce corticosteroid hormone release. This conclusion is supported by data that systemic blockade of CB1R elevates circulating CORT concentrations without affecting ACTH secretion or pituitary c-fos expression; findings that are consistent with a direct effect of the CB1R antagonist to increase glucocorticoid release from the adrenal gland (105).
Human studies reproducibly demonstrate that exposure of naive or infrequent cannabis users to C. sativa (18) or THC (24, 25, 83, 84, 123) increases the secretion of cortisol, findings that are consistent with the rodent literature in which THC has a predominant effect to increase HPA axis activation (5, 113, 116). As discussed above, it is possible that THC is acting as an antagonist and reducing the inhibitory effects of endogenous CB1R signaling. A few studies report that chronic cannabis users do not exhibit a stimulatory effect of THC on cortisol concentrations (25, 123), suggesting that tolerance develops to this effect of THC. Interestingly, some (82, 137), but not all (4), studies have reported that chronic cannabis users exhibit elevated basal cortisol levels, while other studies have shown that stress-induced activation of the HPA axis is blunted in chronic adult and adolescent cannabis users (137, 145). In adolescents with an early onset of use, chronic cannabis use is associated with altered diurnal cortisol rhythms such that cortisol concentrations are higher than normal at night and blunted in the morning (66). Taken together, the available human data suggest that chronic use of C. sativa can lead to dysregulation of basal, circadian, and stressregulated HPA axis activity. Given that THC interacts with CB1R, these data suggest the hypothesis that CB1R-mediated regulation of the HPA axis is the mechanism that underlies the observed dysregulation. Taking this a step further, these data also suggest the hypothesis that eCB signaling is a modulator of HPA axis activity in humans as well as rodents. These hypotheses need to be examined in a rigorous manner.
Regulation of eCB Concentrations and CB1R Expression by Signaling Molecules of the Hypothalamic-Pituitary-Adrenal Axis
Glucocorticoid regulation of endocannabinoid content in brain
Exogenous administration of CORT to rats increases 2-AG contents in the hypothalamus (53), PFC (57), and CA1 region of the hippocampus (152). This effect of CORT is mimicked by acute restraint stress, which increases brain contents of 2-AG within 30 min following stress (57, 122, 152). The mechanisms by which CORT increases 2-AG concentrations have been explored and are discussed in more detail later.
Exogenous CORT administration also results in rapid increases in AEA in the amygdala, hippocampus, and hypothalamus (53). These changes are transient, returning to baseline before CORT concentrations have subsided. The mechanistic bases for this effect have not been examined, although studies in the raphe nucleus suggest that GR-mediated inhibition of eCB catabolism could be involved (151). Although exogenously administered glucocorticoid increases AEA concentrations, acute stress exposure in rodents decreases amygdalar and PFC concentrations of AEA (56, 100, 114, 122). The acute effect of stress to reduce AEA concentrations is accompanied by an increase in the activity of FAAH. Recent data indicate that acute increases in CRH that occur during stress act through CRH-R1 receptors to increase FAAH activity and thereby decrease AEA concentrations in the amygdala, but not in the PFC (41). However, further studies demonstrate that sustained CORT exposure recruits CRH signaling in the PFC, resulting in sustained increase in FAAH activity and lowering of AEA concentrations (42). Thus, stress affects AEA concentrations via at least two mechanisms. The first effect is mediated by the extrahypothalamic release of CRH that results in increased FAAH activity, increased AEA catabolism, and reduced AEA concentrations. The second is mediated by CORT and involves an increase in AEA through an as yet unknown mechanism.
There is evidence that CORT treatment could also exert long-lasting effects on AEA via effects on FAAH gene expression. Five putative GR-binding elements have been identified in the promoter region of the mouse FAAH gene (150), and in vitro studies demonstrate that GR represses FAAH expression (150). In agreement with these observations, male rats exposed to isolation stress exhibit highly significant reductions (greater than 50% in most regions) in FAAH mRNA expression in cortical regions and throughout the dorsal and ventral striatum (128). FAAH protein is reduced by 40% in dorsal root ganglion cells from rats chronically treated with CORT (63). Exposure of rats to chronic CORT did not produce a measureable effect on FAAH mRNA expression, but increased the Vmax for FAAH in the amygdala and hippocampus (9). Similarly, repeated restraint in male mice increases the Vmax for FAAH in amygdala and mPFC (122).
These data, together with the discussion below outlining the role of GR to regulate synaptic 2-AG concentrations, demonstrate multiple mechanisms by which the molecules of the HPA axis exert short- and long-term changes in the concentrations of eCBs that are available to activate their target receptors, including CB1R.
Glucocorticoid regulation of CB1R expression
Glucocorticoids regulate eCB signaling through effects on the expression and function of the CB1R itself as well. Early studies revealed that adrenalectomy results in increased CB1R mRNA expression in the caudate putamen of male rats (93), suggesting that glucocorticoids suppresses neuronal CB1R expression. More recent studies support this hypothesis; in particular, prolonged CORT treatment reduces CB1R binding site density in the hippocampus (9, 52) and amygdala (9), brain regions with high GR expression. Intense activation of the HPA axis significantly reduces CB1R mRNA expression in the frontal cortex seven days later (15). Repeated treatment of rats with CORT results in GR-dependent decreases in CB1R protein expression in primary sensory neurons (63). A single exposure of rats to a live cat, which produces strong activation of the HPA axis, also significantly reduces CB1R mRNA expression in the frontal cortex (15). A recent study carried out in dorsal root ganglion cells from rats exposed to chronic water stress suggests an epigenetic mechanism by which chronic changes in CORT concentrations suppress CB1R expression (62). According to this mechanism, CORT-mediated activation of GR and MR increases transcription of DNA (cytosine-5-)-methyltransferase 1, which methylates the promoter region of the gene for the CB1R, reducing transcription and protein expression. Whether this mechanism is operative in other cell types is not known.
There is also evidence that GR activation can increase CB1R expression. For example, CB1R mRNA in interneurons within mouse myenteric plexus is increased by CORT treatment (90) and upregulation of CB1R expression in injured spinal cord is dependent upon GR (153). Murine osteoblasts in culture also respond to high doses of glucocorticoids with an increase in CB1R expression (156) and bone CB1R expression is increased following chronic, high dose CORT treatment in rats (85). Expression of CB1R-encoding mRNA in alveolar cells cultured from fetal rat lung is increased following hydrocortisone treatment (126). Thus, it is likely that glucocorticoids can alter CB1R expression through multiple mechanisms that could differ by cell type and duration of the elevation in CORT among other factors.
Glucocorticoids Affect Synaptic Plasticity via Endogenous CB1R Signaling: Role in Regulation of the HPA Axis
GR activation affects synaptic plasticity as a result of increased endocannabinoid signaling
Glucocorticoids act through GR to inhibit glutamate (77, 78) and GABA (148) release. There is evidence in several brain regions that eCB/CB1R signaling is mobilized by GR activation and that this mechanism couples glucocorticoids to changes in neurotransmitter release (142). In the hypothalamus, glucocorticoids act through a membrane-associated receptor to rapidly mobilize eCBs (29), which act through CB1R to inhibit release of glutamate from neurons synapsing on CRH neurons in the PVN (28, 29). This mechanism is hypothesized to contribute to glucocorticoid-mediated feedback inhibition of HPA axis activation at the level of the hypothalamus (33, 136). In support of this hypothesis, concentrations of 2-AG in PVN harvested from rats exposed to restraint stress (33) and CORT treatment (53) are increased and inhibition of CB1R signaling potentiates stress-induced c-fos expression in PVN neurons (113).
Similar mechanisms are operative in the mPFC (57) and hippocampus (152), although in these brain regions, the CB1R are present primarily on GABAergic terminals, and therefore, the primary effect of CORT-induced CB1R signaling is to inhibit GABA release (Fig. 2). Like in the hypothalamus, recent data indicate that the GR in the frontal cortex are expressed on postsynaptic profiles (3). Excitatory projections from these brain regions accelerate the poststress decline in circulating glucocorticoids likely through activation of inhibitory inputs from the bed nucleus of the stria terminalis to the PVN (31, 35). Suppression of GABA release in the mPFC and hippocampus would increase the outflow of these glutamatergic projection neurons, and thereby shorten the time period during which CRH neurons are active. In the hippocampus, glucocorticoid-mediated increases in 2-AG mediated signaling are GR-dependent and also reduced by nifedipine, suggesting that GR-mediated increased calcium entry drives 2-AG synthesis (152).
Acute stress and CORT treatment elevate tissue concentrations of 2-AG in mPFC and hippocampus through a GR-dependent mechanism (57, 152). These mechanistic studies align with in vivo data discussed earlier that CB1R agonists dampen HPA axis activation and support an important role for CB1R signaling as a critical mediator of CORT-mediated short- and long-loop feedback inhibition of HPA axis activation.
The molecular mechanisms by which GR activation recruits eCB signaling has not been clearly elucidated. In the PVN, Tasker and colleagues have demonstrated requirements for G proteins and postsynaptic neuronal expression of the canonical GR, which leads to the hypotheses that either the membrane GR is regulated by the nuclear GR or is itself the GR protein (104). The glucocorticoid-induced increases in 2-AG in mPFC (57) and hippocampus (152) are inhibited by GR antagonists provides further support for the involvement of GR in the effect. Some hints to the mechanism come from studies of rapid effects of estrogen (38). Canonical, intracellular estrogen receptors have been shown to be present at the membrane where they physically associate with mGluR to potentiate their signaling through extracellular-signal regulated kinase (ERK) pathways. Both estrogen receptors (8) and GR (98) require caveolin to participate in acute regulation of cellular signaling, suggesting that they encounter other signaling molecules within the caveolae. Given that mGluR also couple to the activation of PLC, GR could hypothetically act in a similar manner and potentiate 2-AG synthesis. As an aside, estradiol has been shown to suppress GABA release in the hippocampus of female rats through a mechanism that involves estrogen receptor/mGluR1 mediated activation of eCB signaling (141). However, in contrast to the glucocorticoid mechanism, it is likely that AEA and not 2-AG is the eCB involved.
An alternative mechanism has been suggested based upon the nongenomic effects of the glucocorticoids to alter the metabolism of arachidonic acid. In particular, glucocorticoids suppress the liberation of free arachidonic acid from phospholipids through inhibition of phospholipase A2, while also enhancing the formation of DAG through increased activity of PLC (94). This pattern of changes in lipid metabolism would result in the shunting of arachidonic acid away from metabolism by cyclooxygenase and other enzymes toward the production of the eCBs, particularly 2-AG.
Repeated stress alters endocannabinoid signaling: Contribution to habituation and stress responsiveness
HPA axis responses to repeated, predictable exposure to the same stress eventually habituate; and there is evidence that eCB/CB1R signaling contributes to this form of regulation. Repeated exposure to a short period of restraint stress in rodents enhances 2-AG mobilization. While single exposure to restraint does not affect limbic brain contents of 2-AG, exposure of male mice to increasing numbers of restraint episodes results in progressive increases in 2-AG content in several brain regions, including amygdala and hypothalamus (114, 122). Similar changes are seen in the amygdala of the male rat (55). Recent studies demonstrate that chronic CORT treatment also increases 2-AG content in the mPFC, but not amygdala of male rats (42). Interestingly, this effect of CORT was abolished by coadministration of the CRH-R1 antagonist, antalarmin, suggesting a role for elevated CRH in the ability of repeated elevation of CORT to potentiate 2-AG content in the mPFC. Further mechanistic insights come from studies of (102) Patel and colleagues who demonstrated that repeated stress results in increased 2-AG in the amygdala, likely as a result of reduced MAGL-mediated catabolism of 2-AG (139). Interestingly, MAGL activity was not altered, but less protein was present at the plasma membrane where it is more likely to encounter 2-AG that is involved in CB1R activation (139). As a result, 2-AG-mediated activation of CB1R would be prolonged following repeated restraint stress exposure.
In vivo studies support the hypothesis that CB1R signaling is required for habituation of behavioral responses and HPA axis activation to the stressor. CB1R antagonist treatment prevents habituation to chronic stress-induced behavioral activation (114) and anhedonia (121) in male mice. Inhibition of CB1R signaling in the amygdala prevents habituation to HPA axis activation in rats (55). CB1R activation is also required for the habituation of innate fear behaviors in mice to repeated homotypic stress (72-74).
Taken together, these data suggest that eCB/CB1R plasticity allows for habituation of the brain to repeated, predictable stress exposures. The ability to habituate to a nonthreatening stimulus reduces the risk for the development of negative consequences produced by chronic stress; therefore, the recruitment of eCB/CB1R signaling by repeated stress could be of great importance to mitigating risk for psychiatric disorders in humans.
Thus, eCB/CB1R signaling in the amygdala guards against spurious HPA axis activation and, through effects in other parts of the limbic circuit, promotes the return of the HPA axis to baseline. Although these mechanisms are not essential for the HPA axis response to stress, loss of eCB/CB1R signaling will likely increase the “wear and tear” of stress on the brain due to hyperactivation of the HPA axis response.
Accumulating evidence suggests that loss of CB1R-mediated signaling can also occur during stress, particularly chronic stress states in which CORT concentrations are consistently elevated and do not habituate. Chronic unpredictable stress exposure decreases CB1R density in the hippocampus of male, but not female, rats (125) and decreases CB1R-mediated signaling in PFC (58).
Endocannabinoid Signaling Mediates Multiple Effects of Glucocorticoids on the Brain and Body
Endocannabinoid signaling couples glucocorticoids to other CNS-mediated effects
The studies discussed above describing the role of CORT mobilization of eCB/CB1R signaling in the feedback regulation of HPA axis activation suggests the more generalized hypothesis that eCBs are increased by glucocorticoids and that CB1R signaling couples glucocorticoids to other CNS effects (54). For example, glucocorticoids enhance the memory of emotionally arousing experiences (27, 130, 131), through the activation of GR and MR in brain regions involved in learning and memory, such as the hippocampus, the amygdala, and the prefrontal cortex (26). Blocking GR impairs consolidation (132) and reconsolidation of aversive events (70). CB1R signaling has been demonstrated to be required for CORT-induced enhancement of memory consolidation, perhaps through increased noradrenergic signaling (14), and/or inhibition of GABA signaling in the BLA (139). GR signaling has also been shown to be required for reconsolidation of social reward memories; however, this effect is not sensitive to CB1R antagonism (1).
Stress, through activation of GR signaling, sets the stage for reinstatement of cocaine seeking behavior in rodents (103). Studies in mice (147) and rats (102) demonstrate the requirement for CB1R in stress-induced reinstatement of cocaine seeking behavior. Low doses of dexamethasone reduce motion sickness in rats, an effect that is partially reversed by the CB1R antagonist, AM251 (158).
Endocannabinoid signaling couples glucocorticoids to peripheral effects
Excessive concentrations of circulating glucocorticoids, as occur in Cushing’s syndrome, for example, are associated with adiposity, weight gain, hepatic steatosis, and dyslipidemia (107). A recent study utilized chronic, high dose treatment with CORT to recapitulate these symptoms in mice (10). The increased body weight, adiposity, hepatic steatosis, and dyslipidemia were all reduced or reversed by global CB1R deletion or pharmacological inhibition. Interestingly, another recent study demonstrated that 24-h treatment of adipocytes in culture with dexamethasone increased CB1R expression (115), suggesting that the effects of high glucocorticoids are amplified by enhanced CB1R expression.
Excessive glucocorticoid exposure is known to reduce biomechanical strength of bone and increase the risk of osteoporosis (44). GR activation impairs osteoblast survival and differentiation (124); and glucocorticoid-related osteopenia is accompanied by increased fat cell accumulation in the bone marrow, which has deleterious effects (133). CB1R−/− mice have high bone mass and CB1R antagonists induce osteoclast apoptosis and prevent bone loss in ovariectomized mice (69). The bone deterioration and formation of fatty marrow induced by glucocorticoid treatment are both attenuated by CB1R antisense and antagonist treatments (85). These findings suggest that the glucocorticoid effects on bone are also CB1R mediated. There is evidence, however, that the protective effect of CB1R on bone loss is age dependent: aged CB1R−/− mice have low bone mass (68) and CB1R antagonist treatment worsens methylprednisolone-induced osteoporosis in aged rats (134).
Stress affects the function of the GI tract. Stress has been shown to alter gastric secretion, intestinal motility, epithelial cell permeability, and blood flow to the mucosal layers (86). Acute and chronic stress are also both associated with visceral hypersensitivity to painful stimuli as a result of reduced pain thresholds (89). CB1R are present in cholinergic neurons in the myenteric and submucosal plexi of the enteric nervous system (20, 88). CB1R−/− mice exposed to 4 days of 2-h immobilization and acoustic stress exposure exhibit increased permeability of the colonic barrier; enhanced inflammation; lower IgA secretion and higher bacterial translocation into the mesenteric lymph nodes than wild-type mice (161). These results indicate that CB1R oppose rather than mediate the effects of stress on GI function.
Summary and Conclusions
The eCB system should be considered as a part of the body’s homeostatic processes. Activation of the CB1R promotes return of the HPA axis to nonstressed levels, together with other restorative effects, such as increased feeding and sleep. In fact, CB1R signaling can be described as promoting the states of “fat, dumb, and happy”; and “calm, cool, and collected.” It could be argued that those phrases describe individuals with robust eCB tone. The importance of robust eCB tone became evident in the human trials of the CB1R antagonist, rimonabant. Rimonabant produced significant adverse psychiatric effects, including increased anxiety, depressed mood, and even suicides (17). Therapeutic approaches that enhance eCB signaling have been proposed by many as logical methods to promote homeostasis and as a result, reduce the risk to develop psychiatric and other stress-related disorders. Only time will tell whether such therapies can be developed.
Acknowledgments
C.J.H. was supported during the writing of this review by NIH grant R01 DA038663; and the Research and Education Program, a component of the Advancing a Healthier Wisconsin endowment at the Medical College of Wisconsin.
References
- 1.Achterberg EJ, Trezza V, Vanderschuren LJ. Glucocorticoid receptor antagonism disrupts the reconsolidation of social reward-related memories in rats. Behav Pharmacol. 2014;25:216–225. doi: 10.1097/FBP.0000000000000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Azad SC, Monory K, Marsicano G, Cravatt BF, Lutz B, Zieglgansberger W, Rammes G. Circuitry for associative plasticity in the amygdala involves endocannabinoid signaling. J Neurosci. 2004;24:9953–9961. doi: 10.1523/JNEUROSCI.2134-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bitencourt RM, Alpar A, Cinquina V, Ferreira SG, Pinheiro BS, Lemos C, Ledent C, Takahashi RN, Sialana FJ, Lubec G, Cunha RA, Harkany T, Kofalvi A. Lack of presynaptic interaction between glucocorticoid and CB1 cannabinoid receptors in GABA- and glutamatergic terminals in the frontal cortex of laboratory rodents. Neurochem Int. 2015;90:72–84. doi: 10.1016/j.neuint.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 4.Block RI, Farinpour R, Schlechte JA. Effects of chronic marijuana use on testosterone, luteinizing hormone, follicle stimulating hormone, prolactin and cortisol in men and women. Drug Alcohol Depend. 1991;28:121–128. doi: 10.1016/0376-8716(91)90068-a. [DOI] [PubMed] [Google Scholar]
- 5.Bloom AS, Kiernan CJ. Interaction of ambient temperature with the effects of delta 9-tetrahydrocannabinol on brain catecholamine synthesis and plasma corticosterone levels. Psychopharmacology (Berl) 1980;67:215–219. doi: 10.1007/BF00431259. [DOI] [PubMed] [Google Scholar]
- 6.Bortolato M, Mangieri RA, Fu J, Kim JH, Arguello O, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. Antidepressant-like activity of the fatty acid amide hydrolase inhibitor URB597 in a rat model of chronic mild stress. Biol Psychiatry. 2007;62:1103–1110. doi: 10.1016/j.biopsych.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Bouaboula M, Rinaldi M, Carayon P, Carillon C, Delpech B, Shire D, Le Fur G, Casellas P. Cannabinoid-receptor expression in human leukocytes. Eur J Biochem. 1993;214:173–180. doi: 10.1111/j.1432-1033.1993.tb17910.x. [DOI] [PubMed] [Google Scholar]
- 8.Boulware MI, Kordasiewicz H, Mermelstein PG. Caveolin proteins are essential for distinct effects of membrane estrogen receptors in neurons. J Neurosci. 2007;27:9941–9950. doi: 10.1523/JNEUROSCI.1647-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowles NP, Hill MN, Bhagat SM, Karatsoreos IN, Hillard CJ, McEwen BS. Chronic, noninvasive glucocorticoid administration suppresses limbic endocannabinoid signaling in mice. Neuroscience. 2012;204:83–89. doi: 10.1016/j.neuroscience.2011.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bowles NP, Karatsoreos IN, Li X, Vemuri VK, Wood JA, Li Z, Tamashiro KL, Schwartz GJ, Makriyannis AM, Kunos G, Hillard CJ, McEwen BS, Hill MN. A peripheral endocannabinoid mechanism contributes to glucocorticoid-mediated metabolic syndrome. Proc Natl Acad Sci U S A. 2015;112:285–290. doi: 10.1073/pnas.1421420112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buckley NE, Hansson S, Harta G, Mezey E. Expression of the CB1 and CB2 receptor messenger RNAs during embryonic development in the rat. Neurosci. 1998;82:1131–1149. doi: 10.1016/s0306-4522(97)00348-5. [DOI] [PubMed] [Google Scholar]
- 12.Buckner JD, Zvolensky MJ, Ecker AH, Jeffries ER. Cannabis craving in response to laboratory-induced social stress among racially diverse cannabis users: The impact of social anxiety disorder. J Psychopharmacol. 2016;30:363–369. doi: 10.1177/0269881116629115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bujarski SJ, Feldner MT, Lewis SF, Babson KA, Trainor CD, Leen-Feldner E, Badour CL, Bonn-Miller MO. Marijuana use among traumatic event-exposed adolescents: Posttraumatic stress symptom frequency predicts coping motivations for use. Addict Behav. 2012;37:53–59. doi: 10.1016/j.addbeh.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 14.Campolongo P, Roozendaal B, Trezza V, Hauer D, Schelling G, McGaugh JL, Cuomo V. Endocannabinoids in the rat basolateral amygdala enhance memory consolidation and enable glucocorticoid modulation of memory. Proc Natl Acad Sci U S A. 2009;106:4888–4893. doi: 10.1073/pnas.0900835106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campos AC, Ferreira FR, da Silva WA, Jr, Guimaraes FS. Predator threat stress promotes long lasting anxiety-like behaviors and modulates synaptophysin and CB1 receptors expression in brain areas associated with PTSD symptoms. Neurosci Lett. 2013;533:34–38. doi: 10.1016/j.neulet.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 16.Caulfield MP, Brown DA. Cannabinoid receptor agonists inhibit Ca current in NG108-15 neuroblastoma cells via a pertussis toxin-sensitive mechanism. Br J Pharmacol. 1992;106:231–232. doi: 10.1111/j.1476-5381.1992.tb14321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Christensen R, Kristensen PK, Bartels EM, Bliddal H, Astrup A. Efficacy and safety of the weight-loss drug rimonabant: A meta-analysis of randomised trials. Lancet. 2007;370:1706–1713. doi: 10.1016/S0140-6736(07)61721-8. [DOI] [PubMed] [Google Scholar]
- 18.Cone EJ, Johnson RE, Moore JD, Roache JD. Acute effects of smoking marijuana on hormones, subjective effects and performance in male human subjects. Pharmacol Biochem Behav. 1986;24:1749–1754. doi: 10.1016/0091-3057(86)90515-0. [DOI] [PubMed] [Google Scholar]
- 19.Cota D, Steiner MA, Marsicano G, Cervino C, Herman JP, Grubler Y, Stalla J, Pasquali R, Lutz B, Stalla GK, Pagotto U. Requirement of cannabinoid receptor type 1 for the basal modulation of hypothalamic-pituitary-adrenal axis function. Endocrinology. 2007;148:1574–1581. doi: 10.1210/en.2005-1649. [DOI] [PubMed] [Google Scholar]
- 20.Coutts AA, Irving AJ, Mackie K, Pertwee RG, Anavi-Goffer S. Localisation of cannabinoid CB(1) receptor immunoreactivity in the guinea pig and rat myenteric plexus. J Comp Neurol. 2002;448:410–422. doi: 10.1002/cne.10270. [DOI] [PubMed] [Google Scholar]
- 21.Craft RM, Marusich JA, Wiley JL. Sex differences in cannabinoid pharmacology: A reflection of differences in the endocannabinoid system? LifeSci. 2012;92:476–481. doi: 10.1016/j.lfs.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci USA. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 24.D’Souza DC, Perry E, MacDougall L, Ammerman Y, Cooper T, Wu YT, Braley G, Gueorguieva R, Krystal JH. The psychotomimetic effects of intravenous delta-9-tetrahydrocannabinol in healthy individuals: Implications for psychosis. Neuropsychopharmacology. 2004;29:15581572. doi: 10.1038/sj.npp.1300496. [DOI] [PubMed] [Google Scholar]
- 25.D’Souza DC, Ranganathan M, Braley G, Gueorguieva R, Zimolo Z, Cooper T, Perry E, Krystal J. Blunted psychotomimetic and amnestic effects of delta-9-tetrahydrocannabinol in frequent users of cannabis. Neuropsychopharmacology. 2008;33:2505–2516. doi: 10.1038/sj.npp.1301643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Kloet ER, Sibug RM, Helmerhorst FM, Schmidt MV. Stress, genes and the mechanism of programming the brain for later life. Neurosci Biobehav Rev. 2005;29:271–281. doi: 10.1016/j.neubiorev.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 27.de Quervain DJ, Aerni A, Schelling G, Roozendaal B. Glucocorticoids and the regulation of memory in health and disease. Front Neuroendocrinol. 2009;30:358–370. doi: 10.1016/j.yfrne.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 28.Di S, Malcher-Lopes R, Halmos KC, Tasker JG. Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: A fast feedback mechanism. J Neurosci. 2003;23:4850–4857. doi: 10.1523/JNEUROSCI.23-12-04850.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Di S, Malcher-Lopes R, Marcheselli VL, Bazan NG, Tasker JG. Rapid glucocorticoid-mediated endocannabinoid release and opposing regulation of glutamate and GABA inputs to hypothalamic magnocellular neurons. Endocrinology. 2005;146:4292–4301. doi: 10.1210/en.2005-0610. [DOI] [PubMed] [Google Scholar]
- 30.Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci U S A. 2002;99:1081910824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diorio D, Viau V, Meaney MJ. The role of the medial prefrontal cortex (cingulate gyrus) in the regulation of hypothalamic-pituitary-adrenal responses to stress. J Neurosci. 1993;13:3839–3847. doi: 10.1523/JNEUROSCI.13-09-03839.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evanson NK, Herman JP. Metabotropic glutamate receptor-mediated signaling dampens the HPA axis response to restraint stress. Physiol Behav. 2015;150:2–7. doi: 10.1016/j.physbeh.2015.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evanson NK, Tasker JG, Hill MN, Hillard CJ, Herman JP. Fast feedback inhibition of the HPA axis by glucocorticoids is mediated by endocannabinoid signaling. Endocrinology. 2010;151:4811–4819. doi: 10.1210/en.2010-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fernandez-Ruiz JJ, Munoz RM, Romero J, Villanua MA, Makriyannis A, Ramos JA. Time course of the effects of different cannabimimetics on prolactin and gonadotrophin secretion: Evidence for the presence of CB1 receptors in hypothalamic structures and their involvement in the effects of cannabimimetics. Biochem Pharmacol. 1997;53:1919–1927. doi: 10.1016/s0006-2952(97)00168-8. [DOI] [PubMed] [Google Scholar]
- 35.Forray MI, Gysling K. Role of noradrenergic projections to the bed nucleus of the stria terminalis in the regulation of the hypothalamic-pituitary-adrenal axis. Brain Res Brain Res Rev. 2004;47:145–160. doi: 10.1016/j.brainresrev.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 36.Fox CL, Towe SL, Stephens RS, Walker DD, Roffman RA. Motives for cannabis use in high-risk adolescent users. Psychol Addict Behav. 2011;25:492–500. doi: 10.1037/a0024331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- 38.Frick KM. Molecular mechanisms underlying the memory-enhancing effects of estradiol. Horm Behav. 2015;74:4–18. doi: 10.1016/j.yhbeh.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ganon-Elazar E, Akirav I. Cannabinoid receptor activation in the basolateral amygdala blocks the effects of stress on the conditioning and extinction of inhibitory avoidance. J Neurosci. 2009;29:11078–11088. doi: 10.1523/JNEUROSCI.1223-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, Cassano T, Morgese MG, Debonnel G, Duranti A, Tontini A, Tarzia G, Mor M, Trezza V, Goldberg SR, Cuomo V, Piomelli D. Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci USA. 2005;102:18620–18625. doi: 10.1073/pnas.0509591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gray JM, Vecchiarelli HA, Morena M, Lee TT, Hermanson DJ, Kim AB, McLaughlin RJ, Hassan KI, Kuhne C, Wotjak CT, Deussing JM, Patel S, Hill MN. Corticotropin-releasing hormone drives anandamide hydrolysis in the amygdala to promote anxiety. J Neurosci. 2015;35:3879–3892. doi: 10.1523/JNEUROSCI.2737-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gray JM, Wilson CD, Lee TT, Pittman QJ, Deussing JM, Hillard CJ, McEwen BS, Schulkin J, Karatsoreos IN, Patel S, Hill MN. Sustained glucocorticoid exposure recruits cortico-limbic CRH signaling to modulate endocannabinoid function. Psychoneuroendocrinology. 2016;66:151–158. doi: 10.1016/j.psyneuen.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gulyas AI, Cravatt BF, Bracey MH, Dinh TP, Piomelli D, Boscia F, Freund TF. Segregation of two endocannabinoid-hydrolyzing enzymes into pre- and postsynaptic compartments in the rat hippocampus, cerebellum and amygdala. Eur J Neurosci. 2004;20:441–458. doi: 10.1111/j.1460-9568.2004.03428.x. [DOI] [PubMed] [Google Scholar]
- 44.Hartmann K, Koenen M, Schauer S, Wittig-Blaich S, Ahmad M, Baschant U, Tuckermann JP. Molecular actions of glucocorticoids in cartilage and bone during health, disease, and steroid therapy. Physiol Rev. 2016;96:409–447. doi: 10.1152/physrev.00011.2015. [DOI] [PubMed] [Google Scholar]
- 45.Herkenham M, Groen BG, Lynn AB, De Costa BR, Richfield EK. Neuronal localization of cannabinoid receptors and second messengers in mutant mouse cerebellum. Brain Res. 1991;552:301–310. doi: 10.1016/0006-8993(91)90096-e. [DOI] [PubMed] [Google Scholar]
- 46.Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: A quantitative in vitro autoradiographic study. J Neurosci. 1991;11:563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herman JP, Figueiredo H, Mueller NK, Ulrich-Lai Y, Ostrander MM, Choi DC, Cullinan WE. Central mechanisms of stress integration: Hierarchical circuitry controlling hypothalamo-pituitary-adrenocortical responsiveness. Front Neuroendocrinol. 2003;24:151–180. doi: 10.1016/j.yfrne.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 48.Herman JP, McKlveen JM, Ghosal S, Kopp B, Wulsin A, Makinson R, Scheimann J, Myers B. Regulation of the hypothalamic-pituitary-adrenocortical stress response. Compr Physiol. 2016;6:603–621. doi: 10.1002/cphy.c150015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herman JP, Ostrander MM, Mueller NK, Figueiredo H. Limbic system mechanisms of stress regulation: Hypothalamo-pituitary-adrenocortical axis. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:1201–1213. doi: 10.1016/j.pnpbp.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 50.Hermann H, Marsicano G, Lutz B. Coexpression of the cannabinoid receptor type 1 with dopamine and serotonin receptors in distinct neuronal subpopulations of the adult mouse forebrain. Neuroscience. 2002;109:451–460. doi: 10.1016/s0306-4522(01)00509-7. [DOI] [PubMed] [Google Scholar]
- 51.Hermanson DJ, Gamble-George JC, Marnett LJ, Patel S. Substrate-selective COX-2 inhibition as a novel strategy for therapeutic endocannabinoid augmentation. Trends Pharmacol Sci. 2014;35:358–367. doi: 10.1016/j.tips.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hill MN, Carrier EJ, Ho WS, Shi L, Patel S, Gorzalka BB, Hillard CJ. Prolonged glucocorticoid treatment decreases cannabinoid CB1 receptor density in the hippocampus. Hippocampus. 2008;18:221–226. doi: 10.1002/hipo.20386. [DOI] [PubMed] [Google Scholar]
- 53.Hill MN, Karatsoreos IN, Hillard CJ, McEwen BS. Rapid elevations in limbic endocannabinoid content by glucocorticoid hormones in vivo. Psychoneuroendocrinology. 2010;35:1333–1338. doi: 10.1016/j.psyneuen.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hill MN, McEwen BS. Endocannabinoids: The silent partner of glucocorticoids in the synapse. Proc Natl Acad Sci U S A. 2009;106:4579–4580. doi: 10.1073/pnas.0901519106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hill MN, McLaughlin RJ, Bingham B, Shrestha L, Lee TT, Gray JM, Hillard CJ, Gorzalka BB, Viau V. Endogenous cannabinoid signaling is essential for stress adaptation. Proc Natl Acad Sci U S A. 2010;107:9406–9411. doi: 10.1073/pnas.0914661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hill MN, McLaughlin RJ, Morrish AC, Viau V, Floresco SB, Hillard CJ, Gorzalka BB. Suppression of amygdalar endocannabinoid signaling by stress contributes to activation of the hypothalamic-pituitary-adrenal axis. Neuropsychopharmacology. 2009;34:2733–2745. doi: 10.1038/npp.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hill MN, McLaughlin RJ, Pan B, Fitzgerald ML, Roberts CJ, Lee TT, Karatsoreos IN, Mackie K, Viau V, Pickel VM, McEwen BS, Liu QS, Gorzalka BB, Hillard CJ. Recruitment of prefrontal cortical endocannabinoid signaling by glucocorticoids contributes to termination of the stress response. J Neurosci. 2011;31:10506–10515. doi: 10.1523/JNEUROSCI.0496-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hillard C, Liu Q-S, Liu X, Pan B, Roberts C, Shi L. Cannabinoids, endocannabinoids and stress. In: DiMarzo V, editor. Cannabinoids. Hoboken, NJ: Wiley-Blackwell; 2014. [Google Scholar]
- 59.Hillard CJ. Biochemistry and pharmacology of the endocannabinoids arachidonylethanolamide and 2-arachidonylglycerol. Prostaglandins Other Lipid Mediat. 2000;61:3–18. doi: 10.1016/s0090-6980(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 60.Hillard CJ. The endocannabinoid signaling system in the CNS: A primer. Int Rev Neurobiol. 2015;125:1–47. doi: 10.1016/bs.irn.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hillard CJ, Manna S, Greenberg MJ, DiCamelli R, Ross RA, Stevenson LA, Murphy V, Pertwee RG, Campbell WB. Synthesis and characterization of potent and selective agonists of the neuronal cannabinoid receptor (CB1) J Pharmacol Exp Ther. 1999;289:1427–1433. [PubMed] [Google Scholar]
- 62.Hong S, Zheng G, Wiley JW. Epigenetic regulation of genes that modulate chronic stress-induced visceral pain in the peripheral nervous system. Gastroenterology. 2015;148:148–157 e147. doi: 10.1053/j.gastro.2014.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hong S, Zheng G, Wu X, Snider NT, Owyang C, Wiley JW. Corticosterone mediates reciprocal changes in CB 1 and TRPV1 receptors in primary sensory neurons in the chronically stressed rat. Gastroenterology. 2011;140:627–637. doi: 10.1053/j.gastro.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Howlett AC. Cannabinoid inhibition of adenylate cyclase. Biochemistry of the response in neuroblastoma cell membranes. Mol Pharmacol. 1985;27:429–436. [PubMed] [Google Scholar]
- 65.Huang GZ, Woolley CS. Estradiol acutely suppresses inhibition in the hippocampus through a sex-specific endocannabinoid and mGluR-dependent mechanism. Neuron. 2012;74:801–808. doi: 10.1016/j.neuron.2012.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huizink AC, Mulder EJ. Maternal smoking, drinking or cannabis use during pregnancy and neurobehavioral and cognitive functioning in human offspring. Neurosci Biobehav Rev. 2006;30:24–41. doi: 10.1016/j.neubiorev.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 67.Hyman SM, Sinha R. Stress-related factors in cannabis use and misuse: Implications for prevention and treatment. J Subst Abuse Treat. 2009;36:400–413. doi: 10.1016/j.jsat.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Idris AI, Sophocleous A, Landao-Bassonga E, Canals M, Milligan G, Baker D, van’t Hof RJ, Ralston SH. Cannabinoid receptor type 1 protects against age-related osteoporosis by regulating osteoblast and adipocyte differentiation in marrow stromal cells. Cell Metab. 2009;10:139147. doi: 10.1016/j.cmet.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 69.Idris AI, van ‘t Hof RJ, Greig IR, Ridge SA, Baker D, Ross RA, Ralston SH. Regulation of bone mass, bone loss and osteoclast activity by cannabinoid receptors. Nat Med. 2005;11:774–779. doi: 10.1038/nm1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jin XC, Lu YF, Yang XF, Ma L, Li BM. Glucocorticoid receptors in the basolateral nucleus of amygdala are required for postreactivation reconsolidation of auditory fear memory. Eur J Neurosci. 2007;25:3702–3712. doi: 10.1111/j.1460-9568.2007.05621.x. [DOI] [PubMed] [Google Scholar]
- 71.Joels M, Sarabdjitsingh RA, Karst H. Unraveling the time domains of corticosteroid hormone influences on brain activity: Rapid, slow, and chronic modes. Pharmacol Rev. 2012;64:901–938. doi: 10.1124/pr.112.005892. [DOI] [PubMed] [Google Scholar]
- 72.Kamprath K, Marsicano G, Tang J, Monory K, Bisogno T, Di Marzo V, Lutz B, Wotjak CT. Cannabinoid CB1 receptor mediates fear extinction via habituation-like processes. J Neurosci. 2006;26:6677–6686. doi: 10.1523/JNEUROSCI.0153-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kamprath K, Plendl W, Marsicano G, Deussing JM, Wurst W, Lutz B, Wotjak CT. Endocannabinoids mediate acute fear adaptation via glutamatergic neurons independently of corticotropin-releasing hormone signaling. Genes Brain Behav. 2009;8:203–211. doi: 10.1111/j.1601-183X.2008.00463.x. [DOI] [PubMed] [Google Scholar]
- 74.Kamprath K, Romo-Parra H, Haring M, Gaburro S, Doengi M, Lutz B, Pape HC. Short-term adaptation of conditioned fear responses through endocannabinoid signaling in the central amygdala. Neuropsychopharmacology. 2011;36:652–663. doi: 10.1038/npp.2010.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kano M. Control of synaptic function by endocannabinoid-mediated retrograde signaling. Proc Jpn Acad Ser B Phys Biol Sci. 2014;90:235–250. doi: 10.2183/pjab.90.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kaplan HB, Martin SS, Johnson RJ, Robbins CA. Escalation of marijuana use: Application of a general theory of deviant behavior. J Health Soc Behav. 1986;27:44–61. [PubMed] [Google Scholar]
- 77.Karst H, Berger S, Erdmann G, Schutz G, Joels M. Metaplasticity of amygdalar responses to the stress hormone corticosterone. Proc Natl Acad Sci U S A. 2010;107:14449–14454. doi: 10.1073/pnas.0914381107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Karst H, Berger S, Turiault M, Tronche F, Schutz G, Joels M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci USA. 2005;102:19204–19207. doi: 10.1073/pnas.0507572102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Katona I, Rancz EA, Acsady L, Ledent C, Mackie K, Hajos N, Freund TF. Distribution of CB1 cannabinoid receptors in the amygdala and their role in the control of GABAergic transmission. J Neurosci. 2001;21:9506–9518. doi: 10.1523/JNEUROSCI.21-23-09506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Katona I, Sperlagh B, Sik A, Kafalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci. 1999;19:4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kearn CS, Greenberg MJ, DiCamelli R, Kurzawa K, Hillard CJ. Relationships between ligand affinities for the cerebellar cannabinoid receptor CB1 and the induction of GDP/GTP exchange. J Neurochem. 1999;72:2379–2387. doi: 10.1046/j.1471-4159.1999.0722379.x. [DOI] [PubMed] [Google Scholar]
- 82.King GR, Ernst T, Deng W, Stenger A, Gonzales RM, Nakama H, Chang L. Altered brain activation during visuomotor integration in chronic active cannabis users: Relationship to cortisol levels. J Neurosci. 2011;31:17923–17931. doi: 10.1523/JNEUROSCI.4148-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kleinloog D, Liem-Moolenaar M, Jacobs G, Klaassen E, de Kam M, Hijman R, van Gerven J. Does olanzapine inhibit the psychomimetic effects of Delta(9)-tetrahydrocannabinol? J Psychopharmacol. 2012;26:1307–1316. doi: 10.1177/0269881112446534. [DOI] [PubMed] [Google Scholar]
- 84.Klumpers LE, Cole DM, Khalili-Mahani N, Soeter RP, Te Beek ET, Rombouts SA, van Gerven JM. Manipulating brain connectivity with delta(9)-tetrahydrocannabinol: A pharmacological resting state FMRI study. Neuroimage. 2012;63:1701–1711. doi: 10.1016/j.neuroimage.2012.07.051. [DOI] [PubMed] [Google Scholar]
- 85.Ko JY, Wu RW, Kuo SJ, Chen MW, Yeh DW, Ke HC, Wu SL, Wang FS. Cannabinoid receptor 1 mediates glucocorticoid-induced bone loss in rats by perturbing bone mineral acquisition and marrow adipogenesis. Arthritis Rheum. 2012;64:1204–1214. doi: 10.1002/art.33457. [DOI] [PubMed] [Google Scholar]
- 86.Konturek PC, Brzozowski T, Konturek SJ. Stress and the gut: Pathophysiology, clinical consequences, diagnostic approach and treatment options. J Physiol Pharmacol. 2011;62:591–599. [PubMed] [Google Scholar]
- 87.Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- 88.Kulkarni-Narla A, Brown DR. Localization of CB1-cannabinoid receptor immunoreactivity in the porcine enteric nervous system. Cell Tissue Res. 2000;302:73–80. doi: 10.1007/s004410000261. [DOI] [PubMed] [Google Scholar]
- 89.Lightman SL. The neuroendocrinology of stress: A never ending story. J Neuroendocrinol. 2008;20:880–884. doi: 10.1111/j.1365-2826.2008.01711.x. [DOI] [PubMed] [Google Scholar]
- 90.Lowette K, Tack J, Vanden Berghe P. Role of corticosterone in the murine enteric nervous system during fasting. Am J Physiol Gastrointest Liver Physiol. 2014;307:G905–913. doi: 10.1152/ajpgi.00233.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mackie K. Cannabinoid receptors: Where they are and what they do. J Neuroendocrinol. 2008;20(Suppl 1):10–14. doi: 10.1111/j.1365-2826.2008.01671.x. [DOI] [PubMed] [Google Scholar]
- 92.Mackie K, Hille B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci. 1992;89:3825–3829. doi: 10.1073/pnas.89.9.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mailleux P, Vanderhaeghen JJ. Glucocorticoid regulation of cannabinoid receptor messenger RNA levels in the rat caudate-putamen. An in situ hybridization study. Neurosci Lett. 1993;156:51–53. doi: 10.1016/0304-3940(93)90437-p. [DOI] [PubMed] [Google Scholar]
- 94.Malcher-Lopes R, Franco A, Tasker JG. Glucocorticoids shift arachidonic acid metabolism toward endocannabinoid synthesis: A nongenomic anti-inflammatory switch. Eur J Pharmacol. 2008;583:322–339. doi: 10.1016/j.ejphar.2007.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Malfitano AM, Basu S, Maresz K, Bifulco M, Dittel BN. What we know and do not know about the cannabinoid receptor 2 (CB2) Semin Immunol. 2014;26:369–379. doi: 10.1016/j.smim.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Manzanares J, Corchero J, Fuentes JA. Opioid and cannabinoid receptor-mediated regulation of the increase in adrenocorticotropin hormone and corticosterone plasma concentrations induced by central administration of delta(9)-tetrahydrocannabinol in rats. Brain Res. 1999;839:173–179. doi: 10.1016/s0006-8993(99)01756-4. [DOI] [PubMed] [Google Scholar]
- 97.Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, Bodor AL, Muccioli GG, Hu SS, Woodruff G, Fung S, Lafourcade M, Alexander JP, Long JZ, Li W, Xu C, Moller T, Mackie K, Manzoni OJ, Cravatt BF, Stella N. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci. 2010;13:951–957. doi: 10.1038/nn.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Matthews L, Berry A, Ohanian V, Ohanian J, Garside H, Ray D. Caveolin mediates rapid glucocorticoid effects and couples glucocorticoid action to the antiproliferative program. Mol Endocrinol. 2008;22:1320–1330. doi: 10.1210/me.2007-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.McEwen BS. Steroid hormone actions on the brain: When is the genome involved? Horm Behav. 1994;28:396–405. doi: 10.1006/hbeh.1994.1036. [DOI] [PubMed] [Google Scholar]
- 100.McLaughlin RJ, Hill MN, Bambico FR, Stuhr KL, Gobbi G, Hillard CJ, Gorzalka BB. Prefrontal cortical anandamide signaling coordinates coping responses to stress through a serotonergic pathway. Eur Neuropsychopharmacol. 2012;22:664–671. doi: 10.1016/j.euroneuro.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.McLaughlin RJ, Hill MN, Gorzalka BB. Monoaminergic neurotransmission contributes to cannabinoid-induced activation of the hypothalamic-pituitary-adrenal axis. Eur J Pharmacol. 2009;624:71–76. doi: 10.1016/j.ejphar.2009.09.055. [DOI] [PubMed] [Google Scholar]
- 102.McReynolds JR, Doncheck EM, Vranjkovic O, Ganzman GS, Baker DA, Hillard CJ, Mantsch JR. CB1 receptor antagonism blocks stress-potentiated reinstatement of cocaine seeking in rats. Psychopharmacology(Berl) 2016;233:99–109. doi: 10.1007/s00213-015-4092-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.McReynolds JR, Pena DF, Blacktop JM, Mantsch JR. Neurobiological mechanisms underlying relapse to cocaine use: Contributions of CRF and noradrenergic systems and regulation by glucocorticoids. Stress. 2014;17:22–38. doi: 10.3109/10253890.2013.872617. [DOI] [PubMed] [Google Scholar]
- 104.Nahar J, Haam J, Chen C, Jiang Z, Glatzer NR, Muglia LJ, Dohanich GP, Herman JP, Tasker JG. Rapid nongenomic glucocorticoid actions in male mouse hypothalamic neuroendocrine cells are dependent on the nuclear glucocorticoid receptor. Endocrinology. 2015;156:2831–2842. doi: 10.1210/en.2015-1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Newsom RJ, Osterlund C, Masini CV, Day HE, Spencer RL, Campeau S. Cannabinoid receptor type 1 antagonism significantly modulates basal and loud noise induced neural and hypothalamic-pituitary-adrenal axis responses in male Sprague-Dawley rats. Neuroscience. 2012;204:64–73. doi: 10.1016/j.neuroscience.2011.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Niederhoffer N, Hansen HH, Fernandez-Ruiz JJ, Szabo B. Effects of cannabinoids on adrenaline release from adrenal medullary cells. Br J Pharmacol. 2001;134:1319–1327. doi: 10.1038/sj.bjp.0704359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nieman LK. Cushing’s syndrome: Update on signs, symptoms and biochemical screening. Eur J Endocrinol. 2015;173:M33–38. doi: 10.1530/EJE-15-0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MC, Ward AM, Hahn YK, Lichtman AH, Conti B, Cravatt BF. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science. 2011;334:809–813. doi: 10.1126/science.1209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Oropeza VC, Mackie K, Van Bockstaele EJ. Cannabinoid receptors are localized to noradrenergic axon terminals in the rat frontal cortex. Brain Res. 2007;1127:36–44. doi: 10.1016/j.brainres.2006.09.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pagotto U, Marsicano G, Fezza F, Theodoropoulou M, Grubler Y, Stalla J, Arzberger T, Milone A, Losa M, Di Marzo V, Lutz B, Stalla GK. Normal human pituitary gland and pituitary adenomas express cannabinoid receptor type 1 and synthesize endogenous cannabinoids: First evidence for a direct role of cannabinoids on hormone modulation at the human pituitary level. J Clin Endocrinol Metab. 2001;86:2687–2696. doi: 10.1210/jcem.86.6.7565. [DOI] [PubMed] [Google Scholar]
- 111.Pan B, Wang W, Long JZ, Sun D, Hillard CJ, Cravatt BF, Liu QS. Blockade of 2-arachidonoylglycerol hydrolysis by selective monoacylglycerol lipase inhibitor 4-nitrophenyl 4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate (JZL184) Enhances retrograde endocannabinoid signaling. J Pharmacol Exp Ther. 2009;331:591–597. doi: 10.1124/jpet.109.158162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Patel S, Carrier EJ, Ho WS, Rademacher DJ, Cunningham S, Reddy DS, Falck JR, Cravatt BF, Hillard CJ. The postmortal accumulation of brain N-arachidonylethanolamine (anandamide) is dependent upon fatty acid amide hydrolase activity. J Lipid Res. 2005;46:342–349. doi: 10.1194/jlr.M400377-JLR200. [DOI] [PubMed] [Google Scholar]
- 113.Patel S, Roelke CT, Rademacher DJ, Cullinan WE, Hillard CJ. Endocannabinoid signaling negatively modulates stress-induced activation of the hypothalamic-pituitary-adrenal axis. Endocrinology. 2004;145:5431–5438. doi: 10.1210/en.2004-0638. [DOI] [PubMed] [Google Scholar]
- 114.Patel S, Roelke CT, Rademacher DJ, Hillard CJ. Inhibition of restraint stress-induced neural and behavioural activation by endogenous cannabinoid signalling. Eur J Neurosci. 2005;21:1057–1069. doi: 10.1111/j.1460-9568.2005.03916.x. [DOI] [PubMed] [Google Scholar]
- 115.Pereira MJ, Palming J, Svensson MK, Rizell M, Dalenback J, Hammar M, Fall T, Sidibeh CO, Svensson PA, Eriksson JW. FKBP5 expression in human adipose tissue increases following dexamethasone exposure and is associated with insulin resistance. Metabolism. 2014;63:1198–1208. doi: 10.1016/j.metabol.2014.05.015. [DOI] [PubMed] [Google Scholar]
- 116.Pertwee RG. Tolerance to the effect of delta1-tetrahydrocannabinol on corticosterone levels in mouse plasma produced by repeated administration of cannabis extract or delta1-tetrahydrocannabinol. Br J Pharmacol. 1974;51:391–397. doi: 10.1111/j.1476-5381.1974.tb10674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pertwee RG. Ligands that target cannabinoid receptors in the brain: From THC to anandamide and beyond. Addict Biol. 2008;13:147–159. doi: 10.1111/j.1369-1600.2008.00108.x. [DOI] [PubMed] [Google Scholar]
- 118.Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, Mechoulam R, Ross RA. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB and CB. Pharmacol Rev. 2010;62:588–631. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pickel VM, Shobin ET, Lane DA, Mackie K. Cannabinoid-1 receptors in the mouse ventral pallidum are targeted to axonal profiles expressing functionally opposed opioid peptides and contacting N-acylphosphatidylethanolamine-hydrolyzing phospholipase D terminals. Neuroscience. 2012;227C:10–21. doi: 10.1016/j.neuroscience.2012.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Qin Z, Zhou X, Pandey NR, Vecchiarelli HA, Stewart CA, Zhang X, Lagace DC, Brunel JM, Beique JC, Stewart AF, Hill MN, Chen HH. Chronic stress induces anxiety via an amygdalar intracellular cascade that impairs endocannabinoid signaling. Neuron. 2015;85:1319–1331. doi: 10.1016/j.neuron.2015.02.015. [DOI] [PubMed] [Google Scholar]
- 121.Rademacher DJ, Hillard CJ. Interactions between endocannabinoids and stress-induced decreased sensitivity to natural reward. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:633–641. doi: 10.1016/j.pnpbp.2006.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rademacher DJ, Meier SE, Shi L, Ho W-SV, Jarrahian A, Hillard CJ. Effects of acute and repeated restraint stress on endocannabinoid content in the amygdala, ventral striatum and medial prefrontal cortex in mice. Neuropharmacol. 2008;54:108–116. doi: 10.1016/j.neuropharm.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 123.Ranganathan M, Braley G, Pittman B, Cooper T, Perry E, Krystal J, D’Souza DC. The effects of cannabinoids on serum cortisol and prolactin in humans. Psychopharmacology (Berl) 2009;203:737–744. doi: 10.1007/s00213-008-1422-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rauch A, Seitz S, Baschant U, Schilling AF, Illing A, Stride B, Kirilov M, Mandic V, Takacz A, Schmidt-Ullrich R, Ostermay S, Schinke T, Spanbroek R, Zaiss MM, Angel PE, Lerner UH, David JP, Reichardt HM, Amling M, Schutz G, Tuckermann JP. Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor. Cell Metab. 2010;11:517–531. doi: 10.1016/j.cmet.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 125.Reich CG, Taylor ME, McCarthy MM. Differential effects of chronic unpredictable stress on hippocampal CB1 receptors in male and female rats. Behav Brain Res. 2009;203:264–269. doi: 10.1016/j.bbr.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rice W, Shannon JM, Burton F, Fiedeldey D. Expression of a brain-type cannabinoid receptor (CB1) in alveolar Type II cells in the lung: Regulation by hydrocortisone. Eur J Pharmacol. 1997;327:227–232. doi: 10.1016/s0014-2999(97)89665-3. [DOI] [PubMed] [Google Scholar]
- 127.Roberts CJ, Stuhr KL, Hutz MJ, Raff H, Hillard CJ. Endocannabinoid signaling in hypothalamic-pituitary-adrenocortical axis recovery following stress: Effects of indirect agonists and comparison of male and female mice. Pharmacol Biochem Behav. 2014;117:17–24. doi: 10.1016/j.pbb.2013.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Robinson SA, Loiacono RE, Christopoulos A, Sexton PM, Malone DT. The effect of social isolation on rat brain expression of genes associated with endocannabinoid signalling. Brain Res. 2010;1343:153–167. doi: 10.1016/j.brainres.2010.04.031. [DOI] [PubMed] [Google Scholar]
- 129.Rodriguez de Fonseca F, Cebeira M, Martin M, Fernandez-Ruiz JJ. Cannabinoid receptors in rat brain areas: Sexual differences, fluctuations during estrous cycle and changes after gonadectomy and sex steroid replacement. Life Sci. 1994;54:159–170. doi: 10.1016/0024-3205(94)00585-0. [DOI] [PubMed] [Google Scholar]
- 130.Roozendaal B, de Quervain DJ, Ferry B, Setlow B, McGaugh JL. Basolateral amygdala-nucleus accumbens interactions in mediating glucocorticoid enhancement of memory consolidation. J Neurosci. 2001;21:2518–2525. doi: 10.1523/JNEUROSCI.21-07-02518.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Roozendaal B, Schelling G, McGaugh JL. Corticotropin-releasing factor in the basolateral amygdala enhances memory consolidation via an interaction with the beta-adrenoceptor-cAMP pathway: Dependence on glucocorticoid receptor activation. J Neurosci. 2008;28:6642–6651. doi: 10.1523/JNEUROSCI.1336-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Roozendaal B, Williams CL, McGaugh JL. Glucocorticoid receptor activation in the rat nucleus of the solitary tract facilitates memory consolidation: Involvement of the basolateral amygdala. Eur J Neurosci. 1999;11:1317–1323. doi: 10.1046/j.1460-9568.1999.00537.x. [DOI] [PubMed] [Google Scholar]
- 133.Sadie-Van Gijsen H, Crowther NJ, Hough FS, Ferris WF. The interrelationship between bone and fat: From cellular see-saw to endocrine reciprocity. Cell Mol Life Sci. 2013;70:2331–2349. doi: 10.1007/s00018-012-1211-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Samir SM, Malek HA. Effect of cannabinoid receptors 1 modulation on osteoporosis in a rat model of different ages. J Physiol Pharmacol. 2014;65:687–694. [PubMed] [Google Scholar]
- 135.Shen M, Piser TM, Seybold VS, Thayer SA. Cannabinoid receptor agonists inhibit glutamatergic synaptic transmission in rat hippocampal cultures. J Neurosci. 1996;16:4322–4334. doi: 10.1523/JNEUROSCI.16-14-04322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Solomon MB, Loftspring M, de Kloet AD, Ghosal S, Jankord R, Flak JN, Wulsin AC, Krause EG, Zhang R, Rice T, McKlveen J, Myers B, Tasker JG, Herman JP. Neuroendocrine function After hypothalamic depletion of glucocorticoid receptors in male and female mice. Endocrinology. 2015;156:2843–2853. doi: 10.1210/en.2015-1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Somaini L, Manfredini M, Amore M, Zaimovic A, Raggi MA, Leonardi C, Gerra ML, Donnini C, Gerra G. Psychobiological responses to unpleasant emotions in cannabis users. Eur Arch Psychiatry Clin Neurosci. 2012;262:47–57. doi: 10.1007/s00406-011-0223-5. [DOI] [PubMed] [Google Scholar]
- 138.Steiner MA, Wotjak CT. Role of the endocannabinoid system in regulation of the hypothalamic-pituitary-adrenocortical axis. Prog Brain Res. 2008;170:397–432. doi: 10.1016/S0079-6123(08)00433-0. [DOI] [PubMed] [Google Scholar]
- 139.Sumislawski JJ, Ramikie TS, Patel S. Reversible gating of endocannabinoid plasticity in the amygdala by chronic stress: A potential role for monoacylglycerol lipase inhibition in the prevention of stress-induced behavioral adaptation. Neuropsychopharmacology. 2011;36:2750–2761. doi: 10.1038/npp.2011.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Szallasi A, Di Marzo V. New perspectives on enigmatic vanilloid receptors. Trends Neurosci. 2000;23:491–497. doi: 10.1016/s0166-2236(00)01630-1. [DOI] [PubMed] [Google Scholar]
- 141.Tabatadze N, Huang G, May RM, Jain A, Woolley CS. Sex differences in molecular signaling at inhibitory synapses in the hippocampus. J Neurosci. 2015;35:11252–11265. doi: 10.1523/JNEUROSCI.1067-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tasker JG, Herman JP. Mechanisms of rapid glucocorticoid feedback inhibition of the hypothalamic-pituitary-adrenal axis. Stress. 2011;14:398–406. doi: 10.3109/10253890.2011.586446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tsou K, Nogueron MI, Muthian S, Sanudo-Pena MC, Hillard CJ, Deutsch DG, Walker JM. Fatty acid amide hydrolase is located preferentially in large neurons in the rat central nervous system as revealed by immunohistochemistry. Neurosci Lett. 1998;254:137–140. doi: 10.1016/s0304-3940(98)00700-9. [DOI] [PubMed] [Google Scholar]
- 144.Ueda N, Tsuboi K, Uyama T. N-acylethanolamine metabolism with special reference to N-acylethanolamine-hydrolyzing acid amidase (NAAA) Prog Lipid Res. 2010;49:299–315. doi: 10.1016/j.plipres.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 145.van Leeuwen AP, Verhulst FC, Reijneveld SA, Vollebergh WA, Ormel J, Huizink AC. Can the gateway hypothesis, the common liability model and/or, the route of administration model predict initiation of cannabis use during adolescence? A survival analysis-the TRAILS study. J Adolesc Health. 2011;48:73–78. doi: 10.1016/j.jadohealth.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 146.Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, Stella N, Makriyannis A, Piomelli D, Davison JS, Marnett LJ, Di Marzo V, Pittman QJ, Patel KD, Sharkey KA. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310:329–332. doi: 10.1126/science.1115740. [DOI] [PubMed] [Google Scholar]
- 147.Vaughn LK, Mantsch JR, Vranjkovic O, Stroh G, Lacourt M, Kreutter M, Hillard CJ. Cannabinoid receptor involvement in stress-induced cocaine reinstatement: Potential interaction with noradrenergic pathways. Neuroscience. 2012;204:117–124. doi: 10.1016/j.neuroscience.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Verkuyl JM, Karst H, Joels M. GABAergic transmission in the rat paraventricular nucleus of the hypothalamus is suppressed by corticosterone and stress. Eur J Neurosci. 2005;21:113–121. doi: 10.1111/j.1460-9568.2004.03846.x. [DOI] [PubMed] [Google Scholar]
- 149.Wade MR, Degroot A, Nomikos GG. Cannabinoid CB1 receptor antagonism modulates plasma corticosterone in rodents. Eur J Pharmacol. 2006;551:162–167. doi: 10.1016/j.ejphar.2006.08.083. [DOI] [PubMed] [Google Scholar]
- 150.Waleh NS, Cravatt BF, Apte-Deshpande A, Terao A, Kilduff TS. Transcriptional regulation of the mouse fatty acid amide hydrolase gene. Gene. 2002;291:203–210. doi: 10.1016/s0378-1119(02)00598-x. [DOI] [PubMed] [Google Scholar]
- 151.Wang J, Shen RY, Haj-Dahmane S. Endocannabinoids mediate the glucocorticoid-induced inhibition of excitatory synaptic transmission to dorsal raphe serotonin neurons. J Physiol. 2012;590:5795–5808. doi: 10.1113/jphysiol.2012.238659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wang M, Hill MN, Zhang L, Gorzalka BB, Hillard CJ, Alger BE. Acute restraint stress enhances hippocampal endocannabinoid function via glucocorticoid receptor activation. J Psychopharmacol. 2012;26:56–70. doi: 10.1177/0269881111409606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wang S, Lim G, Mao J, Sung B, Yang L. Central glucocorticoid receptors regulate the upregulation of spinal cannabinoid-1 receptors after peripheral nerve injury in rats. Pain. 2007;131:96–105. doi: 10.1016/j.pain.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 154.Watts AG. The impact of physiological stimuli on the expression of corticotropin-releasing hormone (CRH) and other neuropeptide genes. Front Neuroendocrinol. 1996;17:281–326. doi: 10.1006/frne.1996.0008. [DOI] [PubMed] [Google Scholar]
- 155.Wittmann G, Deli L, Kallo I, Hrabovszky E, Watanabe M, Liposits Z, Fekete C. Distribution of type 1 cannabinoid receptor (CB1)-immunoreactive axons in the mouse hypothalamus. J Comp Neurol. 2007;503:270–279. doi: 10.1002/cne.21383. [DOI] [PubMed] [Google Scholar]
- 156.Wu RW, Lin TP, Ko JY, Yeh DW, Chen MW, Ke HC, Wu SL, Wang FS. Cannabinoid receptor 1 regulates ERK and GSK-3beta-dependent glucocorticoid inhibition of osteoblast differentiation in murine MC3T3-E1 cells. Bone. 2011;49:1255–1263. doi: 10.1016/j.bone.2011.08.022. [DOI] [PubMed] [Google Scholar]
- 157.Yoshida T, Fukaya M, Uchigashima M, Miura E, Kamiya H, Kano M, Watanabe M. Localization of diacylglycerol lipase-alpha around postsynaptic spine suggests close proximity between production site of an endocannabinoid, 2-arachidonoyl-glycerol, and presynaptic cannabinoid CB1 receptor. J Neurosci. 2006;26:4740–4751. doi: 10.1523/JNEUROSCI.0054-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zheng Y, Wang XL, Mo FF, Li M. Dexamethasone alleviates motion sickness in rats in part by enhancing the endocannabinoid system. Eur J Pharmacol. 2014;727:99–105. doi: 10.1016/j.ejphar.2014.01.047. [DOI] [PubMed] [Google Scholar]
- 159.Zias J, Stark H, Sellgman J, Levy R, Werker E, Breuer A, Mechoulam R. Early medical use of cannabis. Nature. 1993;363:215. doi: 10.1038/363215a0. [DOI] [PubMed] [Google Scholar]
- 160.Ziegler CG, Mohn C, Lamounier-Zepter V, Rettori V, Bornstein SR, Krug AW, Ehrhart-Bornstein M. Expression and function of endocannabinoid receptors in the human adrenal cortex. Horm Metab Res. 2010;42:88–92. doi: 10.1055/s-0029-1241860. [DOI] [PubMed] [Google Scholar]
- 161.Zoppi S, Madrigal JL, Perez-Nievas BG, Marin-Jimenez I, Caso JR, Alou L, Garcia-Bueno B, Colon A, Manzanares J, Gomez-Lus ML, Menchen L, Leza JC. Endogenous cannabinoid system regulates intestinal barrier function in vivo through cannabinoid type 1 receptor activation. Am J Physiol Gastrointest Liver Physiol. 2012;302:G565–571. doi: 10.1152/ajpgi.00158.2011. [DOI] [PubMed] [Google Scholar]